Revealing the Nature of Chemical Bonding in an ALn2Ag3Te5-Type Alkaline-Metal (A) Lanthanide (Ln) Silver Telluride


 and
and
Abstract
:
1. Introduction
2. Results and Discussion
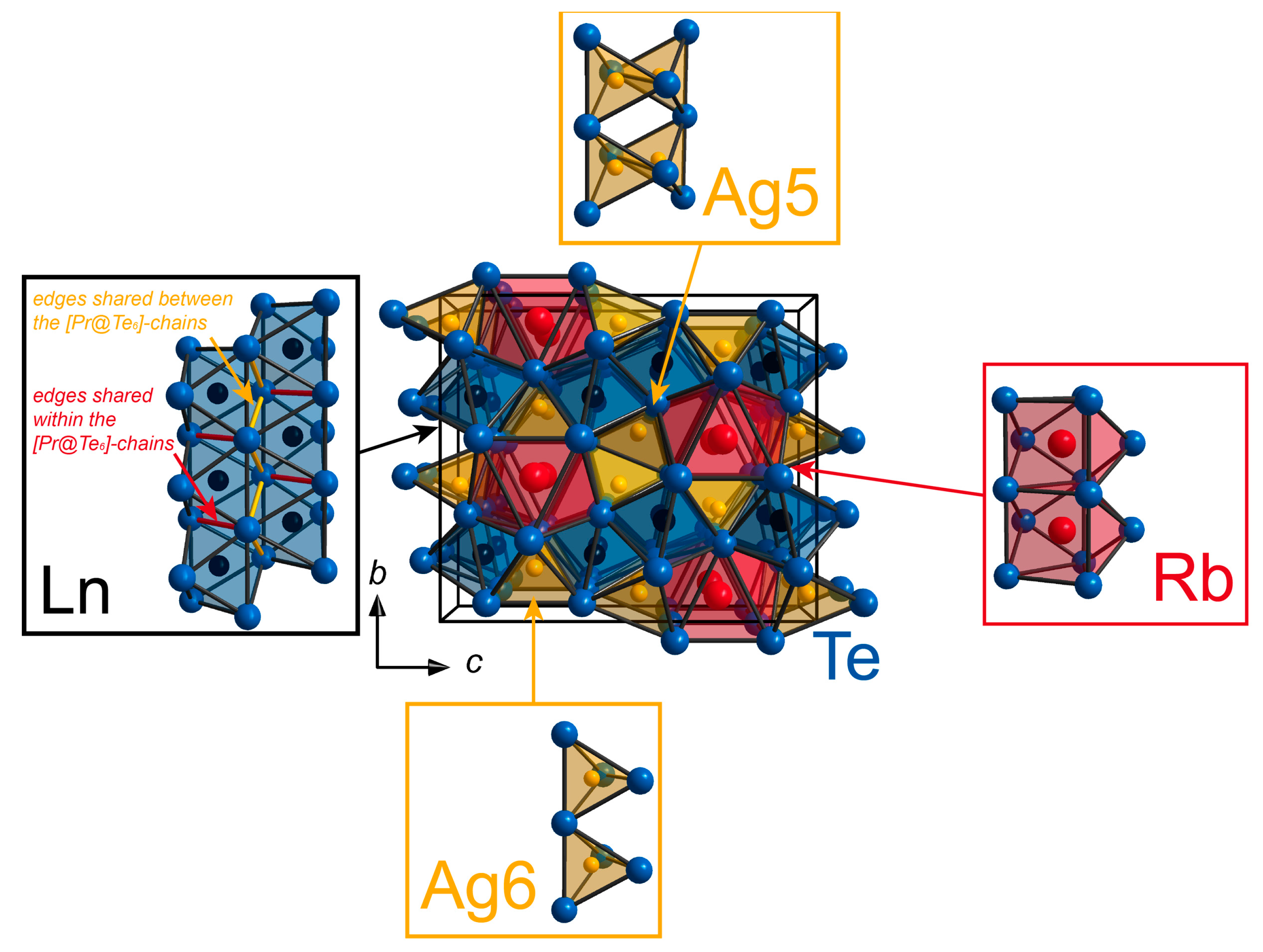
2.1. Structural Details
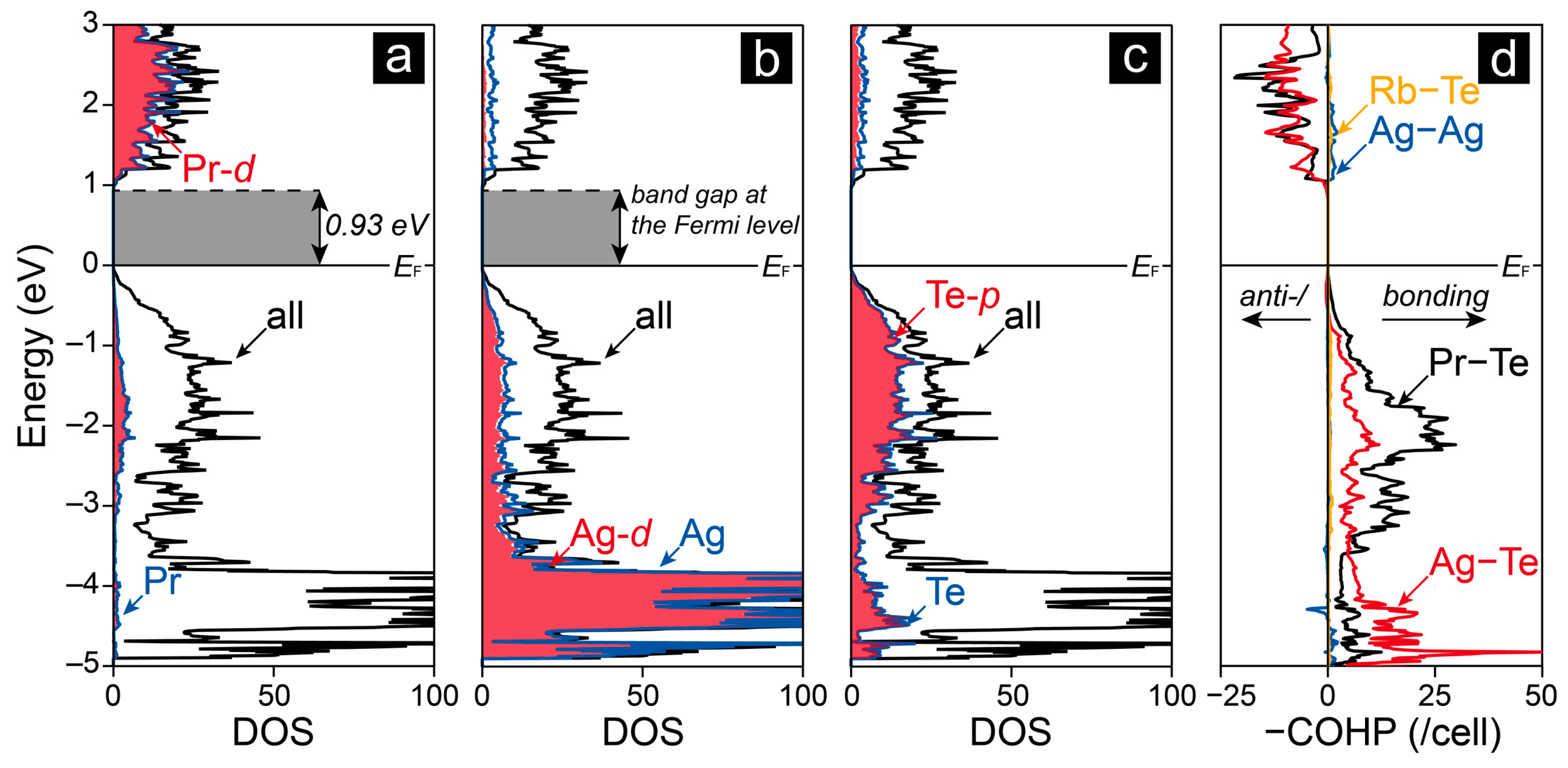
2.2. Electronic Structure and Chemical Bonding Analysis
3. Materials and Methods
3.1. Syntheses
3.2. X-ray Diffraction Studies and Determinations of the Crystal Structures
3.3. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gladisch, F.C.; Steinberg, S. Revealing Tendencies in the Electronic Structures of Polar Intermetallic Compounds. Crystals 2018, 8, 80. [Google Scholar] [CrossRef]
- Miller, G.J. The “Coloring Problem” in Solids: How It Affects Structure, Composition and Properties. Eur. J. Inorg. Chem. 1998, 1998, 523–536. [Google Scholar] [CrossRef]
- Gautier, R.; Zhang, X.; Hu, L.; Yu, L.; Lin, Y.; Sunde, T.O.L.; Chon, D.; Poeppelmeier, K.R.; Zunger, A. Prediction and accelerated laboratory discovery of previously unknown 18-electron ABX compounds. Nat. Chem. 2015, 7, 308–316. [Google Scholar] [CrossRef] [Green Version]
- Curtarolo, S.; Hart, G.L.W.; Nardelli, M.B.; Mingo, N.; Sanvito, S.; Levy, O. The high-throughput highway to computational materials design. Nat. Mater. 2013, 12, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Nesper, R. Bonding Patterns in Intermetallic Compounds. Angew. Chem. Int. Ed. Engl. 1991, 30, 789–817. [Google Scholar] [CrossRef]
- Jones, H. The phase boundaries in binary alloys, part 2: The theory of the α, β phase boundaries. Proc. Phys. Soc. 1937, 49, 250–257. [Google Scholar] [CrossRef]
- Massalski, T.B.; Mizutani, U. Electronic Structure of Hume-Rothery Phases. Prog. Mater. Sci. 1978, 22, 151–262. [Google Scholar] [CrossRef]
- Mizutani, U.; Sato, H. The Physics of the Hume-Rothery Electron Concentration Rule. Crystals 2017, 7, 9. [Google Scholar] [CrossRef]
- Zintl, E. Intermetallische Verbindungen. Angew. Chem. 1939, 52, 1–6. [Google Scholar] [CrossRef]
- Schäfer, H.; Eisenmann, B.; Müller, W. Zintl Phases: Transitions between Metallic and Ionic Bonding. Angew. Chem. Int. Ed. 1973, 12, 694–712. [Google Scholar] [CrossRef]
- Miller, G.J.; Schmidt, M.W.; Wang, F.; You, T.-S. Quantitative Advances in the Zintl-Klemm Formalism. In Structure and Bonding; Fässler, T., Ed.; Springer Publishers: Berlin/Heidelberg, Germany, 2011; Volume 139, pp. 1–55. [Google Scholar]
- Corbett, J.D. Exploratory Synthesis: The Fascinating and Diverse Chemistry of Polar Intermetallic Phases. Inorg. Chem. 2010, 49, 13–28. [Google Scholar] [CrossRef] [PubMed]
- Toberer, E.S.; May, A.F.; Snyder, G.J. Zintl Chemistry for Designing High Efficiency Thermoelectric Materials. Chem. Mater. 2010, 22, 624–634. [Google Scholar] [CrossRef]
- Snyder, G.J.; Toberer, E.S. Complex thermoelectric materials. Nat. Mater. 2008, 7, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Sootsman, J.R.; Chung, D.Y.; Kanatzidis, M.G. New and Old Concepts in Thermoelectric Materials. Angew. Chem. Int. Ed. 2009, 48, 8616–8639. [Google Scholar] [CrossRef] [PubMed]
- Wuttig, M.; Yamada, N. Phase-change materials for rewriteable data storage. Nat. Mater. 2007, 6, 824–832. [Google Scholar] [CrossRef] [PubMed]
- Lencer, D.; Salinga, M.; Grabowski, B.; Hickel, T.; Neugebauer, J.; Wuttig, M. A map for phase-change materials. Nat. Mater. 2008, 7, 972–977. [Google Scholar] [CrossRef]
- Wuttig, M.; Raoux, S. The Science and Technology of Phase Change Materials. Z. Anorg. Allg. Chem. 2012, 638, 2455–2465. [Google Scholar] [CrossRef]
- Böttcher, P. Tellurium-Rich Tellurides. Angew. Chem. Int. Ed. Engl. 1988, 27, 759–772. [Google Scholar] [CrossRef]
- Papoian, G.A.; Hoffmann, R. Hypervalent Bonding in One, Two, and Three Dimensions: Extending the Zintl-Klemm Concept to Nonclassical Electron-Rich Networks. Angew. Chem. Int. Ed. 2000, 39, 2408–2448. [Google Scholar] [CrossRef]
- Göbgen, K.C.; Gladisch, F.C.; Steinberg, S. The Mineral Stützite: a Zintl-Phase or Polar Intermetallic? A Case Study Using Experimental and Quantum-Chemical Techniques. Inorg. Chem. 2018, 57, 412–421. [Google Scholar] [CrossRef]
- Gladisch, F.C.; Steinberg, S. Revealing the Nature of Bonding in Rare-Earth Transition-Metal Tellurides by Means of Methods Based on First Principles. Eur. J. Inorg. Chem. 2017, 2017, 3395–3400. [Google Scholar] [CrossRef]
- Malliakas, C.D.; Kanatzidis, M.G. Charge Density Waves in the Square Nets of Tellurium of AMRETe4 (A = K, Na; M = Cu, Ag; RE = La, Ce). J. Am. Chem. Soc. 2007, 129, 10675–10677. [Google Scholar] [CrossRef]
- Patschke, R.; Heising, J.; Kanatzidis, M.; Brazis, P.; Kannewurf, C.R. KCuCeTe4: A New Intergrowth Rare Earth Telluride with an Incommensurate Superstructure Associated with a Distorted Square Net of Tellurium. Chem. Mater. 1998, 10, 695–697. [Google Scholar] [CrossRef]
- Patschke, R.; Brazis, P.; Kannewurf, C.R.; Kanatzidis, M. K2Ag3CeTe4: A Semiconducting Tunnel Framework Made from the Covalent “Link-Up” of [Ag2CeTe4]3− Layers with Ag. Inorg. Chem. 1998, 37, 6562–6563. [Google Scholar] [CrossRef]
- Patschke, R.; Brazis, P.; Kannewurf, C.R.; Kanatzidis, M. Rb2Cu3CeTe5: A quaternary semiconducting compound with a two-dimensional polytelluride framework. J. Mater. Chem. 1998, 8, 2587–2589. [Google Scholar] [CrossRef]
- Meng, C.-Y.; Chen, H.; Wang, P.; Chen, L. Syntheses, Structures, and Magnetic and Thermoelectric Properties of Double-Tunnel Tellurides: AxRE2Cu6−xTe6 (A = K–Cs; RE = La–Nd). Chem. Mater. 2011, 23, 4910–4919. [Google Scholar] [CrossRef]
- Meng, C.-Y.; Chen, H.; Wang, P. Syntheses, Structures, and Physical Properties of CsRE2Ag3Te5 (RE = Pr, Nd, Sm, Gd–Er) and RbRE2Ag3Te5 (RE = Sm, Gd–Dy). Inorg. Chem. 2014, 53, 6893–6903. [Google Scholar] [CrossRef]
- Babo, J.-M.; Choi, E.S.; Albrecht-Schmitt, T.E. Synthesis, Structure, Magnetism, and Optical Properties of Cs2Cu3DyTe4. Inorg. Chem. 2012, 51, 11730–11735. [Google Scholar] [CrossRef]
- Janka, O.; Pöttgen, R. Reactive Fluxes. In Handbook of Solid State Chemistry; Dronskowski, R., Kikkawa, S., Stein, A., Eds.; Wiley-VCH: Weinheim, Germany, 2017. [Google Scholar]
- Cotton, S. Lanthanide and Actinide Chemistry; John Wiley & Sons Ltd.: Chichester, England, 2006. [Google Scholar]
- Cordero, B.; Gómez, V.; Platero-Prats, A.; Revés, M.; Echeverría, J.; Cremades, E.; Barragán, F.; Alvarez, S. Covalent radii revisited. Dalton Trans. 2008, 2008, 2832–2838. [Google Scholar] [CrossRef]
- Steinberg, S.; Brgoch, J.; Miller, G.J.; Meyer, G. Identifying a Structural Preference in Reduced Rare-Earth Metal Halides by Combining Experimental and Computational Techniques. Inorg. Chem. 2012, 51, 11356–11364. [Google Scholar] [CrossRef] [Green Version]
- Steinberg, S.; Card, N.; Mudring, A.-V. From the Ternary Eu(Au/In)2 and EuAu4(Au/In)2 with Remarkable Au/In Distributions to a New Structure Type: The Gold-Rich Eu5Au16(Au/In)6 Structure. Inorg. Chem. 2015, 54, 8187–8196. [Google Scholar] [CrossRef]
- Smetana, V.; Steinberg, S.; Mudryk, Y.; Pecharsky, V.; Miller, G.J.; Mudring, A.-V. Cation-Poor Complex Metallic Alloys in Ba(Eu)-Au-Al(Ga) Systems: Identifying the Keys that Control Structural Arrangements and Atom Distributions at the Atomic Level. Inorg. Chem. 2015, 54, 10296–10308. [Google Scholar] [CrossRef]
- Steinberg, S.; Dronskowski, R. The Crystal Orbital Hamilton Population (COHP) Method as a Tool to Visualize and Analyze Chemical Bonding in Intermetallic Compounds. Crystals 2018, 8, 225. [Google Scholar] [CrossRef]
- Smetana, V.; Steinberg, S.; Mudring, A.-V. Layered Structures and Disordered Polyanionic Nets in the Cation-Poor Polar Intermetallics CsAu1.4Ga2.8 and CsAu2Ga2.6. Cryst. Growth Des. 2017, 17, 693–700. [Google Scholar] [CrossRef]
- Davaasuren, B.; Dashjav, E.; Rothenberger, A. Synthesis and Characterization of the Ternary Telluroargentate K4[Ag18Te11]. Z. Anorg. Allg. Chem. 2014, 640, 2939–2944. [Google Scholar] [CrossRef]
- Jansen, M. Homoatomic d10–d10 Interactions: Their Effects on Structure and Chemical and Physical Properties. Angew. Chem. Int. Ed. Engl. 1987, 26, 1098–1110. [Google Scholar] [CrossRef]
- Pyykkö, P. Strong Closed-Shell Interactions in Inorganic Chemistry. Chem. Rev. 1997, 97, 597–636. [Google Scholar] [CrossRef]
- Dreele, R.B.; Eyring, L.; Bowman, A.L.; Yamell, J.L. Refinement of the crystal structure of Pr7O12 by powder neutron diffraction. Acta Crystallogr. Sect. B 1975, 31, 971–974. [Google Scholar] [CrossRef]
- Wang, R.; Steinfink, H.; Bradley, W.F. The crystal structure of lanthanum telluride and tellurium-deficient neodymium telluride. Inorg. Chem. 1966, 5, 142–145. [Google Scholar] [CrossRef]
- Stöwe, K. Crystal structure, magnetic properties and band gap measurements of NdTe(2−x) (x = 0.11(1)). Z. Kristallogr. 2001, 216, 215–224. [Google Scholar] [CrossRef]
- Adenis, C.; Langer, V.; Lindqvist, O. Reinvestigation of the structure of tellurium. Acta Crystallogr. Sect. C 1989, 45, 941–942. [Google Scholar] [CrossRef]
- WinXPow Version 2.23; STOE & Cie GmbH: Darmstadt, Germany, 2008.
- Match! Vers. 3.8.0.137; Crystal Impact GbR: Bonn, Germany, 2019.
- SAINT+ and SADABS; Bruker AXS Inc.: Madison, WI, USA, 2009.
- XPREP, Version 6.03; Bruker AXS Inc.: Madison, WI, USA, 2014.
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A Found. Crystallogr. 2008, 64, 112–122. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B Condens. Matter Mater. Phys. 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [Green Version]
- Kresse, G.; Marsman, M.; Furthmüller, J. Vienna Ab Initio Simulation Package (VASP), The Guide; Computational Materials Physics, Faculty of Physics, Universität Wien: Vienna, Austria, 2014. [Google Scholar]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B Condens. Matter Mater. Phys. 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B Condens. Matter Mater. Phys. 1993, 47, 558–561. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B Condens. Matter Mater. Phys. 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Dronskowski, R.; Blöchl, P.E. Crystal Orbital Hamilton Populations (COHP). Energy-Resolved Visualization of Chemical Bonding in Solids Based on Density-Functional Calculations. J. Phys. Chem. 1993, 97, 8617–8624. [Google Scholar] [CrossRef]
- Andersen, O.K. Linear methods in band theory. Phys. Rev. B Condens. Matter Mater. Phys. 1975, 12, 3060–3083. [Google Scholar] [CrossRef] [Green Version]
- Andersen, O.K.; Jepsen, O. Explicit, First-Principles Tight-Binding Theory. Phys. Rev. Lett. 1984, 53, 2571–2574. [Google Scholar] [CrossRef]
- Krier, G.; Jepsen, O.; Burkhardt, A.; Andersen, O.K. TB-LMTO-ASA Program, 4.7 ed.; Max-Planck-Institut für Festkörperforschung: Stuttgart, Germany, 1995. [Google Scholar]
- von Barth, U.; Hedin, L. A local exchange-correlation potential for the spin polarized case: I. J. Phys. C Solid State Phys. 1972, 5, 1629–1642. [Google Scholar] [CrossRef]
- Lambrecht, W.R.L.; Andersen, O.K. Minimal basis sets in the linear muffin-tin orbital method: Application to the diamond-stucture crystals C, Si, and Ge. Phys. Rev. B Condens. Matter Mater. Phys. 1986, 34, 2439–2449. [Google Scholar] [CrossRef]
- Steinberg, S.; Bell, T.; Meyer, G. Electron Counting Rules and Electronic Structure in Tetrameric Transition-Metal (T)-Centered Rare-Earth (R) Cluster Complex Halides (X). Inorg. Chem. 2015, 54, 1026–1037. [Google Scholar] [CrossRef]
- Bigun, I.; Steinberg, S.; Smetana, V.; Mudryk, Y.; Kalychak, Y.; Havela, L.; Pecharsky, V.; Mudring, A.-V. Magnetocaloric Behavior in Ternary Europium Indides EuT5In: Probing the Design Capability of First-Principles-Based Methods on the Multifacted Magnetic Materials. Chem. Mater. 2017, 29, 2599–2614. [Google Scholar] [CrossRef]
- Provino, A.; Steinberg, S.; Smetana, V.; Paramanik, U.; Manfrinetti, P.; Dhar, S.K.; Mudring, A.-V. Gold in the Layered Structures of R3Au7Sn3: From Relativity to Versatility. Cryst. Growth Des. 2016, 16, 5657–5668. [Google Scholar] [CrossRef]
- Li, W.-L.; Ertural, C.; Bogdanovski, D.; Li, J.; Dronskowski, R. Chemical Bonding of Crystalline LnB6 (Ln = La–Lu) and Its Relationship with Ln2B8 Gas-Phase Complexes. Inorg. Chem. 2018, 57, 12999–13008. [Google Scholar] [CrossRef]
- Blöchl, P.E.; Jepsen, O.; Andersen, O.K. Improved tetrahedron method for Brillouin-zone integrations. Phys. Rev. B Condens. Matter Mater. Phys. 1994, 49, 16223–16233. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
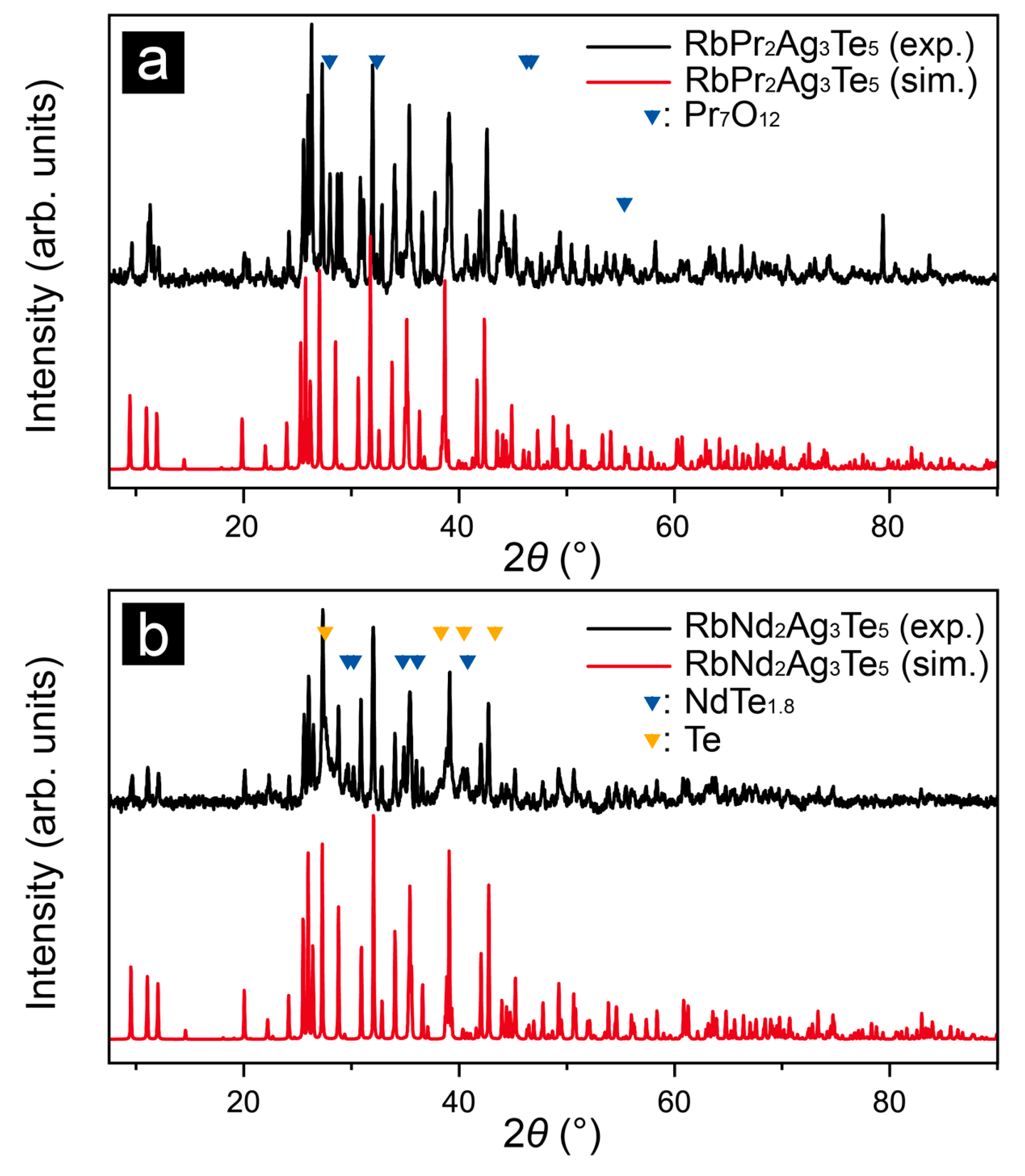{kind=link}
| Formula | RbPr2Ag3Te5 | RbNd2Ag3Te5 |
|---|---|---|
| from wt. | 1328.90 | 1335.56 |
| space group | Cmcm (no. 63) | |
| a (Å) | 4.618(2) | 4.607(2) |
| b (Å) | 16.014(7) | 16.031(7) |
| c (Å) | 18.644(8) | 18.594(8) |
| volume (Å3) | 1378.7(10) | 1373.1(10) |
| Z | 4 | |
| density (calc.), g/cm3 | 6.402 | 6.461 |
| μ (mm−1) | 24.946 | 25.513 |
| F (000) | 2224 | 2232 |
| θ range (°) | 2.19−25.76 | 2.19−25.67 |
| index ranges | −5 ≤ h ≤ 5 −18 ≤ k ≤ 19 −22 ≤ l ≤ 22 | −5 ≤ h ≤ 5 −19 ≤ k ≤ 18 −20 ≤ l ≤ 22 |
| reflections collected | 4203 | 3900 |
| independent reflections | 774 | 762 |
| refinement method | full-matrix least-squares on F2 | |
| data/restraints/parameters | 774/0/37 | 762/0/37 |
| goodness-of-fit on F2 | 1.16 | 1.15 |
| final R indices [I > 2σ(I)] | R1 = 0.036; wR2 = 0.081 | R1 = 0.066; wR2 = 0.156 |
| R indices (all data) | R1 = 0.040; wR2 = 0.082 | R1 = 0.068; wR2 = 0.158 |
| Rint | 0.068 | 0.098 |
| largest diff. peak and hole, e−/Å3 | 1.99 and −3.90 | 3.25 and −6.84 |
| Atom | Position | x | y | z | Ueq, Å2 |
|---|---|---|---|---|---|
| RbPr2Ag3Te5 | |||||
| Pr1 | 8f | 0 | 0.3096(1) | 0.5945(1) | 0.0114(2) |
| Te2 | 8f | ½ | 0.4421(1) | 0.6232(1) | 0.0124(3) |
| Te3 | 8f | 0 | 0.3289(1) | 0.4264(1) | 0.0110(3) |
| Te4 | 4c | 0 | 0.2389(1) | ¾ | 0.0128(3) |
| Ag5 | 8f | 0 | 0.0864(1) | 0.5275(1) | 0.0208(3) |
| Ag6 | 4c | ½ | 0.3360(1) | ¾ | 0.0229(4) |
| Rb7 | 4c | 0 | 0.5587(1) | ¾ | 0.0227(5) |
| RbNd2Ag3Te5 | |||||
| Nd1 | 8f | 0 | 0.3095(1) | 0.5945(1) | 0.0071(4) |
| Te2 | 8f | ½ | 0.4417(1) | 0.6230(1) | 0.0084(4) |
| Te3 | 8f | 0 | 0.3281(1) | 0.4265(1) | 0.0071(4) |
| Te4 | 4c | 0 | 0.2390(1) | ¾ | 0.0082(5) |
| Ag5 | 8f | 0 | 0.0865(1) | 0.5281(1) | 0.0161(5) |
| Ag6 | 4c | ½ | 0.3359(2) | ¾ | 0.0185(6) |
| Rb7 | 4c | 0 | 0.5587(2) | ¾ | 0.0182(7) |
| Interaction | Distance (Å) | No. of Bonds | –ICOHP/bond (eV/bond) | Ave. –ICOHP/bond (eV/bond) | Cum. –ICOHP/cell | % |
|---|---|---|---|---|---|---|
| Pr1‒Te2 | 3.182(1) | 16 | 1.103 | 1.123 | 53.892 | 43.7 |
| Pr1‒Te3 | 3.150(2) | 8 | 1.363 | |||
| Pr1‒Te3 | 3.225(1) | 16 | 0.936 | |||
| Pr1‒Te4 | 3.112(2) | 8 | 1.295 | |||
| Rb7‒Te2 | 3.796(2) | 16 | 0.045 | 0.045 | 1.425 | 1.1 |
| Rb7‒Te3 | 3.749(2) | 8 | 0.038 | |||
| Rb7‒Te4 | 3.696(2) | 8 | 0.050 | |||
| Ag5‒Ag5 | 2.952(2) | 4 | 0.455 | 0.455 | 1.819 | 1.5 |
| Ag5‒Te2 | 2.846(2) | 8 | 1.385 | 1.381 | 66.265 | 53.7 |
| Ag5‒Te2 | 2.919(2) | 8 | 0.930 | |||
| Ag5‒Te3 | 2.812(1) | 16 | 1.412 | |||
| Ag6‒Te2 | 2.913(2) | 8 | 1.323 | |||
| Ag6‒Te4 | 2.783(2) | 8 | 1.821 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Göbgen, K.C.; Fries, K.S.; Gladisch, F.C.; Dronskowski, R.; Steinberg, S. Revealing the Nature of Chemical Bonding in an ALn2Ag3Te5-Type Alkaline-Metal (A) Lanthanide (Ln) Silver Telluride. Inorganics 2019, 7, 70. https://doi.org/10.3390/inorganics7060070
Göbgen KC, Fries KS, Gladisch FC, Dronskowski R, Steinberg S. Revealing the Nature of Chemical Bonding in an ALn2Ag3Te5-Type Alkaline-Metal (A) Lanthanide (Ln) Silver Telluride. Inorganics. 2019; 7(6):70. https://doi.org/10.3390/inorganics7060070
Chicago/Turabian StyleGöbgen, Kai C., Kai S. Fries, Fabian C. Gladisch, Richard Dronskowski, and Simon Steinberg. 2019. "Revealing the Nature of Chemical Bonding in an ALn2Ag3Te5-Type Alkaline-Metal (A) Lanthanide (Ln) Silver Telluride" Inorganics 7, no. 6: 70. https://doi.org/10.3390/inorganics7060070
APA StyleGöbgen, K. C., Fries, K. S., Gladisch, F. C., Dronskowski, R., & Steinberg, S. (2019). Revealing the Nature of Chemical Bonding in an ALn2Ag3Te5-Type Alkaline-Metal (A) Lanthanide (Ln) Silver Telluride. Inorganics, 7(6), 70. https://doi.org/10.3390/inorganics7060070