A Review of the MSCA ITN ECOSTORE—Novel Complex Metal Hydrides for Efficient and Compact Storage of Renewable Energy as Hydrogen and Electricity

 ,
,  , ,
, ,  , , ,
, , , 
Abstract
:| Table of Content | |
| 1. Introduction……… | 3 |
| 2. Promising Metal Hydrides for Battery Application……… | 4 |
| 2.1. Metal Hydrides for Li-ion and Post Li Batteries……… | 4 |
| 2.1.1. Metal Hydrides as Negative Electrode Material with Liquid Electrolyte……… | 5 |
| 2.1.2. Promising Complex Metal Hydrides as Solid-State Electrolytes……… | 8 |
| Lithium Borohydride……… | 9 |
| Lithium Nitride and Lithium Hydride……… | 9 |
| Binary Phases of Hydrides with Lithium Halides ……… | 11 |
| LiBH4-Li2NH: Li5(BH4)3NH—thorough characterization of a cluster complex hydride ……… | 11 |
| Argyrodite Structure Materials ……… | 12 |
| 2.1.3. Application of Metal Hydrides in Solid-State Cells……… | 13 |
| Solid-State Half-Cell ……… | 13 |
| A Full Solid-State Li-ion Cell ……… | 15 |
| 2.1.4. Conclusions……… | 15 |
| 2.2. Na-Based Closo-Borates for Na Batteries……… | 16 |
| 2.2.1. Na-Based Closo-Borates Solid Electrolyte……… | 16 |
| The Na2B12H12 Polymorphism ……… | 16 |
| Nax+2y(CB11H12)x(B12H12)y Solid Electrolyte ……… | 17 |
| 2.2.2. Conclusions……… | 20 |
| 3. Advances in Hydrogen Storage Materials……… | 20 |
| 3.1. Pure Metal Hydrides (Mg, Pd, Ti)……… | 21 |
| 3.2. Amide and Imide Based Systems for H2 Storage……… | 22 |
| 3.2.1. Insights into Alkali-Based Amides and Imides Including Boron……… | 23 |
| LiBH4-LiNH2 ……… | 23 |
| LiNH2-Li2NH ……… | 25 |
| 3.2.2. Insights into the Structure and Reaction Mechanism of Metal Amide—Metal Hydride Composite Systems……… | 25 |
| Ammonolysis of Alkali and Alkaline-Earth Metal Amides ……… | 26 |
| K-Mg-N-H System ……… | 26 |
| KNH2-KH System ……… | 27 |
| Rb-Mg-N-H and Rb-N-H Systems ……… | 27 |
| 3.2.3. Conclusions……… | 27 |
| 3.3. Eutectic Metal Borohydride Systems……… | 28 |
| 3.3.1. Experimental Study and Assessment of Eutectic Borohydride Systems……… | 29 |
| LiBH4-NaBH4 ……… | 29 |
| LiBH4-KBH4 ……… | 30 |
| NaBH4-KBH4 ……… | 30 |
| LiBH4-NaBH4-KBH4 ……… | 30 |
| Other Systems ……… | 31 |
| 3.3.2. Thermodynamic Properties of Eutectic Borohydride Systems……… | 31 |
| 3.3.3. Hydrogen Sorage Properties of Eutectic Metal Borohydride Systems……… | 33 |
| 3.3.4. Conclusions ……… | 33 |
| 3.4. Kinetic Tailoring of 2LiBH4 + MgH2/2LiH + MgB2 with Cost Effective 3TiCl3·AlCl3……… | 34 |
| 3.5. Role of Nanoconfinement in Enhancing the Properties of Hydrogen Storage Materials……… | 39 |
| 3.5.1. Nanoconfinement Approaches……… | 40 |
| 3.5.2. Confined Borohydrides……… | 41 |
| 3.5.3. Conclusions……… | 43 |
| 3.6. Rare Earth Borohydrides……… | 43 |
| 3.6.1. Synthesis of Rare Earth Borohydrides (REB)……… | 44 |
| Solvent Free Synthesis of REB ……… | 44 |
| Solvent-Based Synthesis of REB ……… | 44 |
| 3.6.2. Crystal Structures of Monometalic REB……… | 45 |
| 3.6.3. Crystal Structures of Bimetallic REB ……… | 46 |
| 3.6.4. Reactive Hydride Composites with REB……… | 49 |
| 3.6.5. Conclusions……… | 51 |
| 4. Final Conclusion and Outlook……… | 51 |
| References……… | 53 |
1. Introduction
2. Promising Metal Hydrides for Battery Application
2.1. Metal Hydrides for Li-ion and Post Li Batteries
2.1.1. Metal Hydrides as Negative Electrode Material with Liquid Electrolyte
2.1.2. Promising Complex Metal Hydrides as Solid-State Electrolytes
Lithium Borohydride
Lithium Nitride and Lithium Hydride
Binary Phases of Hydrides with Lithium Halides
LiBH4-Li2NH: Li5(BH4)3NH—thorough characterization of a cluster complex hydride
Argyrodite Structure Materials
2.1.3. Application of Metal Hydrides in Solid-State Cells
Solid-State Half-Cell
A Full Solid-State Li-ion Cell
2.1.4. Conclusions
2.2. Na-Based Closo-Borates for Na Batteries
2.2.1. Na-Based Closo-Borates Solid Electrolyte
The Na2B12H12 Polymorphism
Nax+2y(CB11H12)x(B12H12)y Solid Electrolyte
2.2.2. Conclusions
3. Advances in Hydrogen Storage Materials
3.1. Pure Metal Hydrides (Mg, Pd, Ti)
3.2. Amide and Imide Based Systems for H2 Storage
3.2.1. Insights into Alkali-Based Amides and Imides Including Boron
LiBH4-LiNH2
LiNH2-Li2NH
3.2.2. Insights into the Structure and Reaction Mechanism of Metal Amide—Metal Hydride Composite Systems
Ammonolysis of Alkali and Alkaline-Earth Metal Amides
K-Mg-N-H System
KNH2-KH System
Rb-Mg-N-H and Rb-N-H Systems
3.2.3. Conclusions
3.3. Eutectic Metal Borohydride Systems
3.3.1. Experimental Study and Assessment of Eutectic Borohydride Systems
LiBH4-NaBH4
LiBH4-KBH4
NaBH4-KBH4
LiBH4-NaBH4-KBH4
Other Systems
3.3.2. Thermodynamic Properties of Eutectic Borohydride Systems
3.3.3. Hydrogen Sorage Properties of Eutectic Metal Borohydride Systems
3.3.4. Conclusions
3.4. Kinetic Tailoring of 2LiBH4 + MgH2/2LiH + MgB2 with Cost Effective 3TiCl3·AlCl3
→ 0.072LiCl(s) + 0.024Mg(s) + 0.006AlB2(s) + 0.018TiB2(s) + 0.036H2(g)
ΔG1bar, 25 °C = –10.9 kJ mol−1
ΔG1bar, 25 °C = –7.6 kJ mol−1
3.5. Role of Nanoconfinement in Enhancing the Properties of Hydrogen Storage Materials
3.5.1. Nanoconfinement Approaches
3.5.2. Confined Borohydrides
3.5.3. Conclusions
3.6. Rare Earth Borohydrides
3.6.1. Synthesis of Rare Earth Borohydrides (REB)
Solvent Free Synthesis of REB
Solvent-Based Synthesis of REB
3.6.2. Crystal Structures of Monometalic REB
3.6.3. Crystal Structures of Bimetallic REB
3.6.4. Reactive Hydride Composites with REB
3.6.5. Conclusions
4. Final Conclusions and Outlook
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yoshida, T.; Kojima, K. Toyota MIRAI fuel cell vehicle and progress toward a future hydrogen society. Interface Mag. 2015, 24, 45–49. [Google Scholar] [CrossRef]
- Pillot, C. The rechargeable battery market and main trends 2014–2015. Avicenne Energy 2015. Available online: http://www.avicenne.com/pdf/Fort_Lauderdale_Tutorial_C_Pillot_March2015.pdf (accessed on 21 February 2020).
- Poupin, L.; Humphries, T.D.; Paskevicius, M.; Buckley, C.E. An experimental high temperature thermal battery coupled to a low temperature metal hydride for solar thermal energy storage. Sustain. Energy Fuels 2020, 4, 285–292. [Google Scholar] [CrossRef]
- Zhao-Karger, Z.; Fichtner, M. Beyond intercalation chemistry for rechargeable mg batteries: A short review and perspective. Front. Chem. 2018, 6, 656. [Google Scholar] [CrossRef] [PubMed]
- Elia, G.A.; Marquardt, K.; Hoeppner, K.; Fantini, S.; Lin, R.; Knipping, E.; Peters, W.; Drillet, J.F.; Passerini, S.; Hahn, R. An overview and future perspectives of aluminum batteries. Adv. Mater. 2016, 28, 7564–7579. [Google Scholar] [CrossRef] [PubMed]
- Janek, J.; Zeier, W.G. A solid future for battery development. Nat. Energy 2016, 1, 300. [Google Scholar] [CrossRef]
- Tarascon, J.M. Key challenges in future Li-battery research. Philos. Trans. A Math. Phys. Eng. Sci. 2010, 368, 3227–3241. [Google Scholar] [CrossRef] [Green Version]
- Tarascon, J.M.; Armand, M. Issues and challenges facing rechargeable lithium batteries. Nature 2001, 414, 359–367. [Google Scholar] [CrossRef]
- S. C. 50th A. P. Team. Sony History, 1996th ed.; Sony Corporation, 1996; Available online: https://www.sony.net/SonyInfo/CorporateInfo/History/SonyHistory/2-13.html (accessed on 21 February 2020).
- Li, C.; Zhang, H.P.; Fu, L.J.; Liu, H.; Wu, Y.P.; Ram, E.; Holze, R.; Wu, H.Q. Cathode materials modified by surface coating for lithium ion batteries. Electrochim. Acta 2006, 51, 3872–3883. [Google Scholar] [CrossRef]
- Kraytsberg, A.; Ein-Eli, Y. Higher, stronger, better… A review of 5 volt cathode materials for advanced lithium-ion batteries. Adv. Energy Mater. 2012, 2, 922–939. [Google Scholar] [CrossRef]
- Shetti, N.P.; Dias, S.; Reddy, K.R. Nanostructured organic and inorganic materials for Li-ion batteries: A review. Mater. Sci. Semicond. Process. 2019, 104, 104684. [Google Scholar] [CrossRef]
- Liu, M.; Deng, N.; Ju, J.; Fan, L.; Wang, L.; Li, Z.; Zhao, H.; Yang, G.; Kang, W.; Yan, J. A review: Electrospun nanofiber materials for lithium-sulfur batteries. Adv. Funct. Mater. 2019, 29. [Google Scholar] [CrossRef]
- Wang, H.; Yang, Y.; Liang, Y.; Robinson, J.T.; Li, Y.; Jackson, A.; Cui, Y.; Dai, H. Graphene-Wrapped sulfur particles as a rechargeable lithium-sulfur battery cathode material with high capacity and cycling stability. Nano Lett. 2011, 11, 2644–2647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myung, S.T.; Maglia, F.; Park, K.J.; Yoon, C.S.; Lamp, P.; Kim, S.J.; Sun, Y.K. Nickel-Rich layered cathode materials for automotive lithium-ion batteries: Achievements and perspectives. ACS Energy Lett. 2017, 2, 196–223. [Google Scholar] [CrossRef]
- Oumellal, Y.; Rougier, A.; Nazri, G.A.; Tarascon, J.M.; Aymard, L. Metal hydrides for lithium-ion batteries. Nat. Mater. 2008, 7, 916–921. [Google Scholar] [CrossRef]
- Aymard, L.; Oumellal, Y.; Bonnet, J.P. Metal hydrides: An innovative and challenging conversion reaction anode for lithium-ion batteries. Beilstein J. Nanotechnol. 2015, 6, 1821–1839. [Google Scholar] [CrossRef] [Green Version]
- Braga, M.H.; Grundish, N.S.; Murchison, A.J.; Goodenough, J.B. Alternative strategy for a safe rechargeable battery. Energy Environ. Sci. 2017, 10, 331–336. [Google Scholar] [CrossRef]
- Varzi, A.; Raccichini, R.; Passerini, S.; Scrosati, B. Challenges and prospects of the role of solid electrolytes in the revitalization of lithium metal batteries. J. Mater. Chem. A 2016, 4, 17251–17259. [Google Scholar] [CrossRef] [Green Version]
- Sartori, S.; Cuevas, F.; Latroche, M. Metal hydrides used as negative electrode materials for Li-ion batteries. Appl. Phys. A Mater. Sci. Process. 2016, 122, 135. [Google Scholar] [CrossRef]
- Brutti, S.; Mulas, G.; Piciollo, E.; Panero, S.; Reale, P. Magnesium hydride as a high capacity negative electrode for lithium ion batteries. J. Mater. Chem. 2012, 22, 14531–14537. [Google Scholar] [CrossRef]
- Yang, S.; Wang, H.; Ouyang, L.; Liu, J.; Zhu, M. Improvement in the Electrochemical lithium storage performance of MgH2. Inorganics 2018, 6, 2. [Google Scholar] [CrossRef] [Green Version]
- Oumellal, Y.; Zaidi, W.; Bonnet, J.P.; Cuevas, F.; Latroche, M.; Zhang, J.; Bobet, J.L.; Rougier, A.; Aymard, L. Reactivity of TiH2 hydride with lithium ion: Evidence for a new conversion mechanism. Int. J. Hydrog. Energy 2012, 37, 7831–7835. [Google Scholar] [CrossRef]
- Teprovich, J.A.; Zhang, J.; Colón-Mercado, H.; Cuevas, F.; Peters, B.; Greenway, S.; Zidan, R.; Latroche, M. Li-Driven electrochemical conversion reaction of AlH3, LiAlH4, and NaAlH4. J. Phys. Chem. C 2015, 119, 4666–4674. [Google Scholar] [CrossRef]
- Silvestri, L.; Forgia, S.; Farina, L.; Meggiolaro, D.; Panero, S.; La Barbera, A.; Brutti, S.; Reale, P. Lithium alanates as negative electrodes in lithium-ion batteries. ChemElectroChem 2015, 2, 877–886. [Google Scholar] [CrossRef]
- Silvestri, L.; Farina, L.; Meggiolaro, D.; Panero, S.; Padella, F.; Brutti, S.; Reale, P. Reactivity of sodium alanates in lithium batteries. J. Phys. Chem. C 2015, 119, 28766–28775. [Google Scholar] [CrossRef]
- Ptashnik, V.B.; Dunaeva, T.Y.; Baikov, Y.M. The Electrical Conductivity of Solid Lithium Hydride Doped with Sulphur. Phys. Status Solidi B 1982, 110, K121–K124. [Google Scholar] [CrossRef]
- Berti, N.; Hadjixenophontos, E.; Cuevas, F.; Zhang, J.; Lacoste, A.; Dubot, P.; Schmitz, G.; Latroche, M. Thin films as model system for understanding the electrochemical reaction mechanisms in conversion reaction of MgH2 with lithium. J. Power Sources 2018, 402, 99–106. [Google Scholar] [CrossRef]
- Huen, P.; Ravnsbæk, D.B. Insight into poor cycling stability of MgH2 anodes. J. Electrochem. Soc. 2017, 164, A3138–A3143. [Google Scholar] [CrossRef]
- Huang, L.; Aymard, L.; Bonnet, J.-P. MgH 2-TiH 2 mixture as an anode for lithium-ion batteries: Synergic enhancement of the conversion electrode electrochemical performance. J. Mater. Chem. A 2015, 3, 15091–15096. [Google Scholar] [CrossRef]
- Ito, M.; Setoyama, D.; Matsunaga, J.; Muta, H.; Kurosaki, K.; Uno, M.; Yamanaka, S. Electrical and thermal properties of titanium hydrides. J. Alloys Compd. 2006, 420, 25–28. [Google Scholar] [CrossRef]
- Wipf, H.; Kappesser, B.; Werner, R. Hydrogen diffusion in titanium and zirconium hydrides. J. Alloys Compd. 2000, 310, 190–195. [Google Scholar] [CrossRef]
- Berti, N.; Cuevas, F.; Zhang, J.; Latroche, M. Enhanced reversibility of the electrochemical Li conversion reaction with MgH 2-TiH 2 nanocomposites. Int. J. Hydrog. Energy 2017. [Google Scholar] [CrossRef]
- Berti, N. MgH2-TiH2 Hydrides as Negatives Electrodes of Li-Ion Batteries. Ph.D. Thesis, Université Paris-Est, Paris, France, 2017. [Google Scholar]
- Oumellal, Y.; Rougier, A.; Tarascon, J.-M.; Aymard, L. 2LiH + M (M = Mg, Ti): New concept of negative electrode for rechargeable lithium-ion batteries. J. Power Sources 2009, 192, 698–702. [Google Scholar] [CrossRef]
- Oumellal, Y.; Zlotea, C.; Bastide, S.; Cachet-Vivier, C.; Leonel, E.; Sengmany, S.; Leroy, E.; Aymard, L.; Bonnet, J.P.; Latroche, M. Bottom-Up preparation of MgH(2) nanoparticles with enhanced cycle life stability during electrochemical conversion in Li-ion batteries. Nanoscale 2014, 6, 14459–14466. [Google Scholar] [CrossRef] [PubMed]
- Huen, P.; Peru, F.; Charalambopoulou, G.; Steriotis, T.A.; Jensen, T.R.; Ravnsbaek, D.B. Nanoconfined NaAlH4 conversion electrodes for Li batteries. ACS Omega 2017, 2, 1956–1967. [Google Scholar] [CrossRef]
- Zhang, B.; Xia, G.; Sun, D.; Fang, F.; Yu, X. Magnesium hydride nanoparticles self-assembled on graphene as anode material for high-performance lithium-ion batteries. ACS Nano 2018, 12, 3816–3824. [Google Scholar] [CrossRef] [Green Version]
- Silvestri, L.; Paolone, A.; Cirrincione, L.; Stallworth, P.; Greenbaum, S.; Panero, S.; Brutti, S.; Reale, P. NaAlH4 nanoconfinement in a mesoporous carbon for application in lithium ion batteries. J. Electrochem. Soc. 2017, 164, A1120–A1125. [Google Scholar] [CrossRef]
- Fichtner, M. Nanoconfinement effects in energy storage materials. Phys. Chem. Chem. Phys. 2011, 13, 21186–21195. [Google Scholar] [CrossRef]
- Nielsen, T.K.; Besenbacher, F.; Jensen, T.R. Nanoconfined hydrides for energy storage. Nanoscale 2011, 3, 2086–2098. [Google Scholar] [CrossRef]
- Winter, M.; Besenhard, J.O.; Spahr, M.E.; Novak, P. Insertion electrode materials for rechargeable lithium batteries. Adv. Mater. 1998, 10, 725–763. [Google Scholar] [CrossRef]
- Kaskhedikar, N.A.; Maier, J. Lithium storage in carbon nanostructures. Adv. Mater. 2009, 21, 2664–2680. [Google Scholar] [CrossRef]
- Hu, Y.S.; Adelhelm, P.; Smarsly, B.M.; Hore, S.; Antonietti, M.; Maier, J. Synthesis of hierarchically porous carbon monoliths with highly ordered microstructure and their application in rechargeable lithium batteries with high-rate capability. Adv. Funct. Mater. 2007, 17, 1873–1878. [Google Scholar] [CrossRef]
- Mrgudich, J.N. Conductivity of silver iodide pellets for solid-electrolyte batteries. J. Electrochem. Soc. 1960, 107, 475–479. [Google Scholar] [CrossRef]
- Tubandt, C.; Lorenz, E. Molekularzustand und elektrisches leitvermögen kristallisierter salze. Z. Phys. Chem. 1914, 87, 513–542. [Google Scholar] [CrossRef]
- Bachman, J.C.; Muy, S.; Grimaud, A.; Chang, H.H.; Pour, N.; Lux, S.F.; Paschos, O.; Maglia, F.; Lupart, S.; Lamp, P.; et al. Inorganic solid-state electrolytes for lithium batteries: Mechanisms and properties governing ion conduction. Chem. Rev. 2016, 116, 140–162. [Google Scholar] [CrossRef]
- Zhang, Z.; Shao, Y.; Lotsch, B.; Hu, Y.S.; Li, H.; Janek, J.; Nazar, L.F.; Nan, C.W.; Maier, J.; Armand, M.; et al. New horizons for inorganic solid state ion conductors. Energy Environ. Sci. 2018, 11. [Google Scholar] [CrossRef] [Green Version]
- de Klerk, N.J.J.; Rosłoń, I.; Wagemaker, M. Diffusion mechanism of li argyrodite solid electrolytes for li-ion batteries and prediction of optimized halogen doping: The effect of Li vacancies, halogens, and halogen disorder. Chem. Mater. 2016, 28, 7955–7963. [Google Scholar] [CrossRef] [Green Version]
- Braga, M.H.; Ferreira, J.A.; Stockhausen, V.; Oliveira, J.E.; El-Azab, A. Novel Li3ClO based glasses with superionic properties for lithium batteries. J. Mater. Chem. A 2014, 2, 5470–5480. [Google Scholar] [CrossRef] [Green Version]
- Jalem, R.; Yamamoto, Y.; Shiiba, H.; Nakayama, M.; Munakata, H.; Kasuga, T.; Kanamura, K. Concerted migration mechanism in the li ion dynamics of garnet-type Li7La3Zr2O12. Chem. Mater. 2013, 25, 425–430. [Google Scholar] [CrossRef]
- Weber, D.A.; Senyshyn, A.; Weldert, K.S.; Wenzel, S.; Zhang, W.B.; Kaiser, R.; Berendts, S.; Janek, J.; Zeier, W.G. Structural insights and 3D diffusion pathways within the lithium superionic conductor Li10GeP2S12. Chem. Mater. 2016, 28, 5905–5915. [Google Scholar] [CrossRef]
- Kraft, M.A.; Culver, S.P.; Calderon, M.; Bocher, F.; Krauskopf, T.; Senyshyn, A.; Dietrich, C.; Zevalkink, A.; Janek, J.; Zeier, W.G. Influence of lattice polarizability on the ionic conductivity in the lithium superionic argyrodites Li6PS5X (X = Cl, Br, I). J. Am. Chem. Soc. 2017, 139, 10909–10918. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.; Lu, P.; Liu, Z.; Qi, Y.; Hector, L.G., Jr.; Li, H.; Harris, S.J. Direct calculation of Li-ion transport in the solid electrolyte interphase. J. Am. Chem. Soc. 2012, 134, 15476–15487. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Zhu, Y.; Mo, Y. Origin of fast ion diffusion in super-ionic conductors. Nat. Commun. 2017, 8, 15893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlesinger, H.I.; Brown, H.C. Metallo borohydrides. III. Lithium borohydride. J. Am. Chem. Soc. 1940, 62, 3429–3435. [Google Scholar] [CrossRef]
- Zuttel, A.; Rentsch, S.; Fischer, P.; Wenger, P.; Sudan, P.; Mauron, P.; Emmenegger, C. Hydrogen storage properties of LiBH4. J. Alloys Compd. 2003, 356, 515–520. [Google Scholar] [CrossRef]
- Zuttel, A.; Borgschulte, A.; Orimo, S.I. Tetrahydroborates as new hydrogen storage materials. Scr. Mater. 2007, 56, 823–828. [Google Scholar] [CrossRef]
- Matsuo, M.; Nakamori, Y.; Orimo, S.; Maekawa, H.; Takamura, H. Lithium superionic conduction in lithium borohydride accompanied by structural transition. Appl. Phys. Lett. 2007, 91, 42–45. [Google Scholar] [CrossRef]
- Yao, Y.S.; Klug, D.D. High-Pressure phases of lithium borohydride LiBH4: A first-principles study. Phys. Rev. B 2012, 86, 64107. [Google Scholar] [CrossRef]
- Ikeshoji, T.; Tsuchida, E.; Morishita, T.; Ikeda, K.; Matsuo, M.; Kawazoe, Y.; Orimo, S. Fast-Ionic conductivity of Li+ in LiBH4. Phys. Rev. B 2011, 83, 144301. [Google Scholar] [CrossRef]
- Skripov, A.V.; Soloninin, A.V.; Filinchuk, Y.; Chernyshov, D. Nuclear magnetic resonance study of the rotational motion and the phase transition in LiBH4. J. Phys. Chem. C 2008, 112, 18701–18705. [Google Scholar] [CrossRef]
- Ley, M.B.; Boulineau, S.; Janot, R.l.; Filinchuk, Y.; Jensen, T.R. New Li ion conductors and solid state hydrogen storage materials: LiM(BH4)3Cl, M = La, Gd. J. Phys. Chem. C 2012, 116, 21267–21276. [Google Scholar] [CrossRef]
- Trück, J.; Hadjixenophontos, E.; Joshi, Y.; Richter, G.; Stender, P.; Schmitz, G. Ionic conductivity of melt-frozen LiBH4 films. RSC Adv. 2019, 9, 38855–38859. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Hattori, K.; Yamazaki, T.; Takada, K.; Matsuo, M.; Orimo, S.-I.; Maekawa, H.; Takamura, H. All-solid-state lithium battery with LiBH4 solid electrolyte. J. Power Sources 2013, 226, 61–64. [Google Scholar] [CrossRef]
- Takahashi, K.; Maekawa, H.; Takamura, H. Effects of intermediate layer on interfacial resistance for all-solid-state lithium batteries using lithium borohydride. Solid State Ion. 2014, 262, 179–182. [Google Scholar] [CrossRef]
- Li, W.; Wu, G.; Araújo, C.M.; Scheicher, R.H.; Blomqvist, A.; Ahuja, R.; Xiong, Z.; Feng, Y.; Chen, P. Li+ ion conductivity and diffusion mechanism in α-Li3N and β-Li3N. Energy Environ. Sci. 2010, 3, 1524–1530. [Google Scholar] [CrossRef]
- Li, W.; Wu, G.T.; Xiong, Z.T.; Feng, Y.P.; Chen, P. Li+ ionic conductivities and diffusion mechanisms in Li-based imides and lithium amide. Phys. Chem. Chem. Phys. 2012, 14, 1596–1606. [Google Scholar] [CrossRef]
- Knutz, B.; Skaarup, S. Cycling of Li/Li3n/Tis2 solid-state cells. Solid State Ion. 1983, 9–10, 371–374. [Google Scholar] [CrossRef]
- Paik, B.; Wolczyk, A. Lithium imide (Li2NH) as a solid-state electrolyte for electrochemical energy storage applications. J. Phys. Chem. C 2019, 123, 1619–1625. [Google Scholar] [CrossRef]
- Wolczyk, A.; Pinatel, E.R.; Chierotti, M.R.; Nervi, C.; Gobetto, R.; Baricco, M. Solid-State NMR and thermodynamic investigations on LiBH4LiNH2 system. Int. J. Hydrog. Energy 2016, 41, 14475–14483. [Google Scholar] [CrossRef] [Green Version]
- Wolczyk, A.; Paik, B.; Sato, T.; Nervi, C.; Brighi, M.; Gharib Doust, S.P.; Chierotti, M.; Matsuo, M.; Li, G.; Gobetto, R.; et al. Li5(BH4)3NH: Lithium-rich mixed anion complex hydride. J. Phys. Chem. C 2017, 121, 11069–11075. [Google Scholar] [CrossRef]
- Boukamp, B.A.; Huggins, R.A. Ionic-Conductivity in lithium imide. Phys. Lett. A 1979, 72, 464–466. [Google Scholar] [CrossRef]
- David, W.I.; Jones, M.O.; Gregory, D.H.; Jewell, C.M.; Johnson, S.R.; Walton, A.; Edwards, P.P. A mechanism for non-stoichiometry in the lithium amide/lithium imide hydrogen storage reaction. J. Am. Chem. Soc. 2007, 129, 1594–1601. [Google Scholar] [CrossRef] [PubMed]
- Maekawa, H.; Matsuo, M.; Takamura, H.; Ando, M.; Noda, Y.; Karahashi, T.; Orimo, S. Halide-Stabilized LiBH4, a room-temperature lithium fast-ion conductor. J. Am. Chem. Soc. 2009, 131, 894–895. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, M.; Takamura, H.; Maekawa, H.; Li, H.W.; Orimo, S. Stabilization of lithium superionic conduction phase and enhancement of conductivity of LiBH4 by LiCl addition. Appl. Phys. Lett. 2009, 94, 084103. [Google Scholar] [CrossRef]
- Miyazaki, R.; Karahashi, T.; Kumatani, N.; Noda, Y.; Ando, M.; Takamura, H.; Matsuo, M.; Orimo, S.; Maekawa, H. Room temperature lithium fast-ion conduction and phase relationship of LiI stabilized LiBH4. Solid State Ion. 2011, 192, 143–147. [Google Scholar] [CrossRef]
- Sveinbjörnsson, D.; Myrdal, J.S.G.; Blanchard, D.; Bentzen, J.J.; Hirata, T.; Mogensen, M.B.; Norby, P.; Orimo, S.-I.; Vegge, T. Effect of heat treatment on the lithium ion conduction of the LiBH4-LiI solid solution. J. Phys. Chem. C 2013, 117, 3249–3257. [Google Scholar] [CrossRef]
- Rude, L.H.; Zavorotynska, O.; Arnbjerg, L.M.; Ravnsbæk, D.B.; Malmkjær, R.A.; Grove, H.; Hauback, B.C.; Baricco, M.; Filinchuk, Y.; Besenbacher, F.; et al. Bromide substitution in lithium borohydride, LiBH4-LiBr. Int. J. Hydrog. Energy 2011, 36, 15664–15672. [Google Scholar] [CrossRef] [Green Version]
- Gulino, V.; Brighi, M.; Dematteis, E.M.; Murgia, F.; Nervi, C.; Cerny, R.; Baricco, M. Phase Stability and fast ion conductivity in the hexagonal LiBH4-LiBr-LiCl solid solution. Chem. Mater. 2019, 31, 5133–5144. [Google Scholar] [CrossRef] [Green Version]
- Myrdal, J.S.G.; Blanchard, D.; Sveinbjornsson, D.; Vegge, T. Li-Ion conduction in the LiBH4: LiI system from density functional theory calculations and quasi-elastic neutron scattering. J. Phys. Chem. C 2013, 117, 9084–9091. [Google Scholar] [CrossRef]
- Matsuo, M.; Sato, T.; Miura, Y.; Oguchi, H.; Zhou, Y.; Maekawa, H.; Takamura, H.; Orimo, S.-I. Synthesis and lithium fast-ion conductivity of a new complex hydride Li3(NH2)2I with double-layered structure. Chem. Mater. 2010, 22, 2702–2704. [Google Scholar] [CrossRef]
- Gamba, N.S.; Larochette, P.A.; Gennari, F.C. Li4(NH2)3Cl amide-chloride: A new synthesis route, and hydrogen storage kinetic and thermodynamic properties. RSC Adv. 2016, 6, 15622–15629. [Google Scholar] [CrossRef]
- Anderson, P.A.; Chater, P.A.; Hewett, D.R.; Slater, P.R. Hydrogen storage and ionic mobility in amide-halide systems. Faraday Discuss. 2011, 151, 271–284. [Google Scholar] [CrossRef] [PubMed]
- Davies, R.A.; Anderson, P.A. Synthesis and characterization of two new amide chloride compounds: Potential H-2 storage materials. Int. J. Hydrog. Energy 2015, 40, 3001–3005. [Google Scholar] [CrossRef] [Green Version]
- Matsuo, M.; Remhof, A.; Martelli, P.; Caputo, R.; Ernst, M.; Miura, Y.; Sato, T.; Oguchi, H.; Maekawa, H.; Takamura, H.; et al. Complex hydrides with (BH(4))(-) and (NH(2))(-) anions as new lithium fast-ion conductors. J. Am. Chem. Soc. 2009, 131, 16389–16391. [Google Scholar] [CrossRef]
- Blomqvist, A.; Araujo, C.M.; Scheicher, R.H.; Srepusharawoot, P.; Li, W.; Chen, P.; Ahuja, R. Hydrogen as promoter and inhibitor of superionicity: A case study on Li-N-H systems. Phys. Rev. B 2010, 82. [Google Scholar] [CrossRef]
- Hewett, D.R. Mixed Anion Amides for Hydrogen Storage. Ph.D. Thesis, University of Birmingham, Birmingham, UK, 2012. [Google Scholar]
- Orimo, S.; Nakamori, Y.; Kitahara, G.; Miwa, K.; Ohba, N.; Towata, S.; Züttel, A. Dehydriding and rehydriding reactions of LiBH4. J. Alloys Compd. 2005, 404–406, 427–430. [Google Scholar] [CrossRef]
- Lindemann, I.; Domenech Ferrer, R.; Dunsch, L.; Filinchuk, Y.; Cerny, R.; Hagemann, H.; D’Anna, V.; Lawson Daku, L.M.; Schultz, L.; Gutfleisch, O. Al3Li4(BH4)13: A complex double-cation borohydride with a new structure. Chemistry 2010, 16, 8707–8712. [Google Scholar] [CrossRef]
- Noritake, T.; Aoki, M.; Towata, S.; Ninomiya, A.; Nakamori, Y.; Orimo, S. Crystal structure analysis of novel complex hydrides formed by the combination of LiBH4 and LiNH2. Appl. Phys. A Mater. Sci. Process. 2006, 83, 277–279. [Google Scholar] [CrossRef]
- Aoki, M.; Miwa, K.; Noritake, T.; Kitahara, G.; Nakamori, Y.; Orimo, S.; Towata, S. Destabilization of LiBH4 by mixing with LiNH2. Appl. Phys. A Mater. Sci. Process. 2005, 80, 1409–1412. [Google Scholar] [CrossRef]
- Deiseroth, H.J.; Kong, S.T.; Eckert, H.; Vannahme, J.; Reiner, C.; Zaiss, T.; Schlosser, M. Li6PS5X: A class of crystalline Li-rich solids with an unusually high Li+ mobility. Angew. Chem. Int. Ed. Engl. 2008, 47, 755–758. [Google Scholar] [CrossRef]
- Paskevicius, M.; Hansen, B.R.S.; Jorgensen, M.; Richter, B.; Jensen, T.R. Multifunctionality of silver closo-boranes. Nat. Commun. 2017, 8, 15136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Epp, V.; Gun, O.; Deiseroth, H.J.; Wilkening, M. Highly mobile ions: Low-Temperature NMR directly probes extremely fast Li+ hopping in argyrodite-type Li6PS5Br. J. Phys. Chem. Lett. 2013, 4, 2118–2123. [Google Scholar] [CrossRef]
- Chen, M.; Rao, R.P.; Adams, S. High capacity all-solid-state Cu-Li2S/Li6PS5Br/In batteries. Solid State Ion. 2014, 262, 183–187. [Google Scholar] [CrossRef]
- Rao, R.P.; Adams, S. Studies of lithium argyrodite solid electrolytes for all-solid-state batteries. Phys. Status Solidi A Appl. Mater. Sci. 2011, 208, 1804–1807. [Google Scholar] [CrossRef]
- Dao, A.H.; Lopez-Aranguren, P.; Cerny, R.; Guiader, O.; Zhan, J.X.; Cuevas, F.; Latroche, M.; Jordy, C. Improvement of the ionic conductivity on new substituted borohydride argyrodites. Solid State Ion. 2019, 339, 114987. [Google Scholar] [CrossRef]
- Jordy, C.; López-Aranguren, P.; Dao, A.H.; Latroche, M.; Zhang, J.; Cuevas, F. Solid electrolyte for a lithium-ion electrochemical element. Patent WO/2019/057840, 28 March 2019. [Google Scholar]
- Sakuda, A.; Yamauchi, A.; Yubuchi, S.; Kitamura, N.; Idemoto, Y.; Hayashi, A.; Tatsumisago, M. Mechanochemically prepared Li2S-P2S5-LiBH4 solid electrolytes with an argyrodite structure. ACS Omega 2018, 3, 5453–5458. [Google Scholar] [CrossRef]
- Sakuda, A.; Hayashi, A.; Tatsumisago, M. Recent progress on interface formation in all-solid-state batteries. Curr. Opin. Electrochem. 2017, 6, 108–114. [Google Scholar] [CrossRef]
- Sakuda, A.; Takeuchi, T.; Kobayashi, H. Electrode morphology in all-solid-state lithium secondary batteries consisting of LiNi1/3Co1/3Mn1/3O2 and Li2S-P2S5 solid electrolytes. Solid State Ion. 2016, 285, 112–117. [Google Scholar] [CrossRef] [Green Version]
- Latroche, M.; Blanchard, D.; Cuevas, F.; El Kharbachi, A.; Hauback, B.C.; Jensen, T.R.; de Jongh, P.E.; Kim, S.; Nazer, N.S.; Ngene, P.; et al. Full-Cell hydride-based solid-state Li batteries for energy storage. Int. J. Hydrog. Energy 2019, 44, 7875–7887. [Google Scholar] [CrossRef]
- Zeng, L.; Ichikawa, T.; Kawahito, K.; Miyaoka, H.; Kojima, Y. Bulk-Type all-solid-state lithium-ion batteries: remarkable performances of a carbon nanofiber-supported MgH2 composite electrode. ACS Appl. Mater. Interfaces 2017, 9, 2261–2266. [Google Scholar] [CrossRef]
- Zeng, L.; Kawahito, K.; Ikeda, S.; Ichikawa, T.; Miyaoka, H.; Kojima, Y. Metal hydride-based materials towards high performance negative electrodes for all-solid-state lithium-ion batteries. Chem. Commun. (Camb) 2015, 51, 9773–9776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikeda, S.; Ichikawa, T.; Goshome, K.; Yamaguchi, S.; Miyaoka, H.; Kojima, Y. Anode properties of Al2O3-added MgH2 for all-solid-state lithium-ion batteries. J. Solid State Electrochem. 2015, 19, 3639–3644. [Google Scholar] [CrossRef]
- Ikeda, S.; Ichikawa, T.; Kawahito, K.; Hirabayashi, K.; Miyaoka, H.; Kojima, Y. Anode properties of magnesium hydride catalyzed with niobium oxide for an all solid-state lithium-ion battery. Chem. Commun. (Camb.) 2013, 49, 7174–7176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawahito, K.; Zeng, L.; Ichikawa, T.; Miyaoka, H.; Kojima, Y. Electrochemical performance of titanium hydride for bulk-type all-solid-state lithium-ion batteries. Mater. Trans. 2016, 57, 755–757. [Google Scholar] [CrossRef] [Green Version]
- El Kharbachi, A.; Uesato, H.; Kawai, H.; Wenner, S.; Miyaoka, H.; Sørby, M.H.; Fjellvåg, H.; Ichikawa, T.; Hauback, B.C. MgH2-CoO: A conversion-type composite electrode for LiBH4-based all-solid-state lithium ion batteries. RSC Adv. 2018, 8, 23468–23474. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Xu, J.Y.; Lin, Y.; Li, J.; Lai, Y.Q.; Yuan, C.F.; Zhang, J.; Zhu, K. All-solid-state lithium ion battery: Research and industrial prospects. Acta Chim. Sin. 2013, 71, 869–878. [Google Scholar] [CrossRef] [Green Version]
- Mo, F.J.; Chi, X.W.; Yang, S.P.; Wu, F.L.; Song, Y.; Sun, D.L.; Yao, Y.; Fang, F. Stable three-dimensional metal hydride anodes for solid-state lithium storage. Energy Storage Mater. 2019, 18, 423–428. [Google Scholar] [CrossRef]
- Zaïdi, W.; Bonnet, J.P.; Zhang, J.; Cuevas, F.; Latroche, M.; Couillaud, S.; Bobet, J.L.; Sougrati, M.T.; Jumas, J.C.; Aymard, L. Reactivity of complex hydrides Mg2FeH6, Mg2CoH5 and Mg2NiH4 with lithium ion: Far from equilibrium electrochemically driven conversion reactions. Int. J. Hydrog. Energy 2013, 38, 4798–4808. [Google Scholar] [CrossRef]
- Huen, P.; Ravnsbæk, D.B. All-solid-state lithium batteries—The Mg2FeH6-electrode LiBH4-electrolyte system. Electrochem. Commun. 2018, 87, 81–85. [Google Scholar] [CrossRef]
- Dao, A.H.; Berti, N.; López-Aranguren, P.; Zhang, J.; Cuevas, F.; Jordy, C.; Latroche, M. Electrochemical properties of MgH2-TiH2 nanocomposite as active materials for all-solid-state lithium batteries. J. Power Sources 2018, 397, 143–149. [Google Scholar] [CrossRef]
- López-Aranguren, P.; Berti, N.; Dao, A.H.; Zhang, J.; Cuevas, F.; Latroche, M.; Jordy, C. An all-solid-state metal hydride—Sulfur lithium-ion battery. J. Power Sources 2017, 357, 56–60. [Google Scholar] [CrossRef]
- Unemoto, A.; Yasaku, S.; Nogami, G.; Tazawa, M.; Taniguchi, M.; Matsuo, M.; Ikeshoji, T.; Orimo, S. Development of bulk-type all-solid-state lithium-sulfur battery using LiBH4 electrolyte. Appl. Phys. Lett. 2014, 105, 083901. [Google Scholar] [CrossRef]
- Lin, Z.; Liang, C. Lithium-Sulfur batteries: From liquid to solid cells. J. Mater. Chem. A 2015, 3, 936–958. [Google Scholar] [CrossRef]
- Tarascon, J.M. Is lithium the new gold? Nat. Chem. 2010, 2, 510. [Google Scholar] [CrossRef] [PubMed]
- Amnesty International. Available online: https://www.amnesty.org/en/latest/news/2019/03/amnesty-challenges-industry-leaders-to-clean-up-their-batteries/ (accessed on 21 February 2020).
- Caputo, R.; Garroni, S.; Olid, D.; Teixidor, F.; Surinach, S.; Baro, M.D. Can Na2[B12H12] be a decomposition product of NaBH4? Phys. Chem. Chem. Phys. 2010, 12, 15093–15100. [Google Scholar] [CrossRef]
- Wiersema, R.J.; Hawthorne, M.F. Electrochemistry and Boron-11 nuclear magnetic-resonance spectra of monocarbon carboranes. Inorg. Chem. 1973, 12, 785–788. [Google Scholar] [CrossRef]
- Keen, D.A. Disordering phenomena in superionic conductors. J. Phys. Condens. Matter 2002, 14, R819–R857. [Google Scholar] [CrossRef]
- Udovic, T.J.; Matsuo, M.; Unemoto, A.; Verdal, N.; Stavila, V.; Skripov, A.V.; Rush, J.J.; Takamura, H.; Orimo, S. Sodium superionic conduction in Na2B12H12. Chem Commun. (Camb.) 2014, 50, 3750–3752. [Google Scholar] [CrossRef]
- Verdal, N.; Her, J.H.; Stavila, V.; Soloninin, A.V.; Babanova, O.A.; Skripov, A.V.; Udovic, T.J.; Rush, J.J. Complex high-temperature phase transitions in Li2B12H12 and Na2B12H12. J. Solid State Chem. 2014, 212, 81–91. [Google Scholar] [CrossRef]
- Sadikin, Y.; Schouwink, P.; Brighi, M.; Lodziana, Z.; Cerny, R. Modified anion packing of Na2B12H12 in close to room temperature superionic conductors. Inorg Chem. 2017, 56, 5006–5016. [Google Scholar] [CrossRef]
- Skripov, A.V.; Babanova, O.A.; Soloninin, A.V.; Stavila, V.; Verdal, N.; Udovic, T.J.; Rush, J.J. Nuclear magnetic resonance study of atomic motion in A2B12H12 (A = Na, K, Rb, Cs): Anion reorientations and Na+ mobility. J. Phys. Chem. C 2013, 117, 25961–25968. [Google Scholar] [CrossRef]
- Verdal, N.; Udovic, T.J.; Stavila, V.; Tang, W.S.; Rush, J.J.; Skripov, A.V. Anion reorientations in the superionic conducting phase of Na2B12H12. J. Phys. Chem. C 2014, 118, 17483–17489. [Google Scholar] [CrossRef]
- Wang, Y.; Richards, W.D.; Ong, S.P.; Miara, L.J.; Kim, J.C.; Mo, Y.; Ceder, G. Design principles for solid-state lithium superionic conductors. Nat. Mater. 2015, 14, 1026–1031. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.S.; Unemoto, A.; Zhou, W.; Stavila, V.; Matsuo, M.; Wu, H.; Orimo, S.I.; Udovic, T.J. Unparalleled lithium and sodium superionic conduction in solid electrolytes with large monovalent cage-like anions. Energy Environ. Sci. 2015, 8, 3637–3645. [Google Scholar] [CrossRef] [Green Version]
- Brighi, M.; Murgia, F.; Lodziana, Z.; Schouwink, P.; Wolczyk, A.; Cerny, R. A mixed anion hydroborate/carba-hydroborate as a room temperature Na-ion solid electrolyte. J. Power Sources 2018, 404, 7–12. [Google Scholar] [CrossRef]
- Pauling, L. The principles determining the structure of complex ionic crystals. J. Am. Chem. Soc. 1929, 51, 1010–1026. [Google Scholar] [CrossRef]
- Tang, W.S.; Matsuo, M.; Wu, H.; Stavila, V.; Zhou, W.; Talin, A.A.; Soloninin, A.V.; Skoryunov, R.V.; Babanova, O.A.; Skripov, A.V.; et al. Liquid-Like ionic conduction in solid lithium and sodium monocarba-closo-decaborates near or at room temperature. Adv. Energy Mater. 2016, 6, 1502237. [Google Scholar] [CrossRef]
- Duchêne, L.; Kühnel, R.S.; Stilp, E.; Cuervo Reyes, E.; Remhof, A.; Hagemann, H.; Battaglia, C. A sTable 3 V all-solid-state sodium-ion battery based on a closo-borate electrolyte. Energy Environ. Sci. 2017, 10, 2609–2615. [Google Scholar] [CrossRef]
- Sharafi, A.; Meyer, H.M.; Nanda, J.; Wolfenstine, J.; Sakamoto, J. Characterizing the Li-Li7La3Zr2O12 interface stability and kinetics as a function of temperature and current density. J. Power Sources 2016, 302, 135–139. [Google Scholar] [CrossRef] [Green Version]
- Cheng, E.J.; Sharafi, A.; Sakamoto, J. Intergranular Li metal propagation through polycrystalline Li6.25Al0.25La3Zr2O12 ceramic electrolyte. Electrochim. Acta 2017, 223, 85–91. [Google Scholar] [CrossRef] [Green Version]
- Taylor, N.J.; Stangeland-Molo, S.; Haslam, C.G.; Sharafi, A.; Thompson, T.; Wang, M.; Garcia-Mendez, R.; Sakamoto, J. Demonstration of high current densities and extended cycling in the garnet Li7La3Zr2O12 solid electrolyte. J. Power Sources 2018, 396, 314–318. [Google Scholar] [CrossRef]
- Pasquini, L. The effects of nanostructure on the hydrogen sorption properties of magnesium-based metallic compounds: A review. Crystals 2018, 8, 106. [Google Scholar] [CrossRef] [Green Version]
- Selvam, P.; Viswanathan, B.; Swamy, C.S.; Srinivasan, V. Magnesium and magnesium alloy hydrides. Int. J. Hydrog. Energy 1986, 11, 169–192. [Google Scholar] [CrossRef]
- Crivello, J.C.; Dam, B.; Denys, R.V.; Dornheim, M.; Grant, D.M.; Huot, J.; Jensen, T.R.; de Jongh, P.; Latroche, M.; Milanese, C.; et al. Review of magnesium hydride-based materials: Development and optimisation. Appl. Phys. A Mater. Sci. Process. 2016, 122, 5077. [Google Scholar] [CrossRef] [Green Version]
- Webb, C.J. A review of catalyst-enhanced magnesium hydride as a hydrogen storage material. J. Phys. Chem. Solids 2015, 84, 96–106. [Google Scholar] [CrossRef]
- Cesario Asselli, A.A.; Bourbeau Hébert, N.; Huot, J. The role of morphology and severe plastic deformation on the hydrogen storage properties of magnesium. Int. J. Hydrog. Energy 2014, 39, 12778–12783. [Google Scholar] [CrossRef]
- Chu, H.; Qiu, S.; Sun, L.; Huot, J. Enhancement of the initial hydrogenation of Mg by ball milling with alkali metal amides MNH2 (M = Li or Na). Dalton Trans. 2015, 44, 16694–16697. [Google Scholar] [CrossRef]
- Eijt, S.W.H.; Leegwater, H.; Schut, H.; Anastasopol, A.; Egger, W.; Ravelli, L.; Hugenschmidt, C.; Dam, B. Layer-resolved study of the Mg to MgH2 transformation in Mg-Ti films with short-range chemical order. J. Alloys Compd. 2011, 509, S567–S571. [Google Scholar] [CrossRef]
- AlMatrouk, H.S.; Chihaia, V. Theoretical study on the effects of the magnesium hydride doping with cobalt and nickel on the hydrogen release. Int. J. Hydrog. Energy 2015, 40, 5319–5325. [Google Scholar] [CrossRef]
- Barcelo, S.; Rogers, M.; Grigoropoulos, C.P.; Mao, S.S. Hydrogen storage property of sandwiched magnesium hydride nanoparticle thin film. Int. J. Hydrog. Energy 2010, 35, 7232–7235. [Google Scholar] [CrossRef]
- Okamoto, H. Mg-Pd (Magnesium-Palladium). J. Phase Equilibria Diffus. 2010, 31, 407–408. [Google Scholar] [CrossRef]
- Nobuhara, K.; Kasai, H.; Diño, W.A.; Nakanishi, H. H2 dissociative adsorption on Mg, Ti, Ni, Pd and La surfaces. Surf. Sci. 2004, 566–568, 703–707. [Google Scholar] [CrossRef]
- Krozer, A.; Kasemo, B. Unusual kinetics due to interface hydride formation in the hydriding of Pd/Mg sandwich layers. J. Vac. Sci. Technol. A Vac. Surf. Film 1987, 5, 1003–1005. [Google Scholar] [CrossRef]
- Higuchi, K.; Kajioka, H.; Toiyama, K.; Fujii, H.; Orimo, S.; Kikuchi, Y. In situ study of hydriding-dehydriding properties in some Pd/Mg thin films with different degree of Mg crystallization. J. Alloys Compd. 1999, 293–295, 484–489. [Google Scholar] [CrossRef]
- Kim, K.-B.; Shim, J.-H.; Park, S.-H.; Choi, I.-S.; Oh, K.H.; Cho, Y.W. Dehydrogenation reaction pathway of the LiBH4-MgH2 composite under various pressure conditions. J. Phys. Chem. C 2015, 119, 9714–9720. [Google Scholar] [CrossRef]
- Vajo, J.J.; Mertens, F.; Ahn, C.C.; Bowman, R.C.; Fultz, B. Altering hydrogen storage properties by hydride destabilization through alloy formation: LiH and MgH2 destabilized with Si. J. Phys. Chem. B 2004, 108, 13977–13983. [Google Scholar] [CrossRef]
- Stampfer, J.F., Jr.; Holley, C.E., Jr.; Suttle, J.F. The magnesium-hydrogen system. J. Am. Chem. Soc. 1960, 82, 3504–3508. [Google Scholar] [CrossRef] [Green Version]
- Bogdanovic, B. Thermodynamic investigation of the magnesium-hydrogen system. J. Alloys Compd. 1999, 282, 84–92. [Google Scholar] [CrossRef]
- Takeichi, N.; Sakaida, Y.; Kiyobayashi, T.; Takeshita, H.T. Hydrogen absorption and desorption behavior of magnesium hydride: Incubation period and reaction mechanism. Mater. Trans. 2014, 55, 1161–1167. [Google Scholar] [CrossRef] [Green Version]
- Mooij, L.; Dam, B. Hysteresis and the role of nucleation and growth in the hydrogenation of Mg nanolayers. Phys. Chem. Chem. Phys. 2013, 15, 2782–2792. [Google Scholar] [CrossRef]
- Mooij, L.; Dam, B. Nucleation and growth mechanisms of nano magnesium hydride from the hydrogen sorption kinetics. Phys. Chem. Chem. Phys. 2013, 15, 11501–11510. [Google Scholar] [CrossRef] [PubMed]
- Ingason, A.S.; Olafsson, S. Thermodynamics of hydrogen uptake in Mg films studied by resistance measurements. J. Alloys Compd. 2005, 404, 469–472. [Google Scholar] [CrossRef]
- Gharavi, A.G.; Akyildiz, H.; Ozturk, T. Thickness effects in hydrogen sorption of Mg/Pd thin films. J. Alloys Compd. 2013, 580, S175–S178. [Google Scholar] [CrossRef]
- Hadjixenophontos, E.; Zhang, K.; Weigel, A.; Stender, P.; Schmitz, G. Hydrogenation of Pd/Mg films: A quantitative assessment of transport coefficients. Int. J. Hydrog. Energy 2019, 44, 27862–27875. [Google Scholar] [CrossRef]
- Yao, X.; Zhu, Z.H.; Cheng, H.M.; Lu, G.Q. Hydrogen diffusion and effect of grain size on hydrogenation kinetics in magnesium hydrides. J. Mater. Res. 2008, 23, 336–340. [Google Scholar] [CrossRef]
- Fernandez, J.F.; Sanchez, C.R. Rate determining step in the absorption and desorption of hydrogen by magnesium. J. Alloys Compd. 2002, 340, 189–198. [Google Scholar] [CrossRef]
- Han, J.S.; Pezat, M.; Lee, J.Y. Thermal-Desorption of hydrogen from magnesium hydride. Scr. Metall. 1986, 20, 951–956. [Google Scholar] [CrossRef]
- Hadjixenophontos, E.; Roussel, M.; Sato, T.; Weigel, A.; Stender, P.; Orimo, S.I.; Schmitz, G. Imaging the hydrogenation of Mg thin films. Int. J. Hydrog. Energy 2017, 42, 22411–22416. [Google Scholar] [CrossRef]
- Hadjixenophontos, E.; Michalek, L.; Weigel, A.; Schmitz, G. Hydrogen sorption kinetics in MgH2 and TiH2 thin films. Defect Diffus. Forum 2018, 383, 127–132. [Google Scholar] [CrossRef]
- Kumar, S.; Reddy, G.L.N.; Raju, V.S. Hydrogen storage in Pd capped thermally grown Mg films: Studies by nuclear resonance reaction analysis. J. Alloys Compd. 2009, 476, 500–506. [Google Scholar] [CrossRef]
- Reddy, G.L.N.; Kumar, S.; Sunitha, Y.; Kalavathi, S.; Raju, V.S. Intermixing and formation of Pd-Mg intermetallics in Pd/Mg/Si films. J. Alloys Compd. 2009, 481, 714–718. [Google Scholar] [CrossRef]
- Zheng, S.; Wang, K.; Oleshko, V.P.; Bendersky, L.A. Mg-Fe thin films: A phase-separated structure with fast kinetics of hydrogenation. J. Phys. Chem. C 2012, 116, 21277–21284. [Google Scholar] [CrossRef]
- Fry, C.M.P.; Grant, D.M.; Walker, G.S. Catalysis and evolution on cycling of nano-structured magnesium multilayer thin films. Int. J. Hydrog. Energy 2014, 39, 1173–1184. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Cao, Y.; Huang, L.; Gao, M.; Pan, H. Rare earth-Mg-Ni-based hydrogen storage alloys as negative electrode materials for Ni/MH batteries. J. Alloys Compd. 2011, 509, 675–686. [Google Scholar] [CrossRef]
- Jung, H.; Cho, S.; Lee, W. A catalytic effect on hydrogen absorption kinetics in Pd/Ti/Mg/Ti multilayer thin films. J. Alloys Compd. 2015, 635, 203–206. [Google Scholar] [CrossRef]
- Vermeulen, P.; Niessen, R.A.H.; Notten, P.H.L. Hydrogen storage in metastable MgyTi(1-y) thin films. Electrochem. Commun. 2006, 8, 27–32. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhuang, X.; Zhu, Y.; Wan, N.; Li, L.; Dong, J. Synergistic effects of TiH2 and Pd on hydrogen desorption performances of MgH2. Int. J. Hydrog. Energy 2015, 40, 16338–16346. [Google Scholar] [CrossRef]
- Bazzanella, N.; Checchetto, R.; Miotello, A. Atoms and nanoparticles of transition metals as catalysts for hydrogen desorption from magnesium hydride. J. Nanomater. 2011, 2011, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Peng, D.; Ding, Z.; Zhang, L.; Fu, Y.; Wang, J.; Li, Y.; Han, S. Remarkable hydrogen storage properties and mechanisms of the shell-core MgH2@carbon aerogel microspheres. Int. J. Hydrog. Energy 2018, 43, 3731–3740. [Google Scholar] [CrossRef]
- Ding, Z.M.; Fu, Y.K.; Wang, Y.; Bi, J.; Zhang, L.; Peng, D.D.; Li, Y.; Han, S.M. MgCNi3 prepared by powder metallurgy for improved hydrogen storage properties of MgH2. Int. J. Hydrog. Energy 2019, 44, 8347–8356. [Google Scholar] [CrossRef]
- Pavlyuk, V.; Dmytriv, G.; Chumak, I.; Gutfleisch, O.; Lindemann, I.; Ehrenberg, H. High hydrogen content super-lightweight intermetallics from the Li-Mg-Si system. Int. J. Hydrog. Energy 2013, 38, 5724–5737. [Google Scholar] [CrossRef]
- Garroni, S.; Santoru, A.; Cao, H.J.; Dornheim, M.; Klassen, T.; Milanese, C.; Gennari, F.; Pistidda, C. Recent progress and new perspectives on metal amide and imide systems for solid-state hydrogen storage. Energies 2018, 11, 1027. [Google Scholar] [CrossRef] [Green Version]
- Cao, H.J.; Zhang, Y.; Wang, J.H.; Xiong, Z.T.; Wu, G.T.; Chen, P. Materials design and modification on amide-based composites for hydrogen storage. Prog. Nat. Sci. Mater. Int. 2012, 22, 550–560. [Google Scholar] [CrossRef] [Green Version]
- Gomez, Y.A.; Oyarce, A.; Lindbergh, G.; Lagergren, C. Ammonia contamination of a proton exchange membrane fuel cell. J. Electrochem. Soc. 2018, 165, F189–F197. [Google Scholar] [CrossRef]
- Rude, L.H.; Nielsen, T.K.; Ravnsbaek, D.B.; Bosenberg, U.; Ley, M.B.; Richter, B.; Arnbjerg, L.M.; Dornheim, M.; Filinchuk, Y.; Besenbacher, F.; et al. Tailoring properties of borohydrides for hydrogen storage: A review. Phys. Status Solidi A Appl. Mater. Sci. 2011, 208, 1754–1773. [Google Scholar] [CrossRef]
- Chen, P.; Xiong, Z.; Luo, J.; Lin, J.; Tan, K.L. Interaction of hydrogen with metal nitrides and imides. Nature 2002, 420, 302–304. [Google Scholar] [CrossRef]
- Hu, Y.H.; Ruckenstein, E. H2 storage in Li3N temperature-programmed hydrogenation and dehydrogenation. Ind. Eng. Chem. Res. 2003, 42, 5135–5139. [Google Scholar] [CrossRef]
- Leng, H.Y.; Ichikawa, T.; Hino, S.; Hanada, N.; Isobe, S.; Fujii, H. New metal-N-H system composed of Mg(NH2)2 and LiH for hydrogen storage. J. Phys. Chem. B 2004, 108, 8763–8765. [Google Scholar] [CrossRef]
- Luo, W. (LiNH2-MgH2): A viable hydrogen storage system. J. Alloys Compd. 2004, 381, 284–287. [Google Scholar] [CrossRef]
- Nakamori, Y.; Kitahara, G.; Miwa, K.; Towata, S.; Orimo, S. Reversible hydrogen-storage functions for mixtures of Li3N and Mg3N2. Appl. Phys. A 2005, 80, 1–3. [Google Scholar] [CrossRef]
- Nakamori, Y.; Orimo, S. Destabilization of Li-based complex hydrides. J. Alloys Compd. 2004, 370, 271–275. [Google Scholar] [CrossRef]
- Xiong, Z.T.; Wu, G.T.; Hu, H.J.; Chen, P. Ternary imides for hydrogen storage. Adv. Mater. 2004, 16, 1522. [Google Scholar] [CrossRef]
- Xiong, Z.; Hu, J.; Wu, G.; Chen, P.; Luo, W.; Gross, K.; Wang, J. Thermodynamic and kinetic investigations of the hydrogen storage in the Li-Mg-N-H system. J. Alloys Compd. 2005, 398, 235–239. [Google Scholar] [CrossRef]
- Ichikawa, T. Amides, Imides and Mixtures. In Handbook of Hydrogen Storage: New Materials for Future Energy Storage; Hirscher, M., Ed.; Wiley-VCH: Weinheim, Germany, 2010; Chapter 6; pp. 159–185. [Google Scholar] [CrossRef]
- Paskevicius, M.; Jepsen, L.H.; Schouwink, P.; Cerny, R.; Ravnsbaek, D.B.; Filinchuk, Y.; Dornheim, M.; Besenbacher, F.; Jensen, T.R. Metal borohydrides and derivatives—Synthesis, structure and properties. Chem. Soc. Rev. 2017, 46, 1565–1634. [Google Scholar] [CrossRef] [PubMed]
- Dematteis, E.M.; Vaunois, S.; Pistidda, C.; Dornheim, M.; Baricco, M. Reactive hydride composite of Mg2NiH4 with borohydrides eutectic mixtures. Crystals 2018, 8, 90. [Google Scholar] [CrossRef] [Green Version]
- Callini, E.; Atakli, Z.O.K.; Hauback, B.C.; Orimo, S.; Jensen, C.; Dornheim, M.; Grant, D.; Cho, Y.W.; Chen, P.; Hjorvarsson, B.; et al. Complex and liquid hydrides for energy storage. Appl. Phys. A Mater. Sci. Process. 2016, 122, 353. [Google Scholar] [CrossRef]
- Soulie, J.P.; Renaudin, G.; Cerny, R.; Yvon, K. Lithium Boro-hydride LiBH4. Part 1. Crystal structure. ChemInform 2003, 34. [Google Scholar] [CrossRef]
- Møller, K.; Sheppard, D.; Ravnsbæk, D.; Buckley, C.; Akiba, E.; Li, H.-W.; Jensen, T. Complex metal hydrides for hydrogen, thermal and electrochemical energy storage. Energies 2017, 10, 1645. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.B.; Zhou, X.D.; Cai, Q.; James, W.J.; Yelon, W.B. Crystal and electronic structures of LiNH2. Appl. Phys. Lett. 2006, 88, 041914. [Google Scholar] [CrossRef]
- Chater, P.A.; David, W.I.; Anderson, P.A. Synthesis and structure of the new complex hydride Li2BH4NH2. Chem. Commun. (Camb) 2007, 4770–4772. [Google Scholar] [CrossRef]
- Wu, H.; Zhou, W.; Udovic, T.J.; Rush, J.J.; Yildirim, T. Structures and crystal chemistry of Li2BNH6 and Li4BN3H10. Chem. Mat. 2008, 20, 1245–1247. [Google Scholar] [CrossRef]
- Herbst, J.F.; Hector, L.G. Electronic structure and energetics of the quaternary hydride Li4BN3H10. Appl. Phys. Lett. 2006, 88, 231904. [Google Scholar] [CrossRef] [Green Version]
- Siegel, D.J.; Wolverton, C.; Ozolins, V. Reaction energetics and crystal structure of Li4BN3H10 from first principles. Phys. Rev. B 2007, 75. [Google Scholar] [CrossRef] [Green Version]
- Lim, K.L.; Kazemian, H.; Yaakob, Z.; Daud, W.R.W. Solid-State materials and methods for hydrogen storage: A critical review. Chem. Eng. Technol. 2010, 33, 213–226. [Google Scholar] [CrossRef]
- SGTE Substance Database V 4.1. Available online: http://www.crct.polymtl.ca/fact/documentation/sgps_list.htm (accessed on 21 February 2020).
- El Kharbachi, A.; Pinatel, E.; Nuta, I.; Baricco, M. A thermodynamic assessment of LiBH4. Calphad 2012, 39, 80–90. [Google Scholar] [CrossRef] [Green Version]
- Borgschulte, A.; Jones, M.O.; Callini, E.; Probst, B.; Kato, S.; Zuttel, A.; David, W.I.F.; Orimo, S. Surface and bulk reactions in borohydrides and amides. Energy Environ. Sci. 2012, 5, 6823–6832. [Google Scholar] [CrossRef]
- Singer, J.P.; Meyer, M.S.; Speer, R.M.; Fischer, J.E.; Pinkerton, F.E. Determination of the phase behavior of (LiNH2)c(LiBH4)1-c quaternary hydrides through in situ x-ray diffraction. J. Phys. Chem. C 2009, 113, 18927–18934. [Google Scholar] [CrossRef]
- Pinkerton, F.E.; Meisner, G.P.; Meyer, M.S.; Balogh, M.P.; Kundrat, M.D. Hydrogen desorption exceeding ten weight percent from the new quaternary hydride Li3BN2H8. J. Phys. Chem B 2005, 109, 6–8. [Google Scholar] [CrossRef]
- Chen, P.; Xiong, Z.T.; Luo, J.Z.; Lin, J.Y.; Tan, K.L. Interaction between lithium amide and lithium hydride. J. Phys. Chem. B 2003, 107, 10967–10970. [Google Scholar] [CrossRef]
- Shaw, L.L.; Osborn, W.; Markmaitree, T.; Wan, X. The reaction pathway and rate-limiting step of dehydrogenation of the LiHN2 + LiH mixture. J. Power Sources 2008, 177, 500–505. [Google Scholar] [CrossRef]
- Miceli, G.; Cucinotta, C.S.; Bernasconi, M.; Parrinello, M. First principles study of the LiNH2/Li2NH transformation. J. Phys. Chem. C 2010, 114, 15174–15183. [Google Scholar] [CrossRef]
- Hu, J.; Liu, Y.; Wu, G.; Xiong, Z.; Chua, Y.S.; Chen, P. Improvement of hydrogen storage properties of the Li-Mg-N-H system by addition of LiBH4. Chem. Mater. 2008, 20, 4398–4402. [Google Scholar] [CrossRef]
- Wang, J.; Liu, T.; Wu, G.; Li, W.; Liu, Y.; Araújo, C.M.; Scheicher, R.H.; Blomqvist, A.; Ahuja, R.; Xiong, Z. Potassium-Modified Mg(NH2)2/2LiH system for hydrogen storage. Angew. Chem. Int. Ed. 2009, 48, 5828–5832. [Google Scholar] [CrossRef] [PubMed]
- Pistidda, C.; Santoru, A.; Garroni, S.; Bergemann, N.; Rzeszutek, A.; Horstmann, C.; Thomas, D.; Klassen, T.; Dornheim, M. First direct study of the ammonolysis reaction in the most common alkaline and alkaline earth metal hydrides by in situ SR-PXD. J. Phys. Chem. C 2015, 119, 934–943. [Google Scholar] [CrossRef]
- Jacobs, H.; Von Osten, E. Die Kristallstruktur einer neuen modifikation des kaliumamids, KNH2. Z. Nat. B 1976, 31, 385–386. [Google Scholar] [CrossRef]
- Wang, J.; Chen, P.; Pan, H.; Xiong, Z.; Gao, M.; Wu, G.; Liang, C.; Li, C.; Li, B.; Wang, J. Solid-Solid heterogeneous catalysis: The role of potassium in promoting the dehydrogenation of the Mg(NH2)2/2 LiH composite. ChemSusChem 2013, 6, 2181–2189. [Google Scholar] [CrossRef]
- Wang, J.; Wu, G.; Chua, Y.S.; Guo, J.; Xiong, Z.; Zhang, Y.; Gao, M.; Pan, H.; Chen, P. hydrogen sorption from the Mg(NH2)2-KH system and synthesis of an amide-imide complex of KMg(NH)(NH2). ChemSusChem 2011, 4, 1622–1628. [Google Scholar] [CrossRef]
- Napolitano, E.; Dolci, F.; Campesi, R.; Pistidda, C.; Hoelzel, M.; Moretto, P.; Enzo, S. Crystal structure solution of KMg(ND)(ND2): An ordered mixed amide/imide compound. Int. J. Hydrog. Energy 2014, 39, 868–876. [Google Scholar] [CrossRef] [Green Version]
- Palvadeau, P.; Rouxel, J. L’amidure ternaire K2Mg(NH2)4, l’imidure K2Mg(NH)2 et le nitrure double KMgN. CR Seances Acad. Sci. Ser. C 1968, 266, 1605–1607. [Google Scholar]
- Palvadeau, P.; Rouxel, J. Preparation and structural characterization of magnesium and alkaline metal double amides—M2mg(Nh2)4 Derivatives. Bull. Soc. Chim. Fr. 1970, 480–485. [Google Scholar]
- Santoru, A.; Garroni, S.; Pistidda, C.; Milanese, C.; Girella, A.; Marini, A.; Masolo, E.; Valentoni, A.; Bergemann, N.; Le, T.T.; et al. A new potassium-based intermediate and its role in the desorption properties of the K-Mg-N-H system. Phys. Chem. Chem. Phys. 2016, 18, 3910–3920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santoru, A.; Pistidda, C.; Sorby, M.H.; Chierotti, M.R.; Garroni, S.; Pinatel, E.; Karimi, F.; Cao, H.; Bergemann, N.; Le, T.T.; et al. KNH2-KH: A metal amide-hydride solid solution. Chem. Commun. (Camb) 2016, 52, 11760–11763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durojaiye, T.; Hayes, J.; Goudy, A. Rubidium hydride: An exceptional dehydrogenation catalyst for the lithium amide/magnesium hydride system. J. Phys. Chem. C 2013, 117, 6554–6560. [Google Scholar] [CrossRef]
- Hayes, J.; Durojaiye, T.; Goudy, A. Hydriding and dehydriding kinetics of RbH-doped 2LiNH2/MgH2 hydrogen storage system. J. Alloys Compd. 2015, 645, S496–S499. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Liu, Y.; Gu, Y.; Gao, M.; Pan, H. Improved hydrogen-storage thermodynamics and kinetics for an RbF-Doped Mg (NH2) 2-2 LiH system. Chem. Asian J. 2013, 8, 2136–2143. [Google Scholar] [CrossRef]
- Li, C.; Liu, Y.F.; Ma, R.J.; Zhang, X.; Li, Y.; Gao, M.X.; Pan, H.G. Superior dehydrogenation/hydrogenation kinetics and long-term cycling performance of K and Rb cocatalyzed Mg(NH2)(2)-2LiH system. ACS Appl. Mater. Interfaces 2014, 6, 17024–17033. [Google Scholar] [CrossRef]
- Santoru, A.; Pistidda, C.; Brighi, M.; Chierotti, M.R.; Heere, M.; Karimi, F.; Cao, H.; Capurso, G.; Chaudhary, A.L.; Gizer, G.; et al. Insights into the Rb-Mg-N-H System: An ordered mixed amide/imide phase and a disordered amide/hydride solid solution. Inorg Chem. 2018, 57, 3197–3205. [Google Scholar] [CrossRef]
- Durojaiye, T.; Hayes, J.; Goudy, A. Potassium, rubidium and cesium hydrides as dehydrogenation catalysts for the lithium amide/magnesium hydride system. Int. J. Hydrog. Energy 2015, 40, 2266–2273. [Google Scholar] [CrossRef] [Green Version]
- Hayes, J.; Goudy, A. Thermodynamics, kinetics and modeling studies of KH-RbH-and CsH-doped 2LiNH2/MgH2 hydrogen storage systems. Int. J. Hydrog. Energy 2015, 40, 12336–12342. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.X.; Liu, Y.F.; Zhang, X.; Yang, Y.X.; Zhang, Q.H.; Jin, T.; Wang, Y.X.; Gao, M.X.; Sun, L.X.; Pan, H.G. Synthesis of CsH and its effect on the hydrogen storage properties of the Mg(NH2)(2)-2LiH system. Int. J. Hydrog. Energy 2016, 41, 11264–11274. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, Y.; Zhang, M.; Leng, Z.; Gao, M.; Hu, J.; Liu, Y.; Pan, H. Improved overall hydrogen storage properties of a CsH and KH co-doped Mg (NH 2) 2/2LiH system by forming mixed amides of Li-K and Cs-Mg. RSC Adv. 2017, 7, 30357–30364. [Google Scholar] [CrossRef] [Green Version]
- Callister, W.D., Jr.; Rethwisch, D.G. Materials Science and Engineering: An Introduction, 1 ed.; John Wiley & Sons: Hoboken, NJ, USA, 2014. [Google Scholar]
- Blomgren, G.E.; Vanartsdalen, E.R. Fused salts. Annu. Rev. Phys. Chem. 1960, 11, 273–306. [Google Scholar] [CrossRef]
- Sundermeyer, W. Fused salts and their use as reaction media. Angew. Chem. Int. Ed. 1965, 4, 222. [Google Scholar] [CrossRef]
- Habashi, F. Handbook of Extractive Metallurgy; Wiley VCH: Weinheim, Germany, 1997. [Google Scholar]
- Harries, D.N.; Paskevicius, M.; Sheppard, D.A.; Price, T.E.C.; Buckley, C.E. Concentrating solar thermal heat storage using metal hydrides. Proc. IEEE 2012, 100, 539–549. [Google Scholar] [CrossRef]
- Paskevicius, M.; Pitt, M.P.; Brown, D.H.; Sheppard, D.A.; Chumphongphan, S.; Buckley, C.E. First-Order phase transition in the Li2B12H12 system. Phys. Chem. Chem. Phys. 2013, 15, 15825–15828. [Google Scholar] [CrossRef] [Green Version]
- Paskevicius, M.; Ley, M.B.; Sheppard, D.A.; Jensen, T.R.; Buckley, C.E. Eutectic melting in metal borohydrides. Phys. Chem. Chem. Phys. 2013, 15, 19774–19789. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.Y.; Ravnsbæk, D.; Lee, Y.-S.; Kim, Y.; Cerenius, Y.; Shim, J.-H.; Jensen, T.R.; Hur, N.H.; Cho, Y.W. Decomposition reactions and reversibility of the LiBH4-Ca(BH4)2 composite. J. Phys. Chem. C 2009, 113, 15080–15086. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Reed, D.; Paterakis, C.; Contreras Vasquez, L.; Baricco, M.; Book, D. Study of the decomposition of a 0.62LiBH 4-0.38NaBH 4 mixture. Int. J. Hydrog. Energy 2017, 42, 22480–22488. [Google Scholar] [CrossRef]
- Roedern, E.; Hansen, B.R.S.; Ley, M.B.; Jensen, T.R. Effect of eutectic melting, reactive hydride composites, and nanoconfinement on decomposition and reversibility of LiBH4-KBH4. J. Phys. Chem. C 2015, 119, 25818–25825. [Google Scholar] [CrossRef]
- Javadian, P.; Jensen, T.R. Enhanced hydrogen reversibility of nanoconfined LiBH4-Mg(BH4)(2). Int. J. Hydrog. Energy 2014, 39, 9871–9876. [Google Scholar] [CrossRef]
- Doroodian, A.; Dengler, J.E.; Genest, A.; Rosch, N.; Rieger, B. Methylguanidinium borohydride: An ionic-liquid-based hydrogen-storage material. Angew. Chem. Int. Ed. Engl. 2010, 49, 1871–1873. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Gao, H.; Shreeve, J.M. Borohydride ionic liquids and borane/ionic-liquid solutions as hypergolic fuels with superior low ignition-delay times. Angew. Chem. Int. Ed. Engl. 2014, 53, 2969–2972. [Google Scholar] [CrossRef]
- Yan, Y.; Rentsch, D.; Remhof, A. Controllable decomposition of Ca(BH4)2 for reversible hydrogen storage. Phys. Chem. Chem. Phys. 2017, 19, 7788–7792. [Google Scholar] [CrossRef] [PubMed]
- Li, H.W.; Orimo, S.; Nakamori, Y.; Miwa, K.; Ohba, N.; Towata, S.; Zuttel, A. Materials designing of metal borohydrides: Viewpoints from thermodynamical stabilities. J. Alloys Compd. 2007, 446, 315–318. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Peaslee, D.; Sheehan, T.P.; Majzoub, E.H. Decomposition behavior of eutectic LiBH4-Mg(BH4)2 and its confinement effects in ordered nanoporous carbon. J. Phys. Chem. C 2014, 118, 27265–27271. [Google Scholar] [CrossRef]
- Ley, M.B.; Roedern, E.; Jensen, T.R. Eutectic melting of LiBH4-KBH4. Phys. Chem. Chem. Phys. 2014, 16, 24194–24199. [Google Scholar] [CrossRef]
- Javadian, P.; Sheppard, D.A.; Buckley, C.E.; Jensen, T.R. Hydrogen storage properties of nanoconfined LiBH4-NaBH4. Int. J. Hydrog. Energy 2015, 40, 14916–14924. [Google Scholar] [CrossRef] [Green Version]
- Dematteis, E.M.; Roedern, E.; Pinatel, E.R.; Corno, M.; Jensen, T.R.; Baricco, M. A thermodynamic investigation of the LiBH4-NaBH4 system. RSC Adv. 2016, 6, 60101–60108. [Google Scholar] [CrossRef] [Green Version]
- Jensen, S.R.H.; Jepsen, L.H.; Skibsted, J.; Jensen, T.R. Phase diagram for the NaBH4-KBH4 system and the stability of a Na1-xKxBH4 solid solution. J. Phys. Chem. C 2015, 119, 27919–27929. [Google Scholar] [CrossRef]
- Dematteis, E.M.; Pinatel, E.R.; Corno, M.; Jensen, T.R.; Baricco, M. Phase diagrams of the LiBH4-NaBH4-KBH4 system. Phys. Chem. Chem. Phys. 2017, 19, 25071–25079. [Google Scholar] [CrossRef]
- Bardají, E.G.; Zhao-Karger, Z.; Boucharat, N.; Nale, A.; van Setten, M.J.; Lohstroh, W.; Röhm, E.; Catti, M.; Fichtner, M. LiBH4-Mg(BH4)2: A physical mixture of metal borohydrides as hydrogen storage material. J. Phys. Chem. C 2011, 115, 6095–6101. [Google Scholar] [CrossRef]
- Fang, Z.Z.; Kang, X.D.; Wang, P.; Li, H.W.; Orimo, S.I. Unexpected dehydrogenation behavior of LiBH4/Mg(BH4)(2) mixture associated with the in situ formation of dual-cation borohydride. J. Alloys Compd. 2010, 491, L1–L4. [Google Scholar] [CrossRef]
- Lee, H.S.; Lee, Y.S.; Suh, J.Y.; Kim, M.; Yu, J.S.; Cho, Y.W. Enhanced desorption and absorption properties of eutectic LiBH4-Ca(BH4)(2) infiltrated into mesoporous carbon. J. Phys. Chem. C 2011, 115, 20027–20035. [Google Scholar] [CrossRef]
- Ley, M.; Roedern, E.; Thygesen, P.; Jensen, T. Melting behavior and thermolysis of NaBH4-Mg(BH4)2 and NaBH4-Ca(BH4)2 composites. Energies 2015, 8, 2701–2713. [Google Scholar] [CrossRef] [Green Version]
- Huot, J.; Cuevas, F.; Deledda, S.; Edalati, K.; Filinchuk, Y.; Grosdidier, T.; Hauback, B.C.; Heere, M.; Jensen, T.R.; Latroche, M.; et al. Mechanochemistry of metal hydrides: Recent advances. Materials 2019, 12, 2778. [Google Scholar] [CrossRef] [Green Version]
- Hino, S.; Fonnelop, J.E.; Corno, M.; Zavorotynska, O.; Damin, A.; Richter, B.; Baricco, M.; Jensen, T.R.; Sorby, M.H.; Hauback, B.C. Halide substitution in magnesium borohydride. J. Phys. Chem. C 2012, 116, 12482–12488. [Google Scholar] [CrossRef] [Green Version]
- Rude, L.H.; Groppo, E.; Arnbjerg, L.M.; Ravnsbæk, D.B.; Malmkjær, R.A.; Filinchuk, Y.; Baricco, M.; Besenbacher, F.; Jensen, T.R. Iodide substitution in lithium borohydride, LiBH4-LiI. J. Alloys Compd. 2011, 509, 8299–8305. [Google Scholar] [CrossRef] [Green Version]
- Olsen, J.E.; Karen, P.; Sørby, M.H.; Hauback, B.C. Effect of chloride substitution on the order-disorder transition in NaBH4 and Na11BD4. J. Alloys Compd. 2014, 587, 374–379. [Google Scholar] [CrossRef] [Green Version]
- Grove, H.; Rude, L.H.; Jensen, T.R.; Corno, M.; Ugliengo, P.; Baricco, M.; Sorby, M.H.; Hauback, B.C. Halide substitution in Ca(BH4)(2). RSC Adv. 2014, 4, 4736–4742. [Google Scholar] [CrossRef] [Green Version]
- Rude, L.H.; Filinchuk, Y.; Sørby, M.H.; Hauback, B.C.; Besenbacher, F.; Jensen, T.R. Anion substitution in Ca(BH4)2-CaI2: Synthesis, structure and stability of three new compounds. J. Phys. Chem. C 2011, 115, 7768–7777. [Google Scholar] [CrossRef]
- Ravnsbæk, D.B.; Rude, L.H.; Jensen, T.R. Chloride substitution in sodium borohydride. J. Solid State Chem. 2011, 184, 1858–1866. [Google Scholar] [CrossRef]
- Arnbjerg, L.M.; Ravnsbæk, D.B.; Filinchuk, Y.; Vang, R.T.; Cerenius, Y.; Besenbacher, F.; Jørgensen, J.-E.; Jakobsen, H.J.; Jensen, T.R. Structure and dynamics for LiBH4-LiCl solid solutions. Chem. Mater. 2009, 21, 5772–5782. [Google Scholar] [CrossRef] [Green Version]
- Zavorotynska, O.; Corno, M.; Pinatel, E.; Rude, L.H.; Ugliengo, P.; Jensen, T.R.; Baricco, M. Theoretical and experimental study of LiBH4-LiCl solid solution. Crystals 2012, 2, 144–158. [Google Scholar] [CrossRef] [Green Version]
- Pinatel, E.R.; Corno, M.; Ugliengo, P.; Baricco, M. Effects of metastability on hydrogen sorption in fluorine substituted hydrides. J. Alloys Compd. 2014, 615, S706–S710. [Google Scholar] [CrossRef] [Green Version]
- Corno, M.; Pinatel, E.; Ugliengo, P.; Baricco, M. A computational study on the effect of fluorine substitution in LiBH4. J. Alloys Compd. 2011, 509, S679–S683. [Google Scholar] [CrossRef]
- Rude, L.H.; Filso, U.; D’Anna, V.; Spyratou, A.; Richter, B.; Hino, S.; Zavorotynska, O.; Baricco, M.; Sorby, M.H.; Hauback, B.C.; et al. Hydrogen-Fluorine exchange in NaBH4-NaBF4. Phys. Chem. Chem. Phys. 2013, 15, 18185–18194. [Google Scholar] [CrossRef] [Green Version]
- Richter, B.; Ravnsbaek, D.B.; Sharma, M.; Spyratou, A.; Hagemann, H.; Jensen, T.R. Fluoride substitution in LiBH4; destabilization and decomposition. Phys. Chem. Chem. Phys. 2017, 19, 30157–30165. [Google Scholar] [CrossRef]
- Lukas, H.L.; Fries, S.G.; Sundman, B. Computational Thermodynamics: The Calphad Method; Cambridge University Press: Cambridge, UK, 2007. [Google Scholar]
- Adams, R.M. Borax to Boranes; American Chemical Society: Washington, DC, USA, 1961; Volume 32, pp. 60–68. [Google Scholar]
- Semenenko, K.N.; Chavgun, a.P.; Surov, V.N. Interaction of sodium tetrahydroborate with potassium and lithium tetrahydroborates. Russ. J. Inorg. Chem. 1971, 16, 271–273. [Google Scholar]
- Skripov, A.V.; Soloninin, A.V.; Rude, L.H.; Jensen, T.R.; Filinchuk, Y. Nuclear magnetic resonance studies of reorientational motion and Li diffusion in LiBH4-LiI solid solutions. J. Phys. Chem. C 2012, 116, 26177–26184. [Google Scholar] [CrossRef] [Green Version]
- Huff, G.F. Method and composition for subjecting metals to reducing conditions. U.S. Patent No. 2,935,428, 3 May 1960. [Google Scholar]
- Dematteis, E.M.; Baricco, M. Hydrogen Desorption in Mg(BH4)(2)-Ca(BH4)(2) System. Energies 2019, 12, 3230. [Google Scholar] [CrossRef] [Green Version]
- Dematteis, E.M.; Pistidda, C.; Dornheim, M.; Baricco, M. Exploring ternary and quaternary mixtures in the LiBH4-NaBH4-KBH4-Mg(BH4)2-Ca(B4)2 system. ChemPhysChem 2019, 20, 1348–1359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dematteis, E.M.; Santoru, A.; Poletti, M.G.; Pistidda, C.; Klassen, T.; Dornheim, M.; Baricco, M. Phase stability and hydrogen desorption in a quinary equimolar mixture of light-metals borohydrides. Int. J. Hydrog. Energy 2018, 43, 16793–16803. [Google Scholar] [CrossRef]
- Milanese, C.; Garroni, S.; Girella, A.; Mulas, G.; Berbenni, V.; Bruni, G.; Suriñach, S.; Baró, M.D.; Marini, A. Thermodynamic and kinetic investigations on pure and doped NaBH4-MgH2 system. J. Phys. Chem. C 2011, 115, 3151–3162. [Google Scholar] [CrossRef]
- Nakamori, Y.; Miwa, K.; Ninomiya, A.; Li, H.W.; Ohba, N.; Towata, S.I.; Zuttel, A.; Orimo, S.I. Correlation between thermodynamical stabilities of metal borohydrides and cation electronegativites: First-principles calculations and experiments. Phys. Rev. B 2006, 74. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Heere, M.; Contreras Vasquez, L.; Paterakis, C.; Sørby, M.H.; Hauback, B.C.; Book, D. Dehydrogenation and rehydrogenation of a 0.62LiBH4-0.38NaBH4 mixture with nano-sized Ni. Int. J. Hydrog. Energy 2018, 43, 16782–16792. [Google Scholar] [CrossRef]
- Liu, Y. Low Melting Point Alkali Metal Borohydride Mixtures for Hydrogen Storage. Ph.D. Thesis, University Birmingham, Birmingham, UK, 2018. [Google Scholar]
- Chaudhary, A.-L.; Li, G.; Matsuo, M.; Orimo, S.-i.; Deledda, S.; Sørby, M.H.; Hauback, B.C.; Pistidda, C.; Klassen, T.; Dornheim, M. Simultaneous desorption behavior of M borohydrides and Mg2FeH6 reactive hydride composites (M = Mg, then Li, Na, K, Ca). Appl. Phys. Lett. 2015, 107, 073905. [Google Scholar] [CrossRef] [Green Version]
- Afonso, G.; Bonakdarpour, A.; Wilkinson, D.P. Hydrogen storage properties of the destabilized 4NaBH4/5Mg2NiH4 composite system. J. Phys. Chem. C 2013, 117, 21105–21111. [Google Scholar] [CrossRef]
- Bergemann, N.; Pistidda, C.; Uptmoor, M.; Milanese, C.; Santoru, A.; Emmler, T.; Puszkiel, J.; Dornheim, M.; Klassen, T. A new mutually destabilized reactive hydride system: LiBH4-Mg2NiH4. J. Energy Chem. 2019, 34, 240–254. [Google Scholar] [CrossRef] [Green Version]
- Javadian, P.; Zlotea, C.; Ghimbeu, C.M.; Latroche, M.; Jensen, T.R. Hydrogen storage properties of nanoconfined LiBH4-Mg2NiH4 reactive hydride composites. J. Phys. Chem. C 2015, 119, 5819–5826. [Google Scholar] [CrossRef]
- Li, W.; Vajo, J.J.; Cumberland, R.W.; Liu, P.; Hwang, S.J.; Kim, C.; Bowman, R.C. Hydrogenation of magnesium nickel boride for reversible hydrogen storage. J. Phys. Chem. Lett. 2010, 1, 69–72. [Google Scholar] [CrossRef] [Green Version]
- Vajo, J.J.; Li, W.; Liu, P. Thermodynamic and kinetic destabilization in LiBH4/Mg2NiH4: Promise for borohydride-based hydrogen storage. Chem. Commun. (Camb) 2010, 46, 6687–6689. [Google Scholar] [CrossRef] [PubMed]
- Bergemann, N.; Pistidda, C.; Milanese, C.; Emmler, T.; Karimi, F.; Chaudhary, A.L.; Chierotti, M.R.; Klassen, T.; Dornheim, M. Ca(BH4)2-Mg2NiH4: On the pathway to a Ca(BH4)2 system with a reversible hydrogen cycle. Chem. Commun. (Camb) 2016, 52, 4836–4839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paterakis, C.; Guo, S.; Heere, M.; Liu, Y.; Contreras, L.F.; Sørby, M.H.; Hauback, B.C.; Reed, D.; Book, D. Study of the NaBH4-NaBr system and the behaviour of its low temperature phase transition. Int. J. Hydrog. Energy 2017, 42, 22538–22543. [Google Scholar] [CrossRef]
- Bogdanovic, B.; Schwickardi, M. Ti-Doped alkali metal aluminium hydrides as potential novel reversible hydrogen storage materials. J. Alloys Compd. 1997, 253, 1–9. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, W. Improvement on hydrogen storage properties of complex metal hydride. Conf. Proc. 2012, 29–48. [Google Scholar]
- Milanese, C.; Jensen, T.R.; Hauback, B.C.; Pistidda, C.; Dornheim, M.; Yang, H.; Lombardo, L.; Zuettel, A.; Filinchuk, Y.; Ngene, P.; et al. Complex hydrides for energy storage. Int. J. Hydrog. Energy 2019, 44, 7860–7874. [Google Scholar] [CrossRef] [Green Version]
- Moysés Araújo, C.; Scheicher, R.H.; Ahuja, R. Thermodynamic analysis of hydrogen sorption reactions in Li-Mg-N-H systems. Appl. Phys. Lett. 2008, 92, 021907. [Google Scholar] [CrossRef]
- Barkhordarian, G.; Klassen, T.; Dornheim, M.; Bormann, R. Unexpected kinetic effect of MgB2 in reactive hydride composites containing complex borohydrides. J. Alloys Compd. 2007, 440, L18–L21. [Google Scholar] [CrossRef]
- Reilly, J.J.; Wiswall, R.H. Reaction of hydrogen with alloys of magnesium and copper. Inorg. Chem. 1967, 6, 2220. [Google Scholar] [CrossRef]
- Vajo, J.J.; Olson, G.L. Hydrogen storage in destabilized chemical systems. Scr. Mater. 2007, 56, 829–834. [Google Scholar] [CrossRef]
- Vajo, J.J.; Skeith, S.L.; Mertens, F. Reversible storage of hydrogen in destabilized LiBH4. J. Phys. Chem. B 2005, 109, 3719–3722. [Google Scholar] [CrossRef] [PubMed]
- Dornheim, M.; Doppiu, S.; Barkhordarian, G.; Boesenberg, U.; Klassen, T.; Gutfleisch, O.; Bormann, R. Hydrogen storage in magnesium-based hydrides and hydride composites. Scr. Mater. 2007, 56, 841–846. [Google Scholar] [CrossRef]
- Bösenberg, U.; Kim, J.W.; Gosslar, D.; Eigen, N.; Jensen, T.R.; von Colbe, J.M.B.; Zhou, Y.; Dahms, M.; Kim, D.H.; Günther, R. Role of additives in LiBH4-MgH2 reactive hydride composites for sorption kinetics. Acta Mater. 2010, 58, 3381–3389. [Google Scholar] [CrossRef] [Green Version]
- Bosenberg, U.; Vainio, U.; Pranzas, P.K.; von Colbe, J.M.; Goerigk, G.; Welter, E.; Dornheim, M.; Schreyer, A.; Bormann, R. On the chemical state and distribution of Zr-and V-based additives in reactive hydride composites. Nanotechnology 2009, 20, 204003. [Google Scholar] [CrossRef] [PubMed]
- Deprez, E.; Justo, A.; Rojas, T.C.; López-Cartés, C.; Bonatto Minella, C.; Bösenberg, U.; Dornheim, M.; Bormann, R.; Fernández, A. Microstructural study of the LiBH4-MgH2 reactive hydride composite with and without Ti-isopropoxide additive. Acta Mater. 2010, 58, 5683–5694. [Google Scholar] [CrossRef] [Green Version]
- Deprez, E.; Muñoz-Márquez, M.A.; Roldán, M.A.; Prestipino, C.; Palomares, F.J.; Minella, C.B.; Bösenberg, U.; Dornheim, M.; Bormann, R.; Fernández, A. Oxidation state and local structure of Ti-based additives in the reactive hydride composite 2LiBH4 + MgH2. J. Phys. Chem. C 2010, 114, 3309–3317. [Google Scholar] [CrossRef] [Green Version]
- Fan, M.; Sun, L.; Zhang, Y.; Xu, F.; Zhang, J.; Chu, H. The catalytic effect of additive Nb2O5Nb2O5 on the reversible hydrogen storage performances of LiBH4-MgH2LiBH4-MgH2 composite. Int. J. Hydrog. Energy 2008, 33, 74–80. [Google Scholar] [CrossRef]
- Kou, H.Q.; Sang, G.; Zhou, Y.L.; Wang, X.Y.; Huang, Z.Y.; Luo, W.H.; Chen, L.X.; Xiao, X.Z.; Yang, G.Y.; Hu, C.W. Enhanced hydrogen storage properties of LiBH4 modified by NbF5. Int. J. Hydrog. Energy 2014, 39, 11675–11682. [Google Scholar] [CrossRef]
- Liu, B.H.; Zhang, B.J.; Jiang, Y. Hydrogen storage performance of LiBH4+1/2MgH2 composites improved by Ce-based additives. Int. J. Hydrog. Energy 2011, 36, 5418–5424. [Google Scholar] [CrossRef]
- Sridechprasat, P.; Suttisawat, Y.; Rangsunvigit, P.; Kitiyanan, B.; Kulprathipanja, S. Catalyzed LiBH4 and MgH2 mixture for hydrogen storage. Int. J. Hydrog. Energy 2011, 36, 1200–1205. [Google Scholar] [CrossRef]
- Wang, P.; Ma, L.; Fang, Z.; Kang, X.; Wang, P. Improved hydrogen storage property of Li-Mg-B-H system by milling with titanium trifluoride. Energy Environ. Sci. 2009, 2. [Google Scholar] [CrossRef]
- Li, Y.; Izuhara, T.; Takeshita, H.T. Promotional effect of aluminum on MgH2 + LiBH4 hydrogen storage materials. Mater. Trans. 2011, 52, 641–646. [Google Scholar] [CrossRef] [Green Version]
- Bellosta von Colbe, J.; Ares, J.-R.; Barale, J.; Baricco, M.; Buckley, C.; Capurso, G.; Gallandat, N.; Grant, D.M.; Guzik, M.N.; Jacob, I.; et al. Application of hydrides in hydrogen storage and compression: Achievements, outlook and perspectives. Int. J. Hydrog. Energy 2019, 44, 7780–7808. [Google Scholar] [CrossRef]
- Broom, D.P.; Webb, C.J.; Fanourgakis, G.S.; Froudakis, G.E.; Trikalitis, P.N.; Hirscher, M. Concepts for improving hydrogen storage in nanoporous materials. Int. J. Hydrog. Energy 2019, 44, 7768–7779. [Google Scholar] [CrossRef]
- Balde, C.P.; Hereijgers, B.P.; Bitter, J.H.; de Jong, K.P. Facilitated hydrogen storage in NaAlH4 supported on carbon nanofibers. Angew. Chem. Int. Ed. Engl. 2006, 45, 3501–3503. [Google Scholar] [CrossRef] [Green Version]
- Berube, V.; Chen, G.; Dresselhaus, M.S. Impact of nanostructuring on the enthalpy of formation of metal hydrides. Int. J. Hydrog. Energy 2008, 33, 4122–4131. [Google Scholar] [CrossRef]
- Kim, H.; Karkamkar, A.; Autrey, T.; Chupas, P.; Proffen, T. Determination of structure and phase transition of light element nanocomposites in mesoporous silica: Case study of NH3BH3 in MCM-41. J. Am. Chem. Soc. 2009, 131, 13749–13755. [Google Scholar] [CrossRef]
- Nielsen, T.K.; Bosenberg, U.; Gosalawit, R.; Dornheim, M.; Cerenius, Y.; Besenbacher, F.; Jensen, T.R. A reversible nanoconfined chemical reaction. ACS Nano 2010, 4, 3903–3908. [Google Scholar] [CrossRef]
- Sabrina, S.; Kenneth, D.K.; Zhirong, Z.-K.; Eisa Gil, B.; Maximilian, F.; Bjørn, C.H. Small-Angle scattering investigations of Mg-borohydride infiltrated in activated carbon. Nanotechnology 2009, 20, 505702. [Google Scholar]
- Zaluski, L.; Zaluska, A.; StromOlsen, J.O. Nanocrystalline metal hydrides. J. Alloys Compd. 1997, 253, 70–79. [Google Scholar] [CrossRef]
- Nwakwuo, C.C.; Pistidda, C.; Dornheim, M.; Hutchison, J.L.; Sykes, J.M. Microstructural analysis of hydrogen absorption in 2NaH + MgB2. Scr. Mater. 2011, 64, 351–354. [Google Scholar] [CrossRef]
- Garroni, S.; Pistidda, C.; Brunelli, M.; Vaughan, G.B.M.; Surinach, S.; Baro, M.D. Hydrogen desorption mechanism of 2NaBH4 + MgH2 composite prepared by high-energy ball milling. Scr. Mater. 2009, 60, 1129–1132. [Google Scholar] [CrossRef]
- Mao, J.F.; Yu, X.B.; Guo, Z.P.; Liu, H.K.; Wu, Z.; Ni, J. Enhanced hydrogen storage performances of NaBH4-MgH2 system. J. Alloys Compd. 2009, 479, 619–623. [Google Scholar] [CrossRef]
- Pistidda, C.; Napolitano, E.; Pottmaier, D.; Dornheim, M.; Klassen, T.; Baricco, M.; Enzo, S. Structural study of a new B-rich phase obtained by partial hydrogenation of 2NaH + MgB2. Int. J. Hydrog. Energy 2013, 38, 10479–10484. [Google Scholar] [CrossRef] [Green Version]
- Pistidda, C.; Garroni, S.; Minella, C.B.; Dolci, F.; Jensen, T.R.; Nolis, P.; Bosenberg, U.; Cerenius, Y.; Lohstroh, W.; Fichtner, M.; et al. Pressure effect on the 2NaH + MgB2 hydrogen absorption reaction. J. Phys. Chem. C 2010, 114, 21816–21823. [Google Scholar] [CrossRef]
- Heere, M.; Sorby, M.H.; Pistidda, C.; Dornheim, M.; Hauback, B.C. Milling time effect of Reactive hydride composites of NaF-NaH-MgB2 investigated by in situ powder diffraction. Int. J. Hydrog. Energy 2016, 41, 13101–13108. [Google Scholar] [CrossRef]
- Le, T.-T.; Pistidda, C.; Puszkiel, J.; Castro Riglos, M.V.; Karimi, F.; Skibsted, J.; GharibDoust, S.P.; Richter, B.; Emmler, T.; Milanese, C.; et al. Design of a nanometric AlTi additive for MgB2-based reactive hydride composites with superior kinetic properties. J. Phys. Chem. C 2018, 122, 7642–7655. [Google Scholar] [CrossRef] [Green Version]
- Mangan, M.A.; Kral, M.V.; Spanos, G. Correlation between the crystallography and morphology of proeutectoid Widmanstätten cementite precipitates. Acta Mater. 1999, 47, 4263–4274. [Google Scholar] [CrossRef]
- Zhang, M.X.; Kelly, P.M. Edge-to-edge matching model for predicting orientation relationships and habit planes—The improvements. Scr. Mater. 2005, 52, 963–968. [Google Scholar] [CrossRef]
- Bonatto Minella, C.; Pellicer, E.; Rossinyol, E.; Karimi, F.; Pistidda, C.; Garroni, S.; Milanese, C.; Nolis, P.; Baró, M.D.; Gutfleisch, O.; et al. Chemical state, distribution, and role of Ti-and Nb-based additives on the Ca(BH4)2 system. J. Phys. Chem. C 2013, 117, 4394–4403. [Google Scholar] [CrossRef] [Green Version]
- Puszkiel, J.A.; Gennari, F.C.; Larochette, P.A.; Ramallo-López, J.M.; Vainio, U.; Karimi, F.; Pranzas, P.K.; Troiani, H.; Pistidda, C.; Jepsen, J.; et al. Effect of Fe additive on the hydrogenation-dehydrogenation properties of 2LiH + MgB2/2LiBH4 + MgH2 system. J. Power Sources 2015, 284, 606–616. [Google Scholar] [CrossRef]
- Pranzas, P.K.; Bosenberg, U.; Karimi, F.; Munning, M.; Metz, O.; Minella, C.B.; Schmitz, H.W.; Beckmann, F.; Vainio, U.; Zajac, D.; et al. Characterization of hydrogen storage materials and systems with photons and neutrons. Adv. Eng. Mater. 2011, 13, 730–736. [Google Scholar] [CrossRef]
- Gleiter, H. Nanostructured materials: Basic concepts and microstructure. Acta Mater. 2000, 48, 1–29. [Google Scholar] [CrossRef] [Green Version]
- Berube, F.; Kaliaguine, S. Calcination and thermal degradation mechanisms of triblock copolymer template in SBA-15 materials. Microporous Mesoporous Mater. 2008, 115, 469–479. [Google Scholar] [CrossRef]
- Berube, V.; Radtke, G.; Dresselhaus, M.; Chen, G. Size effects on the hydrogen storage properties of nanostructured metal hydrides: A review. Int. J. Energy Res. 2007, 31, 637–663. [Google Scholar] [CrossRef]
- Fichtner, M. Properties of nanoscale metal hydrides. Nanotechnology 2009, 20. [Google Scholar] [CrossRef] [PubMed]
- Stuhr, U.; Wipf, H.; Udovic, T.J.; Weissmuller, J.; Gleiter, H. The vibrational excitations and the position of hydrogen in nanocrystalline palladium. J. Phys. Condens. Matter 1995, 7, 219–230. [Google Scholar] [CrossRef]
- Koch, C.C. Synthesis of nanostructured materials by mechanical milling: Problems and opportunities. Nanostruct. Mater. 1997, 9, 13–22. [Google Scholar] [CrossRef]
- Zhang, D.L. Processing of advanced materials using high-energy mechanical milling. Prog. Mater. Sci. 2004, 49, 537–560. [Google Scholar] [CrossRef]
- Suryanarayana, C. Mechanical alloying and milling. Prog. Mater. Sci. 2001, 46, 1–184. [Google Scholar] [CrossRef]
- Isaacoff, B.P.; Brown, K.A. Progress in top-down control of bottom-up assembly. Nano Lett 2017, 17, 6508–6510. [Google Scholar] [CrossRef] [PubMed]
- Thanh, N.T.; Maclean, N.; Mahiddine, S. Mechanisms of nucleation and growth of nanoparticles in solution. Chem. Rev. 2014, 114, 7610–7630. [Google Scholar] [CrossRef] [PubMed]
- Norberg, N.S.; Arthur, T.S.; Fredrick, S.J.; Prieto, A.L. Size-Dependent Hydrogen storage properties of Mg nanocrystals prepared from solution. J. Am. Chem. Soc. 2011, 133, 10679–10681. [Google Scholar] [CrossRef] [PubMed]
- Grammatikopoulos, P.; Steinhauer, S.; Vernieres, J.; Singh, V.; Sowwan, M. Nanoparticle design by gas-phase synthesis. Adv. Phys. X 2016, 1, 81–100. [Google Scholar] [CrossRef]
- Kalidindi, S.B.; Jagirdar, B.R. Highly monodisperse colloidal magnesium nanoparticles by room temperature digestive ripening. Inorg. Chem. 2009, 48, 4524–4529. [Google Scholar] [CrossRef] [PubMed]
- Vajo, J.J.; Salguero, T.T.; Gross, A.E.; Skeith, S.L.; Olson, G.L. Thermodynamic destabilization and reaction kinetics in light metal hydride systems. J. Alloys Compd. 2007, 446, 409–414. [Google Scholar] [CrossRef]
- Kumar, A.; Jeon, K.W.; Kumari, N.; Lee, I.S. Spatially confined formation and transformation of nanocrystals within nanometer-sized reaction media. Acc. Chem. Res. 2018, 51, 2867–2879. [Google Scholar] [CrossRef]
- Pacula, A.; Mokaya, R. Synthesis and high hydrogen storage capacity of zeolite-like carbons nanocast using as-synthesized zeolite templates. J. Phys. Chem. C 2008, 112, 2764–2769. [Google Scholar] [CrossRef]
- Wang, H.; Gao, Q.; Hu, J. High hydrogen storage capacity of porous carbons prepared by using activated carbon. J. Am. Chem. Soc. 2009, 131, 7016–7022. [Google Scholar] [CrossRef]
- Surrey, A.; Minella, C.B.; Fechler, N.; Antonietti, M.; Grafe, H.J.; Schultz, L.; Rellinghaus, B. Improved hydrogen storage properties of LiBH4 via nanoconfinement in micro- and mesoporous aerogel-like carbon. Int. J. Hydrog. Energy 2016, 41, 5540–5548. [Google Scholar] [CrossRef]
- de Jongh, P.E.; Eggenhuisen, T.M. Melt infiltration: An emerging technique for the preparation of novel functional nanostructured materials. Adv. Mater. 2013, 25, 6672–6690. [Google Scholar] [CrossRef] [PubMed]
- Rouquerol, J.; Avnir, D.; Fairbridge, C.W.; Everett, D.H.; Haynes, J.M.; Pernicone, N.; Ramsay, J.D.F.; Sing, K.S.W.; Unger, K.K. Recommendations for the characterization of porous solids (Technical Report). Pure Appl. Chem. 1994, 66, 1739–1758. [Google Scholar] [CrossRef]
- Liu, Y.X.; Deng, J.G.; Xie, S.H.; Wang, Z.W.; Dai, H.X. Catalytic removal of volatile organic compounds using ordered porous transition metal oxide and supported noble metal catalysts. Chin. J. Catal. 2016, 37, 1193–1205. [Google Scholar] [CrossRef]
- Trinidad, J.; Marco, I.; Arruebarrena, G.; Wendt, J.; Letzig, D.; de Argandona, E.; Goodall, R. Processing of magnesium porous structures by infiltration casting for biomedical applications. Adv. Eng. Mater. 2014, 16, 241–247. [Google Scholar] [CrossRef]
- Banerjee, A.; Chandran, R.B.; Davidson, J.H. Experimental investigation of a reticulated porous alumina heat exchanger for high temperature gas heat recovery. Appl. Therm. Eng. 2015, 75, 889–895. [Google Scholar] [CrossRef] [Green Version]
- Lai, Q.; Paskevicius, M.; Sheppard, D.A.; Buckley, C.E.; Thornton, A.W.; Hill, M.R.; Gu, Q.; Mao, J.; Huang, Z.; Liu, H.K.; et al. Hydrogen storage materials for mobile and stationary applications: Current state of the art. ChemSusChem 2015, 8, 2789–2825. [Google Scholar] [CrossRef]
- Mosegaard, L.; Moller, B.; Jorgensen, J.E.; Bosenberg, U.; Dornheim, M.; Hanson, J.C.; Cerenius, Y.; Walker, G.; Jakobsen, H.J.; Besenbacher, F.; et al. Intermediate phases observed during decomposition of LiBH4. J. Alloys Compd. 2007, 446, 301–305. [Google Scholar] [CrossRef]
- Ngene, P.; Adelhelm, P.; Beale, A.M.; de Jong, K.P.; de Jongh, P.E. LiBH4/SBA-15 nanocomposites prepared by melt infiltration under hydrogen pressure: Synthesis and hydrogen sorption properties. J. Phys. Chem. C 2010, 114, 6163–6168. [Google Scholar] [CrossRef]
- Peru, F.; Garroni, S.; Campesi, R.; Milanese, C.; Marini, A.; Pellicer, E.; Baro, M.D.; Mulas, G. Ammonia-free infiltration of NaBH4 into highly-ordered mesoporous silica and carbon matrices for hydrogen storage. J. Alloys Compd. 2013, 580, S309–S312. [Google Scholar] [CrossRef]
- Adelhelm, P.; de Jongh, P.E. The impact of carbon materials on the hydrogen storage properties of light metal hydrides. J. Mater. Chem. 2011, 21, 2417–2427. [Google Scholar] [CrossRef] [Green Version]
- Ampoumogli, A.; Steriotis, T.; Trikalitis, P.; Bardaji, E.G.; Fichtner, M.; Stubos, A.; Charalambopoulou, G. Synthesis and characterisation of a mesoporous carbon/calcium borohydride nanocomposite for hydrogen storage. Int. J. Hydrog. Energy 2012, 37, 16631–16635. [Google Scholar] [CrossRef]
- Comanescu, C.; Capurso, G.; Maddalena, A. Nanoconfinement in activated mesoporous carbon of calcium borohydride for improved reversible hydrogen storage. Nanotechnology 2012, 23. [Google Scholar] [CrossRef] [PubMed]
- Lai, Q.W.; Wang, T.; Sun, Y.H.; Aguey-Zinsou, K.F. Rational design of nanosized light elements for hydrogen storage: Classes, synthesis, characterization, and properties. Adv. Mater. Technol. 2018, 3. [Google Scholar] [CrossRef]
- Cahen, S.; Eymery, J.B.; Janot, R.; Tarascon, J.M. Improvement of the LiBH4 hydrogen desorption by inclusion into mesoporous carbons. J. Power Sources 2009, 189, 902–908. [Google Scholar] [CrossRef]
- Verdal, N.; Udovic, T.J.; Rush, J.J.; Liu, X.F.; Majzoub, E.H.; Vajo, J.J.; Gross, A.F. Dynamical perturbations of tetrahydroborate anions in LiBH4 due to nanoconfinement in controlled-pore carbon scaffolds. J. Phys. Chem. C 2013, 117, 17983–17995. [Google Scholar] [CrossRef]
- Fang, Z.Z.; Wang, P.; Rufford, T.E.; Kang, X.D.; Lu, G.Q.; Cheng, H.M. Kinetic-and thermodynamic-based improvements of lithium borohydride incorporated into activated carbon. Acta Mater. 2008, 56, 6257–6263. [Google Scholar] [CrossRef]
- Plerdsranoy, P.; Wiset, N.; Milanese, C.; Laipple, D.; Marini, A.; Klassen, T.; Dornheim, M.; Utike, R.G. Improvement of thermal stability and reduction of LiBH4/polymer host interaction of nanoconfined LiBH4 for reversible hydrogen storage. Int. J. Hydrog. Energy 2015, 40, 392–402. [Google Scholar] [CrossRef]
- Gross, A.F.; Vajo, J.J.; Van Atta, S.L.; Olson, G.L. Enhanced hydrogen storage kinetics of LiBH4 in nanoporous carbon scaffolds. J. Phys. Chem. C 2008, 112, 5651–5657. [Google Scholar] [CrossRef]
- Liu, X.F.; Peaslee, D.; Jost, C.Z.; Majzoub, E.H. Controlling the Decomposition pathway of LiBH4 via confinement in highly ordered nanoporous carbon. J. Phys. Chem. C 2010, 114, 14036–14041. [Google Scholar] [CrossRef]
- Suwarno Ngene, P.; Nale, A.; Eggenhuisen, T.M.; Oschatz, M.; Embs, J.P.; Remhof, A.; de Jongh, P.E. Confinement effects for lithium borohydride: Comparing silica and carbon scaffolds. J. Phys. Chem. C Nanomater. Interfaces 2017, 121, 4197–4205. [Google Scholar] [CrossRef]
- Plerdsranoy, P.; Kaewsuwan, D.; Chanlek, N.; Utke, R. Effects of specific surface area and pore volume of activated carbon nanofibers on nanoconfinement and dehydrogenation of LiBH4. Int. J. Hydrog. Energy 2017, 42, 6189–6201. [Google Scholar] [CrossRef]
- Guo, L.L.; Li, Y.; Ma, Y.F.; Liu, Y.; Peng, D.D.; Zhang, L.; Han, S.M. Enhanced hydrogen storage capacity and reversibility of LiBH4 encapsulated in carbon nanocages. Int. J. Hydrog. Energy 2017, 42, 2215–2222. [Google Scholar] [CrossRef]
- Thiangviriya, S.; Utke, R. LiBH4 nanoconfined in activated carbon nanofiber for reversible hydrogen storage. Int. J. Hydrog. Energy 2015, 40, 4167–4174. [Google Scholar] [CrossRef]
- Liu, X.F.; Peaslee, D.; Jost, C.Z.; Baumann, T.F.; Majzoub, E.H. Systematic pore-size effects of nanoconfinement of LiBH4: Elimination of diborane release and tunable behavior for hydrogen storage applications. Chem. Mater. 2011, 23, 1331–1336. [Google Scholar] [CrossRef]
- Urgnani, J.; Torres, F.J.; Palumbo, M.; Baricco, M. Hydrogen release from solid state NaBH4. Int. J. Hydrog. Energy 2008, 33, 3111–3115. [Google Scholar] [CrossRef]
- Ngene, P.; van den Berg, R.; Verkuijlen, M.H.W.; de Jong, K.P.; de Jongh, P.E. Reversibility of the hydrogen desorption from NaBH4 by confinement in nanoporous carbon. Energy Environ. Sci. 2011, 4, 4108–4115. [Google Scholar] [CrossRef] [Green Version]
- Martelli, P.; Caputo, R.; Remhof, A.; Mauron, P.; Borgschulte, A.; Zuttel, A. Stability and Decomposition of NaBH4. J. Phys. Chem. C 2010, 114, 7173–7177. [Google Scholar] [CrossRef]
- Ampoumogli, A.; Steriotis, T.; Trikalitis, P.; Giasafaki, D.; Bardaji, E.G.; Fichtner, M.; Charalambopoulou, G. Nanostructured composites of mesoporous carbons and boranates as hydrogen storage materials. J. Alloys Compd. 2011, 509, S705–S708. [Google Scholar] [CrossRef]
- Chong, L.; Zeng, X.; Ding, W.; Liu, D.J.; Zou, J. NaBH4 in “Graphene Wrapper”: Significantly enhanced hydrogen storage capacity and regenerability through nanoencapsulation. Adv. Mater. 2015, 27, 5070–5074. [Google Scholar] [CrossRef]
- Filinchuk, Y.; Richter, B.; Jensen, T.R.; Dmitriev, V.; Chernyshov, D.; Hagemann, H. Porous and dense magnesium borohydride frameworks: Synthesis, stability, and reversible absorption of guest species. Angew. Chem. Int. Ed. Engl. 2011, 50, 11162–11166. [Google Scholar] [CrossRef]
- Heere, M.; Zavorotynska, O.; Deledda, S.; Sorby, M.H.; Book, D.; Steriotis, T.; Hauback, B.C. Effect of additives, ball milling and isotopic exchange in porous magnesium borohydride. RSC Adv. 2018, 8, 27645–27653. [Google Scholar] [CrossRef] [Green Version]
- Paskevicius, M.; Pitt, M.P.; Webb, C.J.; Sheppard, D.A.; Filsø, U.; Gray, E.M.; Buckley, C.E. In-Situ X-ray diffraction study of γ-Mg(BH4)2 decomposition. J. Phys. Chem. C 2012, 116, 15231–15240. [Google Scholar] [CrossRef]
- Zavorotynska, O.; El-Kharbachi, A.; Deledda, S.; Hauback, B.C. Recent progress in magnesium borohydride Mg(BH4)(2): Fundamentals and applications for energy storage. Int. J. Hydrog. Energy 2016, 41, 14387–14403. [Google Scholar] [CrossRef] [Green Version]
- Lohstroh, W.; Heere, M. Structure and dynamics of borohydrides studied by neutron scattering techniques: A review. J. Phys. Soc. Jpn. 2020, 89. [Google Scholar] [CrossRef]
- Chłopek, K.; Frommen, C.; Léon, A.; Zabara, O.; Fichtner, M. Synthesis and properties of magnesium tetrahydroborate, Mg(BH4)2. J. Mater. Chem. 2007, 17, 3496–3503. [Google Scholar] [CrossRef] [Green Version]
- Dai, B.; Sholl, D.S.; Johnson, J.K. First-Principles study of experimental and hypothetical Mg(BH4)(2) crystal structures. J. Phys. Chem. C 2008, 112, 4391–4395. [Google Scholar] [CrossRef]
- Fichtner, M.; Zhao-Karger, Z.; Hu, J.; Roth, A.; Weidler, P. The kinetic properties of Mg(BH4)2 infiltrated in activated carbon. Nanotechnology 2009, 20. [Google Scholar] [CrossRef]
- Clemencon, D.; Davoisne, C.; Chotard, J.N.; Janot, R. Enhancement of the hydrogen release of Mg(BH4)(2) by concomitant effects of nano-confinement and catalysis. Int. J. Hydrog. Energy 2019, 44, 4253–4262. [Google Scholar] [CrossRef]
- Wahab, M.A.; Jia, Y.; Yang, D.J.; Zhao, H.J.; Yao, X.D. Enhanced hydrogen desorption from Mg(BH4)(2) by combining nanoconfinement and a Ni catalyst. J. Mater. Chem. A 2013, 1, 3471–3478. [Google Scholar] [CrossRef]
- Nale, A.; Catti, M.; Bardají, E.G.; Fichtner, M. On the decomposition of the 0.6LiBH4-0.4Mg(BH4)2 eutectic mixture for hydrogen storage. Int. J. Hydrog. Energy 2011, 36, 13676–13682. [Google Scholar] [CrossRef]
- Zhao-Karger, Z.; Witter, R.; Bardají, E.G.; Wang, D.; Cossement, D.; Fichtner, M. Altered reaction pathways of eutectic LiBH4-Mg(BH4)2 by nanoconfinement. J. Mater. Chem. A 2013, 1, 3379. [Google Scholar] [CrossRef]
- Kim, Y.; Reed, D.; Lee, Y.S.; Lee, J.Y.; Shim, J.H.; Book, D.; Cho, Y.W. Identification of the dehydrogenated product of Ca(BH4)(2). J. Phys. Chem. C 2009, 113, 5865–5871. [Google Scholar] [CrossRef]
- Ampoumogli, A.; Charalambopoulou, G.; Javadian, P.; Richter, B.; Jensen, T.R.; Steriotis, T. Hydrogen desorption and cycling properties of composites based on mesoporous carbons and a LiBH4-Ca(BH4)2 eutectic mixture. J. Alloys Compd. 2015, 645, S480–S484. [Google Scholar] [CrossRef]
- Javadian, P.; GharibDoust, S.P.; Li, H.-W.; Sheppard, D.A.; Buckley, C.E.; Jensen, T.R. Reversibility of LiBH4 facilitated by the LiBH4-Ca(BH4)2 eutectic. J. Phys. Chem. C 2017, 121, 18439–18449. [Google Scholar] [CrossRef]
- Shannon, R.D. Revised effective ionic-radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr. Sect. A 1976, 32, 751–767. [Google Scholar] [CrossRef]
- Wall, F. Rare earth elements. In Critical Metals Handbook; John Wiley & Sons: Hoboken, NJ, USA, 2014; pp. 312–339. [Google Scholar] [CrossRef]
- Frommen, C.; Sørby, M.; Heere, M.; Humphries, T.; Olsen, J.; Hauback, B. Rare earth borohydrides—Crystal structures and thermal properties. Energies 2017, 10, 2115. [Google Scholar] [CrossRef] [Green Version]
- Olsen, J.E.; Frommen, C.; Jensen, T.R.; Riktor, M.D.; Sørby, M.H.; Hauback, B.C. Structure and thermal properties of composites with RE-borohydrides (RE = La, Ce, Pr, Nd, Sm, Eu, Gd, Tb, Er, Yb or Lu) and LiBH4. RSC Adv. 2014, 4, 1570–1582. [Google Scholar] [CrossRef]
- Olsen, J.E.; Frommen, C.; Sørby, M.H.; Hauback, B.C. Crystal structures and properties of solvent-free LiYb(BH4)4-xClx, Yb(BH4)3 and Yb(BH4)2-xClx. RSC Adv. 2013, 3, 10764–10774. [Google Scholar] [CrossRef]
- Frommen, C.; Heere, M.; Riktor, M.D.; Sørby, M.H.; Hauback, B.C. Hydrogen storage properties of rare earth (RE) borohydrides (RE = La, Er) in composite mixtures with LiBH4 and LiH. J. Alloys Compd. 2015, 645, S155–S159. [Google Scholar] [CrossRef] [Green Version]
- Ley, M.B.; Ravnsbæk, D.B.; Filinchuk, Y.; Lee, Y.-S.; Janot, R.; Cho, Y.W.; Skibsted, J.; Jensen, T.R. LiCe(BH4)3Cl, a new lithium-ion conductor and hydrogen storage material with isolated tetranuclear anionic clusters. Chem. Mater. 2012, 24, 1654–1663. [Google Scholar] [CrossRef]
- Sato, T.; Miwa, K.; Nakamori, Y.; Ohoyama, K.; Li, H.-W.; Noritake, T.; Aoki, M.; Towata, S.-I.; Orimo, S.-I. Experimental and computational studies on solvent-free rare-earth metal borohydrides R(BH4)3 (R = Y, Dy, and Gd). Phys. Rev. B 2008, 77, 104–114. [Google Scholar] [CrossRef]
- Ravnsbæk, D.B.; Filinchuk, Y.; Černý, R.; Ley, M.B.; Haase, D.R.; Jakobsen, H.J.; Skibsted, J.; Jensen, T.R. Thermal polymorphism and decomposition of Y(BH4)3. Inorg. Chem. 2010, 49, 3801–3809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frommen, C.; Aliouane, N.; Deledda, S.; Fonnelop, J.E.; Grove, H.; Lieutenant, K.; Llamas-Jansa, I.; Sartori, S.; Sorby, M.H.; Hauback, B.C. Crystal structure, polymorphism, and thermal properties of yttrium borohydride Y(BH4)(3). J. Alloys Compd. 2010, 496, 710–716. [Google Scholar] [CrossRef]
- Ley, M.B.; Paskevicius, M.; Schouwink, P.; Richter, B.; Sheppard, D.A.; Buckley, C.E.; Jensen, T.R. Novel solvates M(BH(4))(3)S(CH(3))(2) and properties of halide-free M(BH(4))(3) (M = Y or Gd). Dalton Trans. 2014, 43, 13333–13342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humphries, T.D.; Ley, M.B.; Frommen, C.; Munroe, K.T.; Jensen, T.R.; Hauback, B.C. Crystal structure and in situ decomposition of Eu(BH4)(2) and Sm(BH4)(2). J. Mater. Chem. A 2015, 3, 691–698. [Google Scholar] [CrossRef] [Green Version]
- Ley, M.B.; Jorgensen, M.; Cerny, R.; Filinchuk, Y.; Jensen, T.R. From M(BH4)3 (M = La, Ce) borohydride frameworks to controllable synthesis of porous hydrides and ion conductors. Inorg. Chem. 2016, 55, 9748–9756. [Google Scholar] [CrossRef]
- GharibDoust, S.P.; Ravnsbæk, D.B.; Černý, R.; Jensen, T.R. Synthesis, structure and properties of bimetallic sodium rare-earth (RE) borohydrides, NaRE(BH4)4, RE = Ce, Pr, Er or Gd. Dalton Trans. 2017, 46, 13421–13431. [Google Scholar] [CrossRef]
- Sharma, M.; Didelot, E.; Spyratou, A.; Lawson Daku, L.M.; Cerny, R.; Hagemann, H. Halide free M(BH4)2 (M = Sr, Ba, and Eu) synthesis, structure, and decomposition. Inorg. Chem. 2016, 55, 7090–7097. [Google Scholar] [CrossRef]
- Wegner, W.; Jaron, T.; Grochala, W. Polymorphism and hydrogen discharge from holmium borohydride, Ho(BH4)3, and KHo(BH4)(4). Int. J. Hydrog. Energy 2014, 39, 20024–20030. [Google Scholar] [CrossRef]
- Grinderslev, J.B.; Moller, K.T.; Bremholm, M.; Jensen, T.R. Trends in synthesis, crystal structure, and thermal and magnetic properties of rare-earth metal borohydrides. Inorg. Chem. 2019, 58, 5503–5517. [Google Scholar] [CrossRef]
- Richter, B.; Grinderslev, J.B.; Moller, K.T.; Paskevicius, M.; Jensen, T.R. From metal hydrides to metal borohydrides. Inorg. Chem. 2018, 57, 10768–10780. [Google Scholar] [CrossRef] [PubMed]
- Heere, M.; Payandeh Gharib Doust, S.H.; Frommen, C.; Humphries, T.D.; Ley, M.B.; Sorby, M.H.; Jensen, T.R.; Hauback, B.C. The influence of LiH on the rehydrogenation behavior of halide free rare earth (RE) borohydrides (RE = Pr, Er). Phys. Chem. Chem. Phys. 2016, 18, 24387–24395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wegner, W.; Jaron, T.; Grochala, W. Preparation of a series of lanthanide borohydrides and their thermal decomposition to refractory lanthanide borides. J. Alloys Compd. 2018, 744, 57–63. [Google Scholar] [CrossRef]
- Payandeh GharibDoust, S.; Heere, M.; Nervi, C.; Sorby, M.H.; Hauback, B.C.; Jensen, T.R. Synthesis, structure, and polymorphic transitions of praseodymium(iii) and neodymium(iii) borohydride, Pr(BH4)3 and Nd(BH4)3. Dalton Trans. 2018, 47, 8307–8319. [Google Scholar] [CrossRef] [PubMed]
- West, A.R. Solid State Chemistry and Its Applications; John Wiley & Sons: Hoboken, NJ, USA, 2014. [Google Scholar]
- Cerny, R.; Schouwink, P. The crystal chemistry of inorganic metal borohydrides and their relation to metal oxides. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2015, 71, 619–640. [Google Scholar] [CrossRef]
- GharibDoust, S.P.; Brighi, M.; Sadikin, Y.; Ravnsbæk, D.B.; Černý, R.; Jensen, T.; Skibsted, J. Synthesis, structure and Li Ion conductivity of LiLa(BH4)3X, X = Cl, Br, I. J. Phys. Chem. C 2017, 121, 19010–19021. [Google Scholar]
- GharibDoust, S.P.; Heere, M.; Sorby, M.H.; Ley, M.B.; Ravnsbaek, D.B.; Hauback, B.C.; Cerny, R.; Jensen, T.R. Synthesis, structure and properties of new bimetallic sodium and potassium lanthanum borohydrides. Dalton Trans. 2016, 45, 19002–19011. [Google Scholar] [CrossRef]
- Lee, Y.-S.; Ley, M.B.; Jensen, T.R.; Cho, Y.W. Lithium ion disorder and conduction mechanism in LiCe (BH4) 3Cl. J. Phys. Chem. C 2016, 120, 19035–19042. [Google Scholar] [CrossRef]
- Brighi, M.; Schouwink, P.; Sadikin, Y.; Cerny, R. Fast ion conduction in garnet-type metal borohydrides Li3K3Ce2(BH4)(12) and Li3K3La2(BH4)(12). J. Alloys Compd. 2016, 662, 388–395. [Google Scholar] [CrossRef]
- Schouwink, P.; Ley, M.B.; Tissot, A.; Hagemann, H.; Jensen, T.R.; Smrcok, L.; Cerny, R. Structure and properties of complex hydride perovskite materials. Nat. Commun. 2014, 5, 5706. [Google Scholar] [CrossRef]
- Møller, K.T.; Jørgensen, M.; Fogh, A.S.; Jensen, T.R. Perovskite alkali metal samarium borohydrides: Crystal structures and thermal decomposition. Dalton Trans. 2017, 46, 11905–11912. [Google Scholar] [CrossRef] [PubMed]
- Schouwink, P.; Didelot, E.; Lee, Y.S.; Mazet, T.; Cerny, R. Structural and magnetocaloric properties of novel gadolinium borohydrides. J. Alloys Compd. 2016, 664, 378–384. [Google Scholar] [CrossRef]
- Roedern, E.; Lee, Y.-S.; Ley, M.B.; Park, K.; Cho, Y.W.; Skibsted, J.; Jensen, T.R. Solid state synthesis, structural characterization and ionic conductivity of bimetallic alkali-metal yttrium borohydrides MY(BH4)4 (M = Li and Na). J. Mater. Chem. A 2016, 4, 8793–8802. [Google Scholar] [CrossRef]
- Jaron, T.; Wegner, W.; Fijalkowski, K.J.; Leszczynski, P.J.; Grochala, W. Facile formation of thermodynamically unstable novel borohydride materials by a wet chemistry route. Chem. A Eur. J. 2015, 21, 5689–5692. [Google Scholar] [CrossRef]
- Ravnsbæk, D.B.; Ley, M.B.; Lee, Y.-S.; Hagemann, H.; D’Anna, V.; Cho, Y.W.; Filinchuk, Y.; Jensen, T.R. A mixed-cation mixed-anion borohydride NaY(BH4)2Cl2. Int. J. Hydrog. Energy 2012, 37, 8428–8438. [Google Scholar] [CrossRef]
- Jaroń, T.; Grochala, W. Probing Lewis acidity of Y (BH4)3 via its reactions with MB4 (M = Li, Na, K, NMe4). Dalton Trans. 2011, 40, 12808–12817. [Google Scholar] [CrossRef]
- Sadikin, Y.; Stare, K.; Schouwink, P.; Ley, M.B.; Jensen, T.R.; Meden, A.; Černý, R. Alkali metal-yttrium borohydrides: The link between coordination of small and large rare-earth. J. Solid State Chem. 2015, 225, 231–239. [Google Scholar] [CrossRef]
- Jaroń, T.; Wegner, W.; Grochala, W. M[Y(BH4)4] and M2Li[Y(BH4)6-xClx](M = Rb, Cs): New borohydride derivatives of yttrium and their hydrogen storage properties. Dalton Trans. 2013, 42, 6886–6893. [Google Scholar] [CrossRef]
- Heere, M.; GharibDoust, S.P.; Sorby, M.H.; Frommen, C.; Jensen, T.R.; Hauback, B.C. In situ investigations of bimetallic potassium erbium borohydride. Int. J. Hydrog. Energy 2017, 42, 22468–22474. [Google Scholar] [CrossRef]
- Wegner, W.; Jaron, T.; Grochala, W. MYb(BH4)4 (M = K, Na) from laboratory X-ray powder data. Acta Cryst. C 2013, 69, 1289–1291. [Google Scholar] [CrossRef]
- Gennari, F.C. Mechanochemical synthesis of erbium borohydride: Polymorphism, thermal decomposition and hydrogen storage. J. Alloys Compd. 2013, 581, 192–195. [Google Scholar] [CrossRef]
- Heere, M.; GharibDoust, S.P.; Brighi, M.; Frommen, C.; Sørby, M.; Černý, R.; Jensen, T.; Hauback, B. Hydrogen sorption in erbium borohydride composite mixtures with LiBH4 and/or LiH. Inorganics 2017, 5, 31. [Google Scholar] [CrossRef]
- Thangadurai, V.; Kaack, H.; Weppner, W.J.F. Novel fast lithium ion conduction in Garnet-type Li5La3M2O12(M = Nb, Ta). J. Am. Ceram. Soc. 2003, 86, 437–440. [Google Scholar] [CrossRef]
- Marks, S.; Heck, J.G.; Habicht, M.H.; Ona-Burgos, P.; Feldmann, C.; Roesky, P.W. [Ln(BH4)2(THF)2] (Ln = Eu, Yb)—A highly luminescent material. Synthesis, properties, reactivity, and NMR studies. J. Am. Chem Soc. 2012, 134, 16983–16986. [Google Scholar] [CrossRef]
- Wegner, W.; van Leusen, J.; Majewski, J.; Grochala, W.; Kogerler, P. Borohydride as magnetic superexchange pathway in late lanthanide borohydrides. Eur. J. Inorg. Chem. 2019, 2019, 1776–1783. [Google Scholar] [CrossRef]
- Payandeh, S.; Asakura, R.; Avramidou, P.; Rentsch, D.; Łodziana, Z.; Černý, R.; Remhof, A.; Battaglia, C. Nido-Borate/Closo-Borate mixed-anion electrolytes for all-solid-state batteries. Chem. Mater. 2020. [Google Scholar] [CrossRef]
- Roedern, E.; Kuhnel, R.S.; Remhof, A.; Battaglia, C. Magnesium ethylenediamine borohydride as solid-state electrolyte for magnesium batteries. Sci. Rep. 2017, 7, 46189. [Google Scholar] [CrossRef] [Green Version]
- Burankova, T.; Roedern, E.; Maniadaki, A.E.; Hagemann, H.; Rentsch, D.; Lodziana, Z.; Battaglia, C.; Remhof, A.; Embs, J.P. Dynamics of the coordination complexes in a solid-state mg electrolyte. J. Phys. Chem. Lett. 2018, 9, 6450–6455. [Google Scholar] [CrossRef]
- Le Ruyet, R.; Berthelot, R.; Salager, E.; Florian, P.; Fleutot, B.; Janot, R. Investigation of Mg(BH4)(NH2)-based composite materials with enhanced Mg2+ ionic conductivity. J. Phys. Chem. C 2019, 123, 10756–10763. [Google Scholar] [CrossRef]




























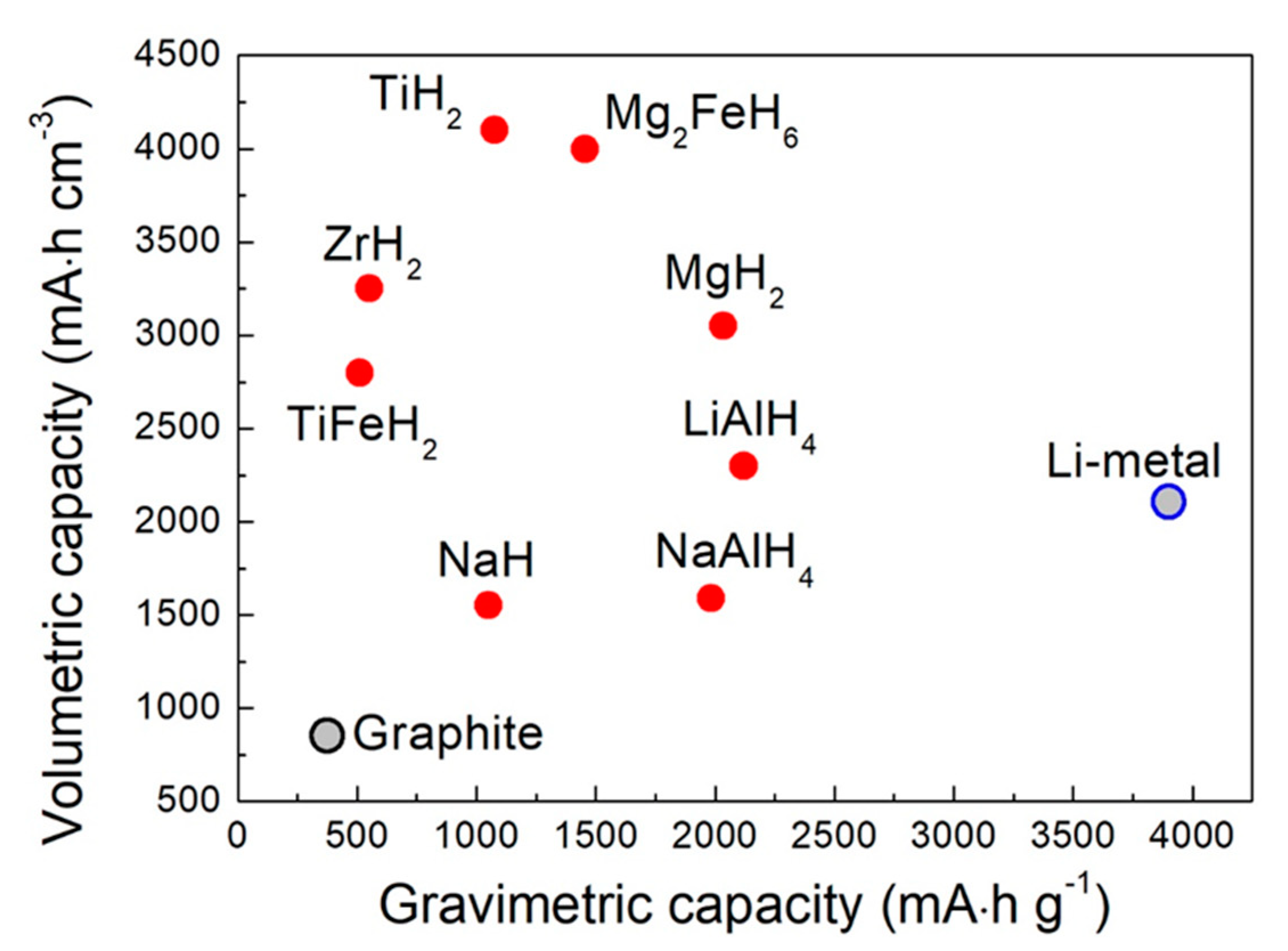{kind=link}
{kind=link}
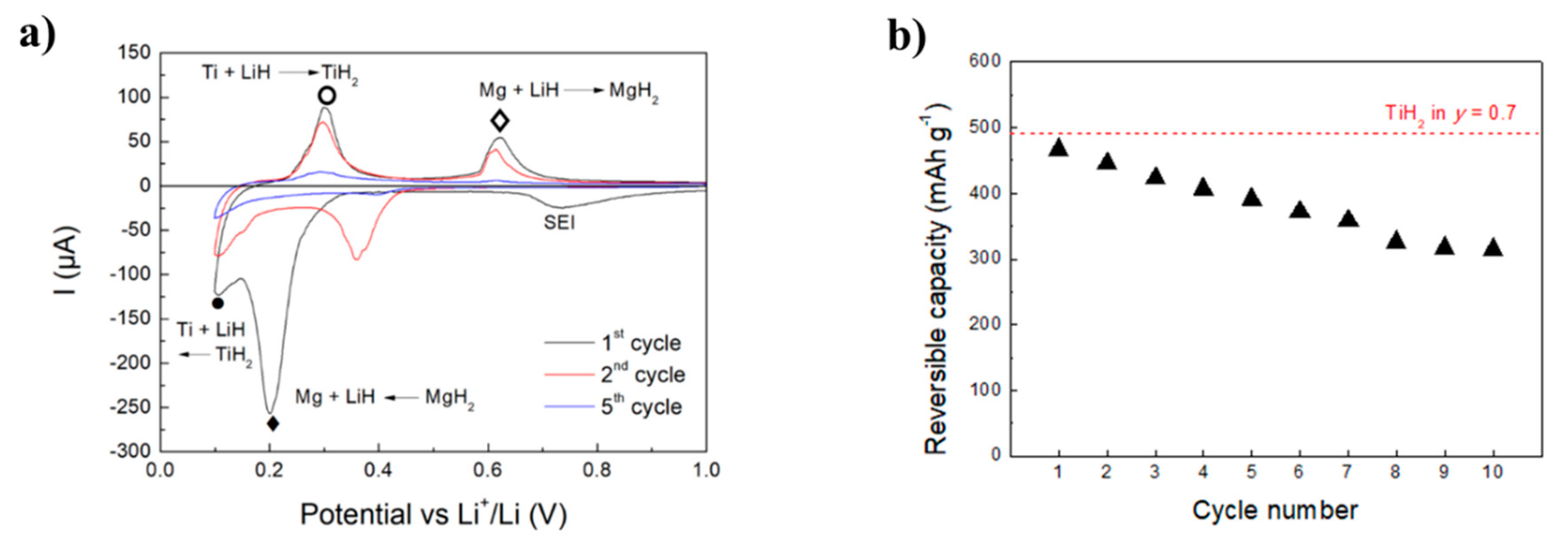{kind=link}
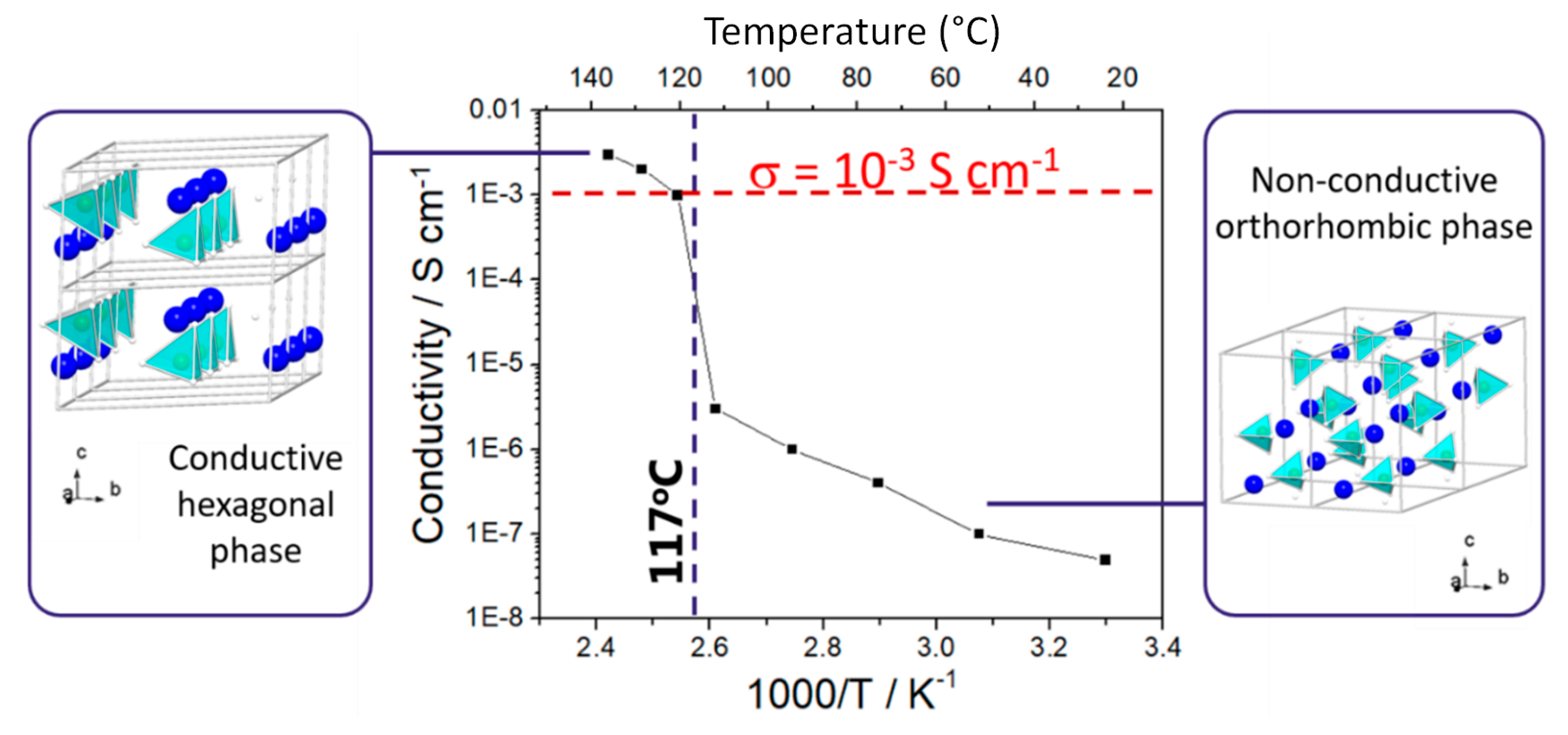{kind=link}
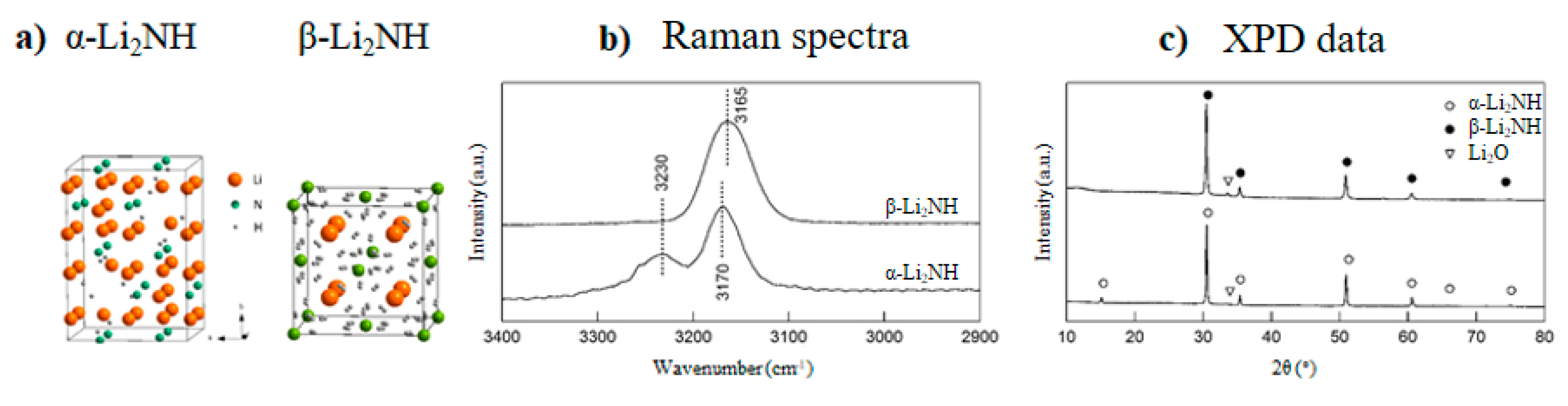{kind=link}
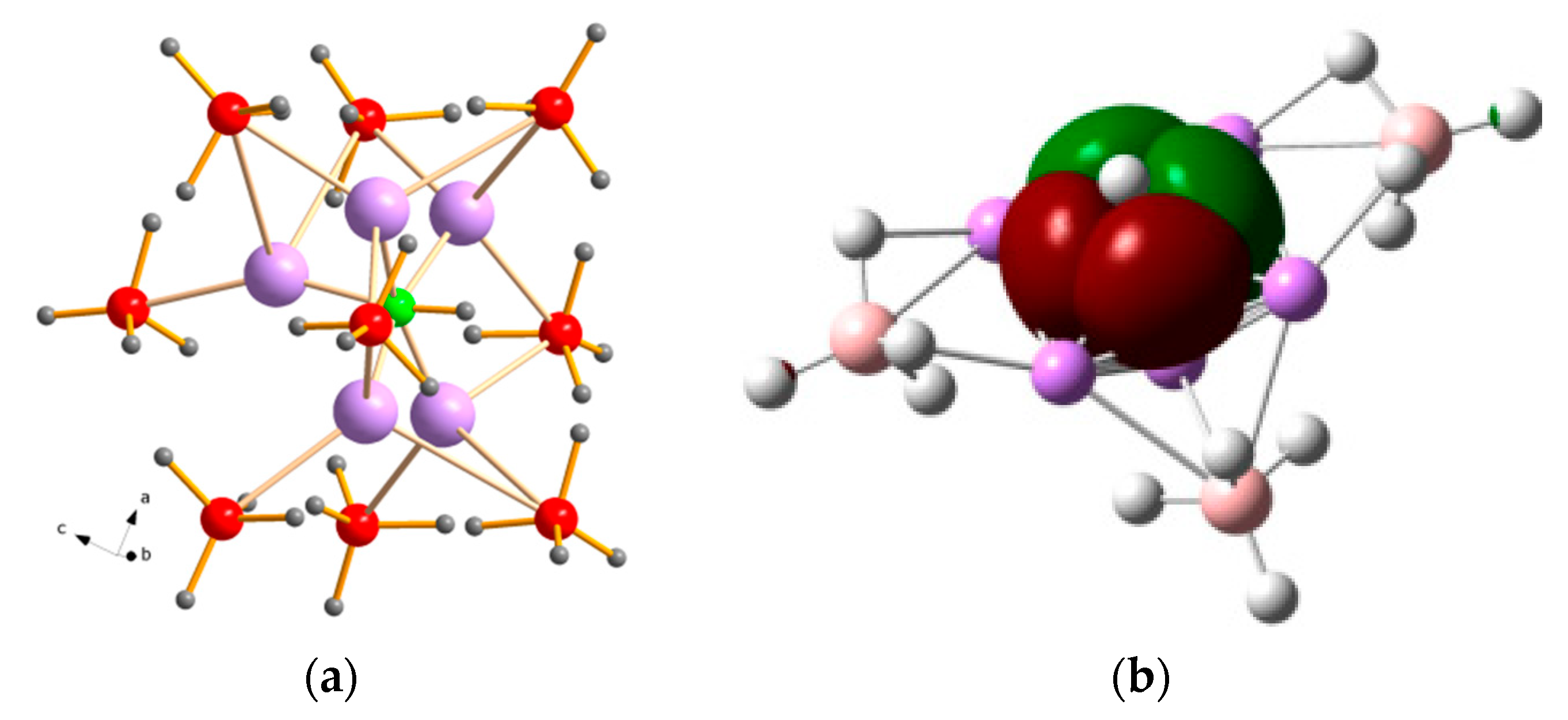{kind=link}
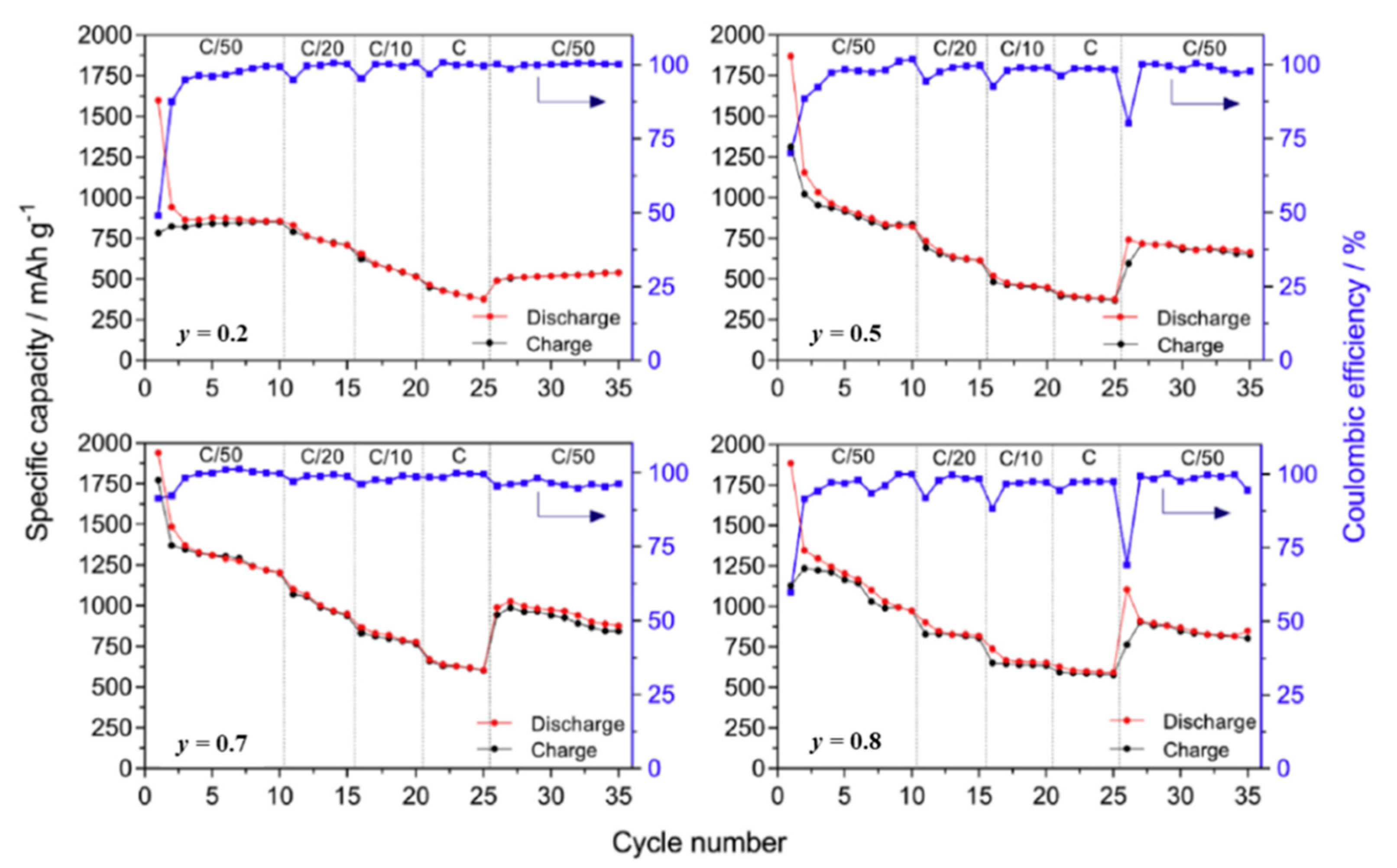{kind=link}
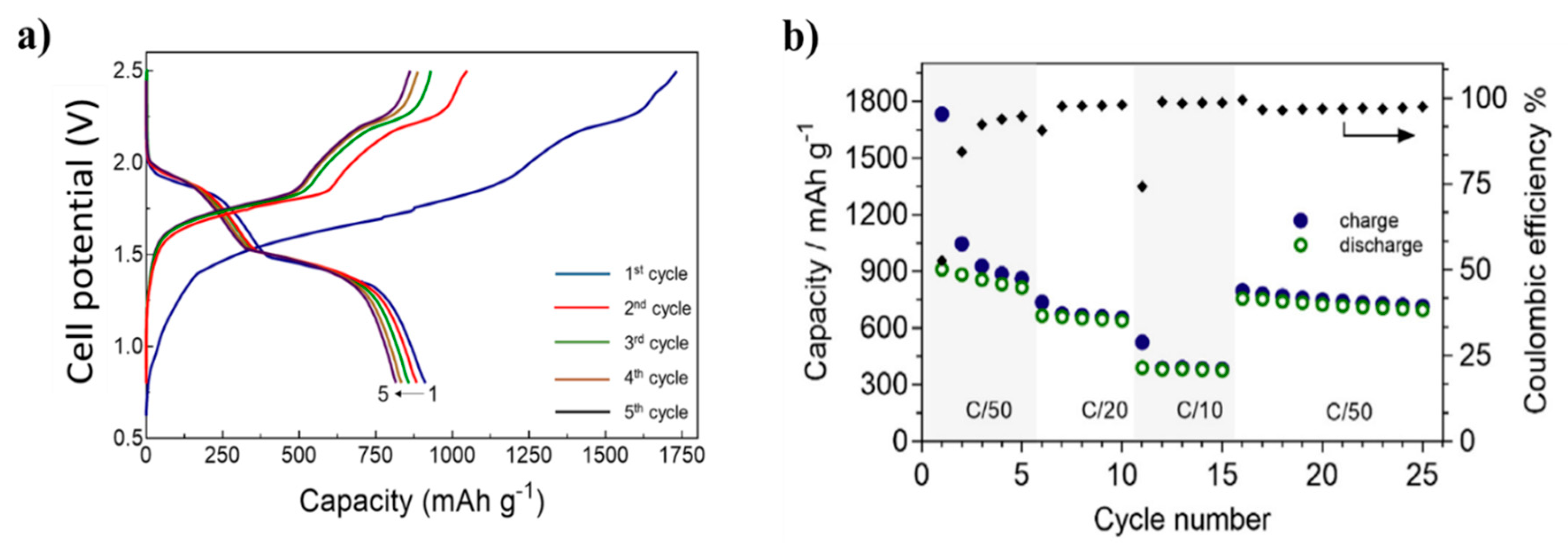{kind=link}
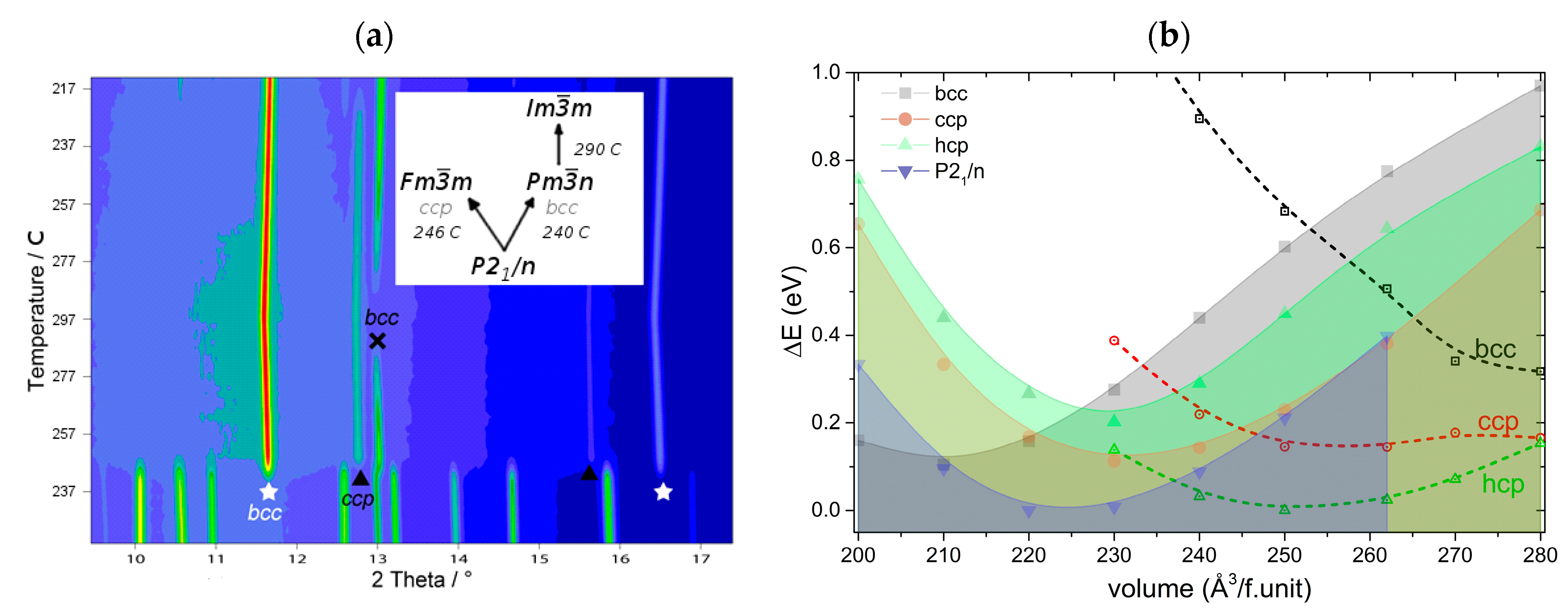{kind=link}
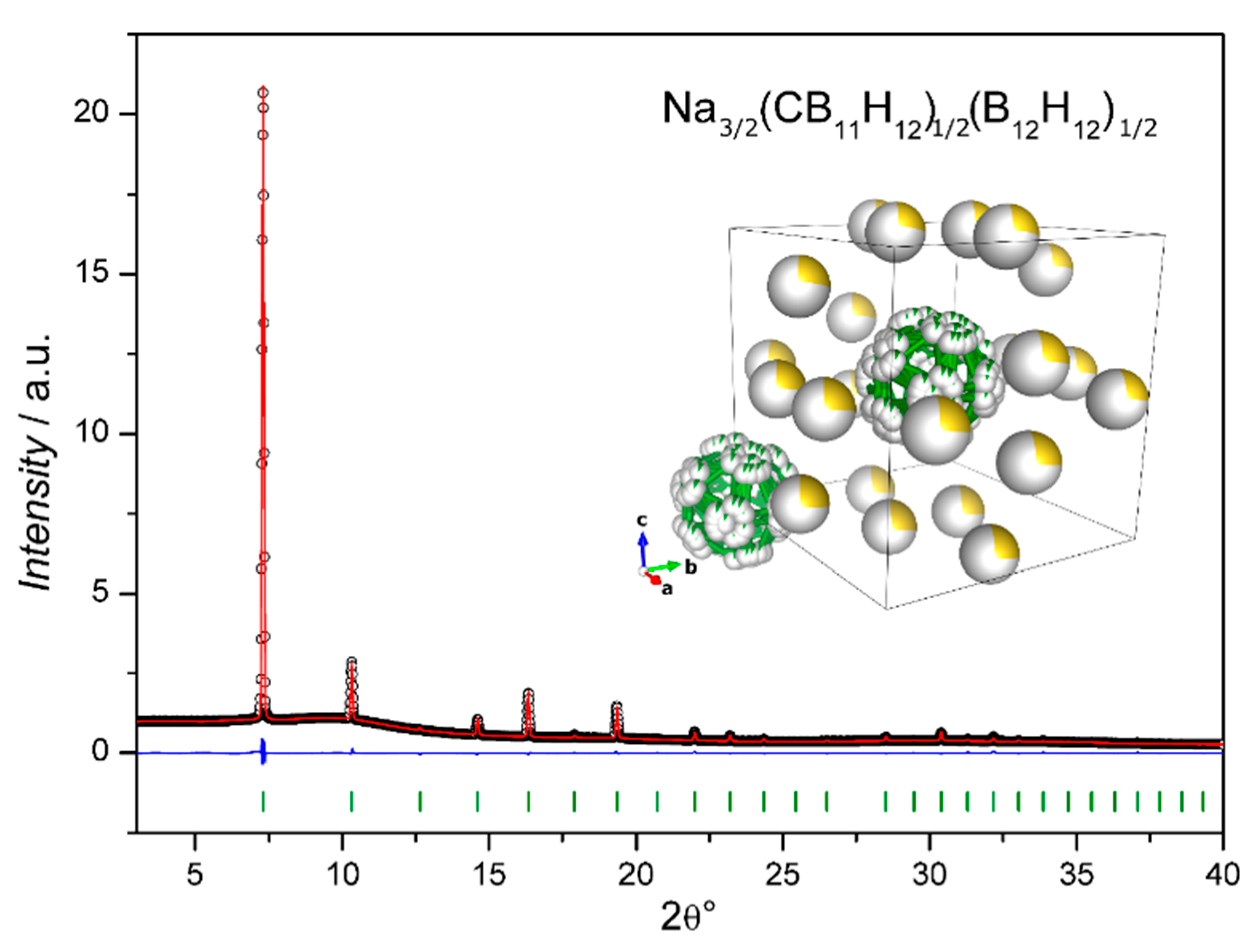{kind=link}
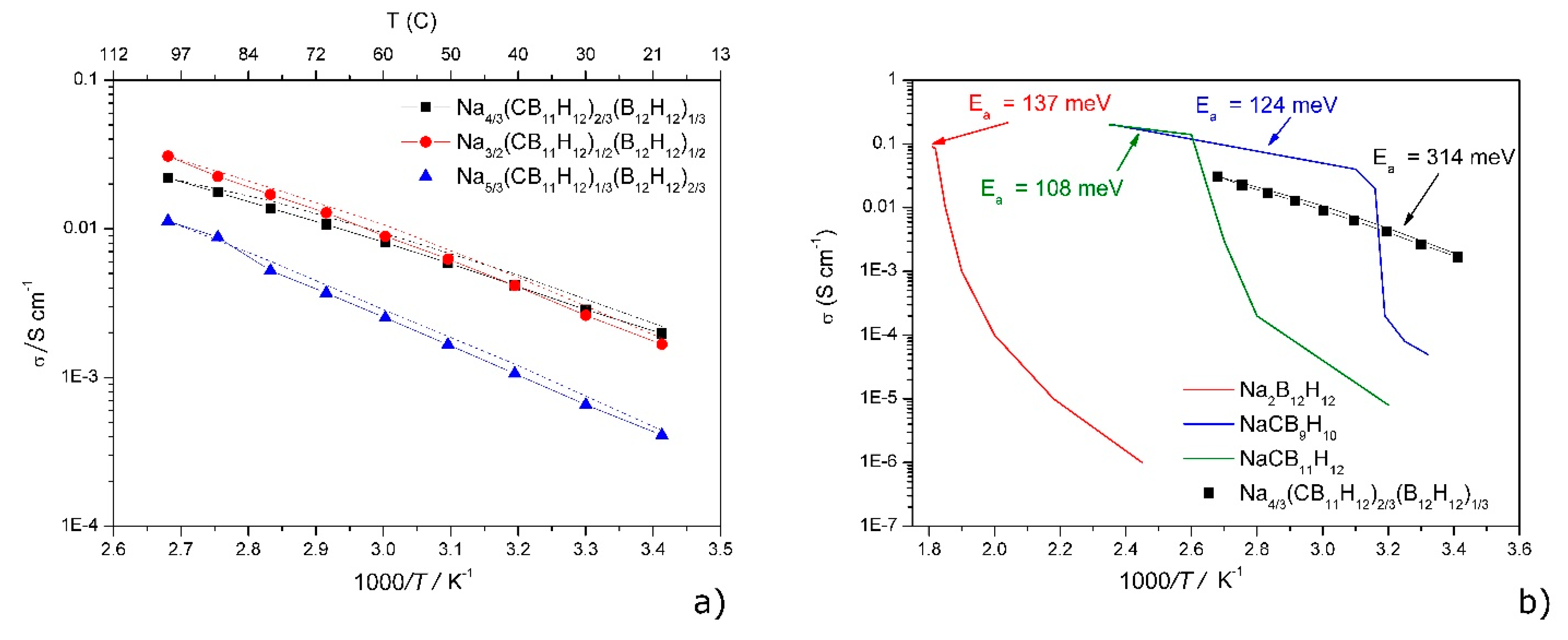{kind=link}
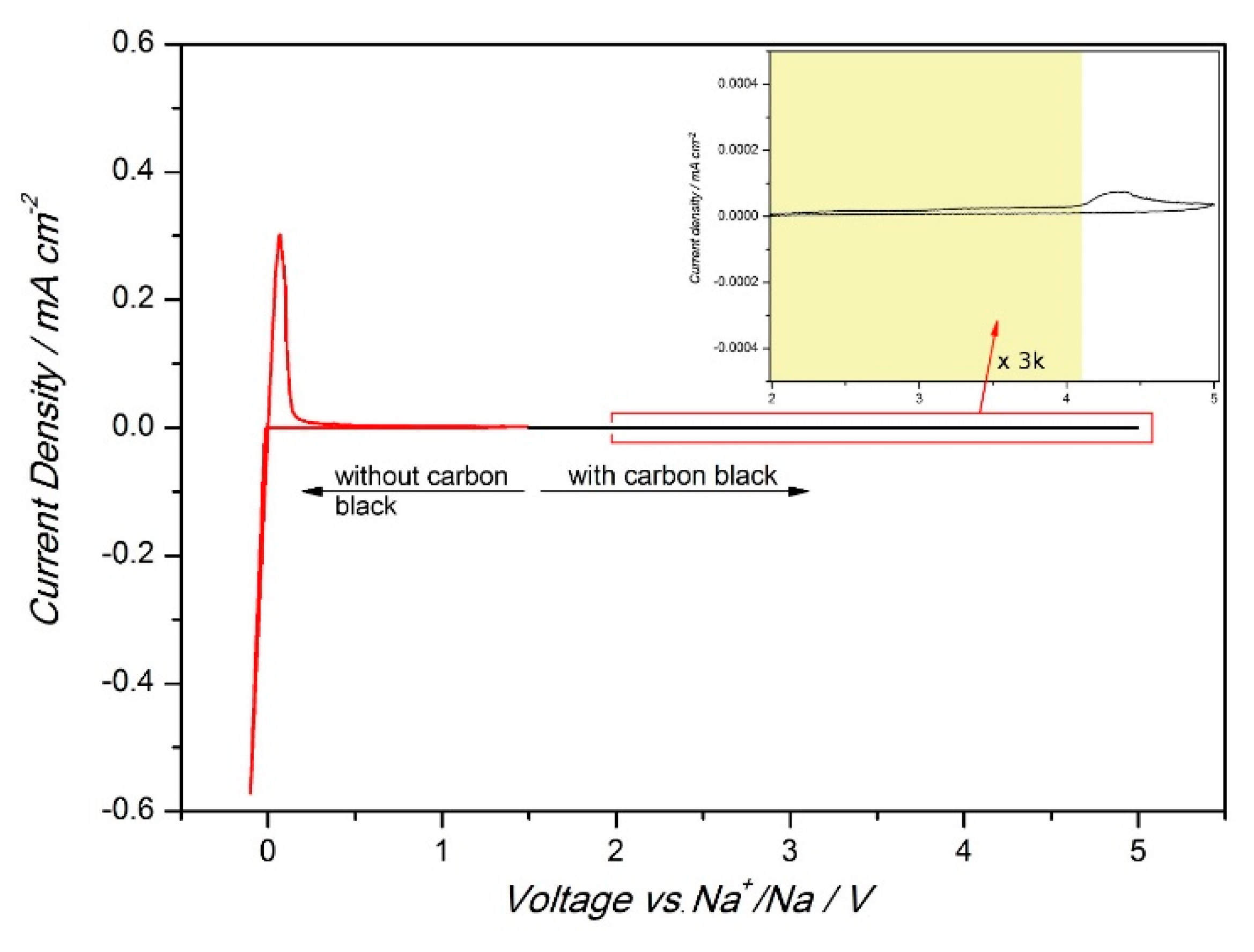{kind=link}
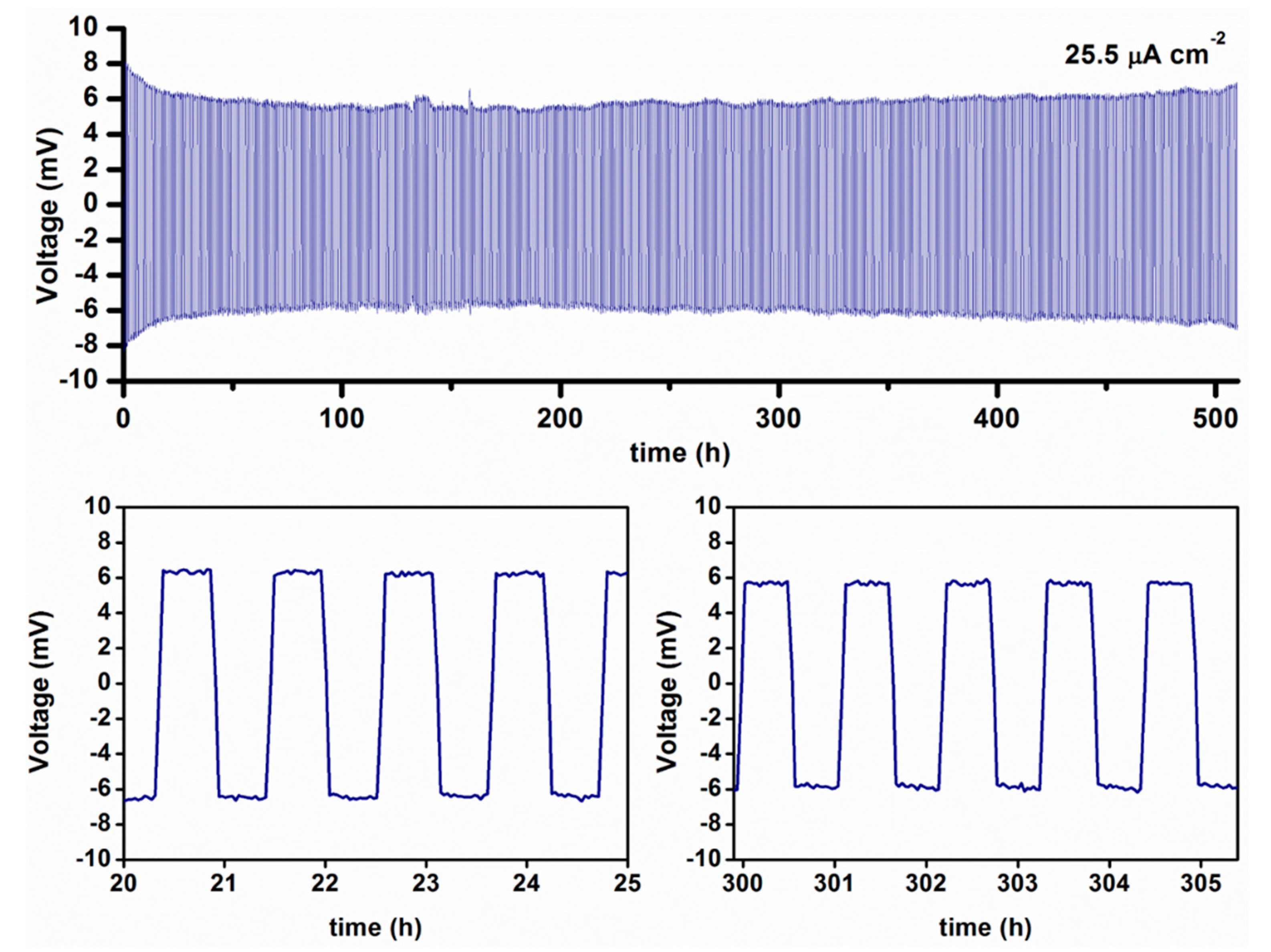{kind=link}
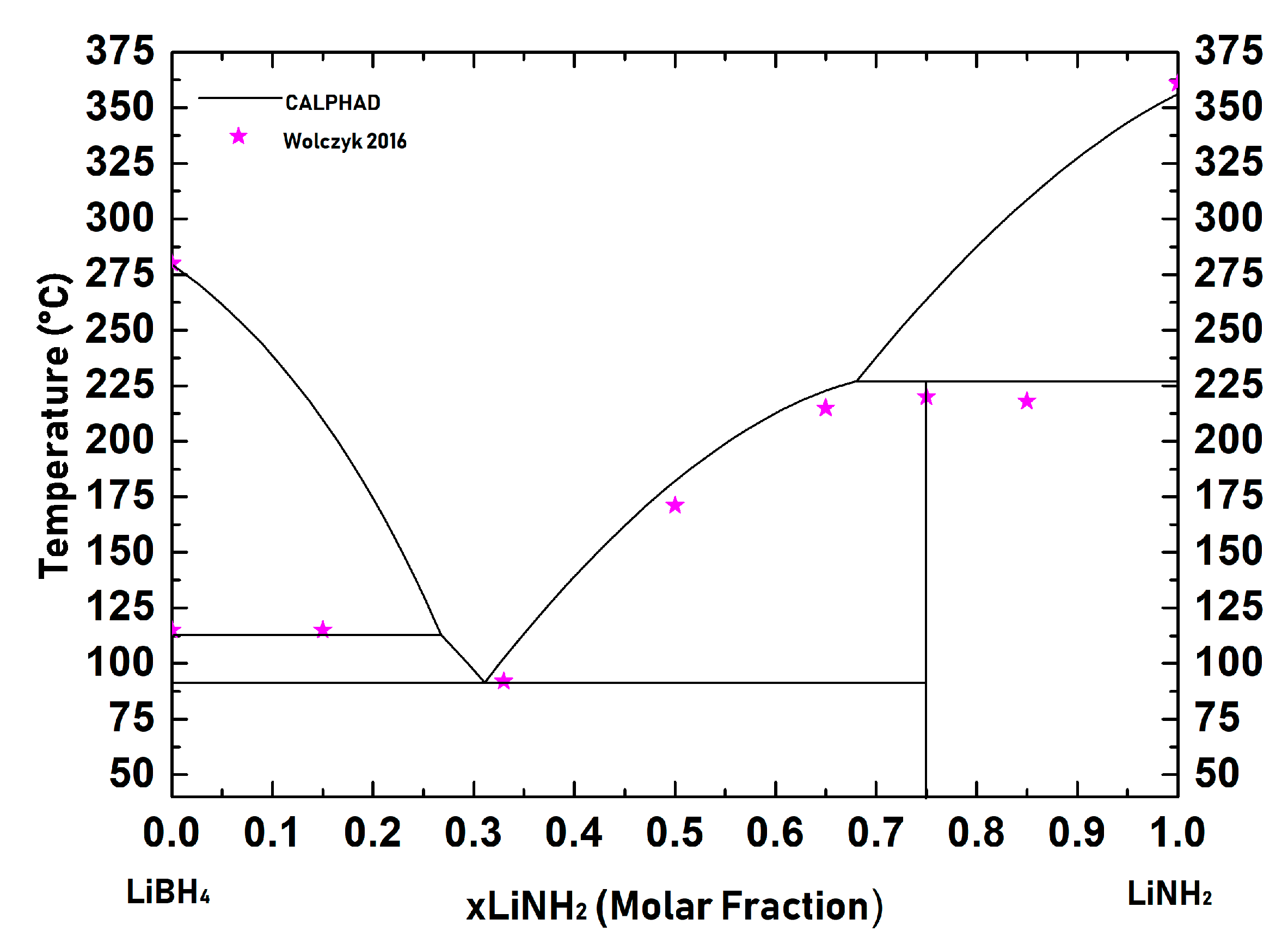{kind=link}
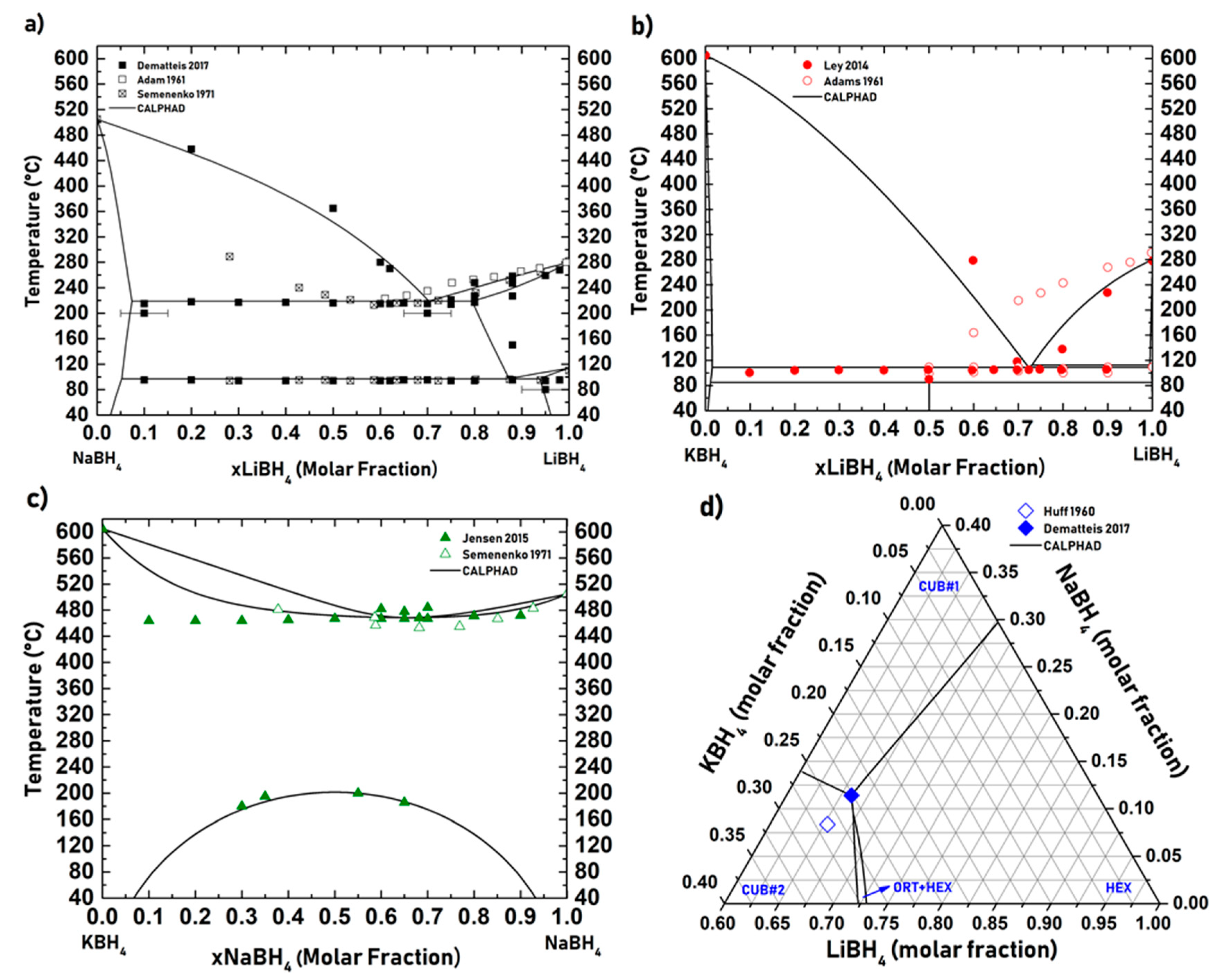{kind=link}
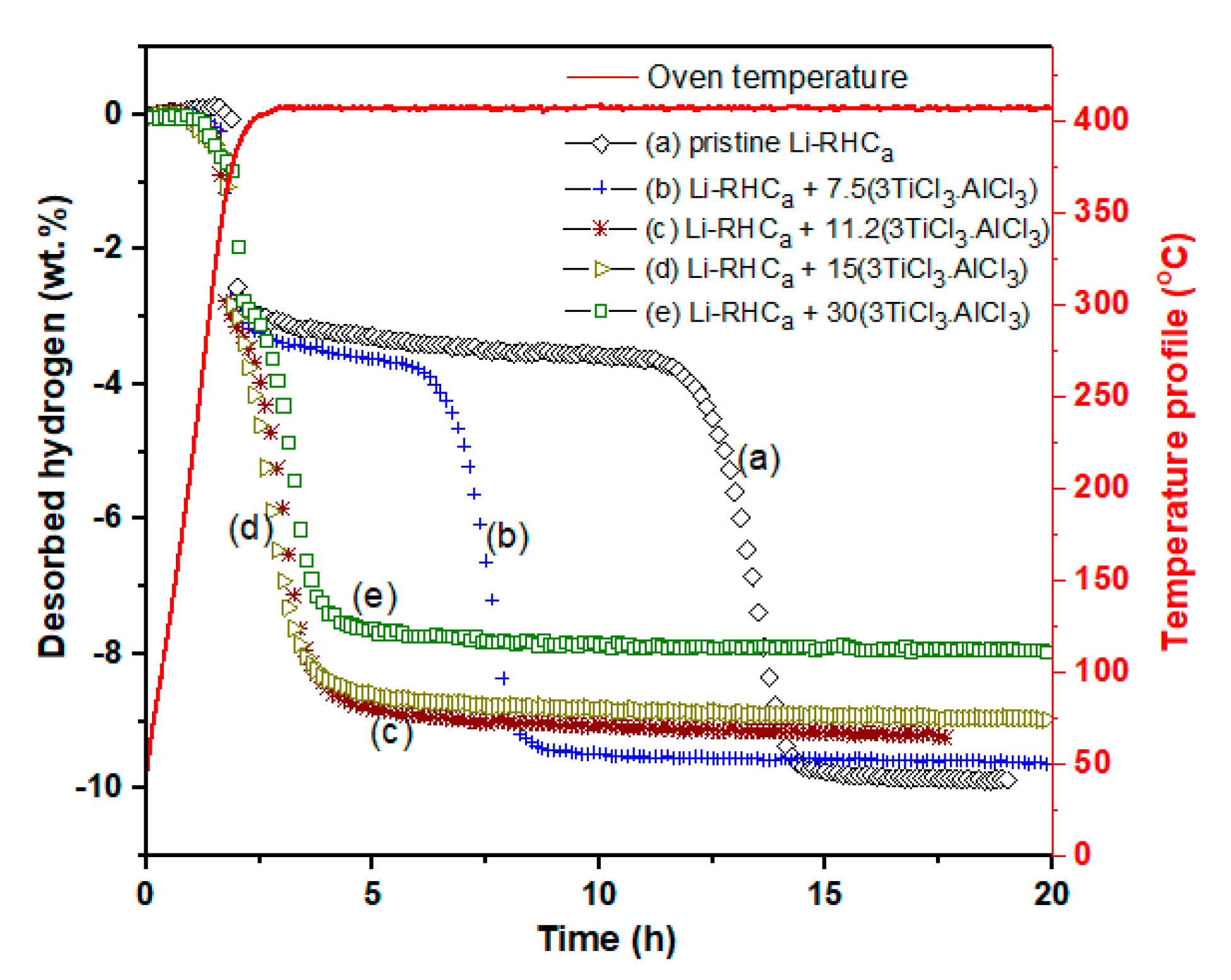{kind=link}
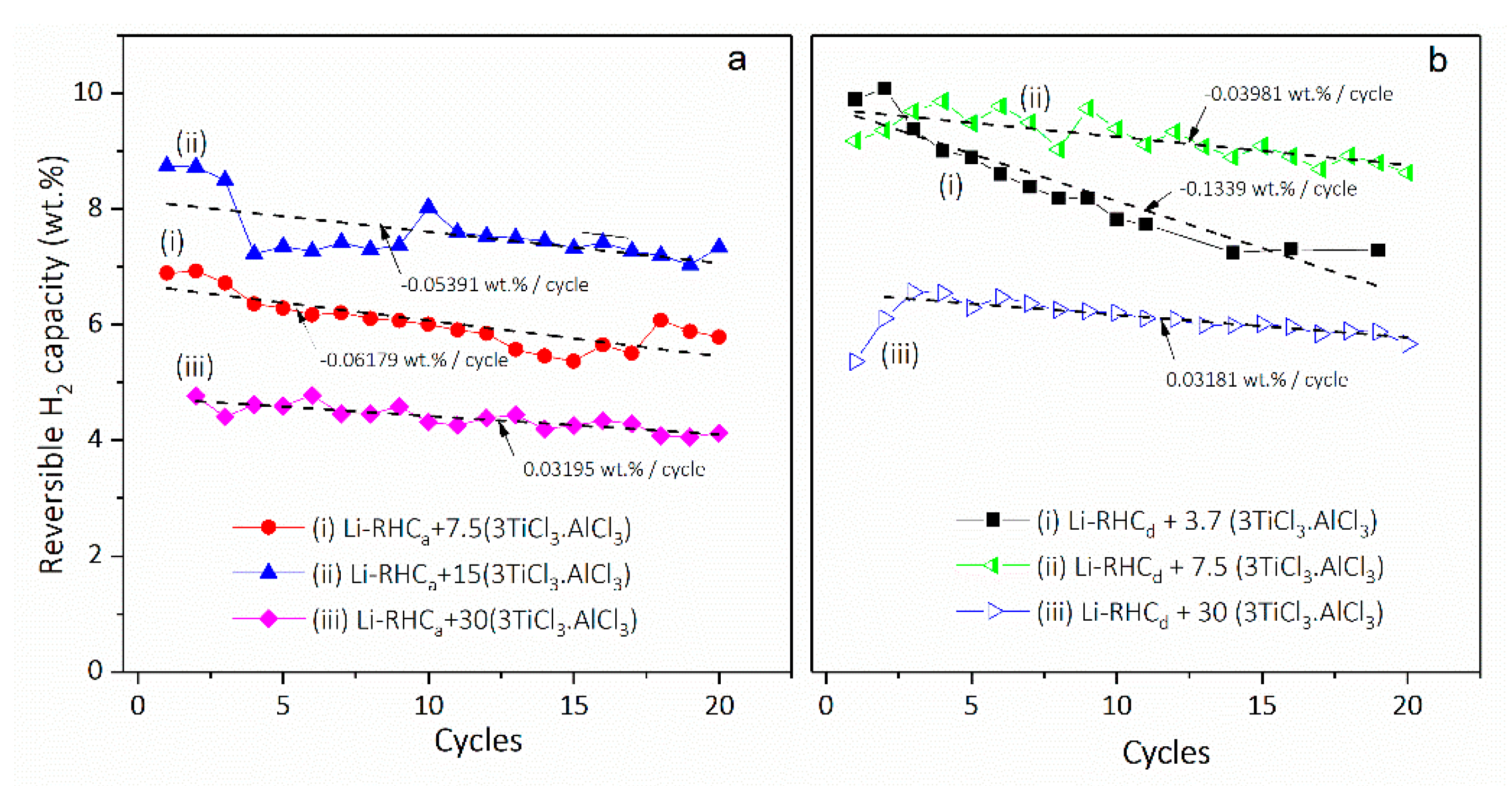{kind=link}
{kind=link}
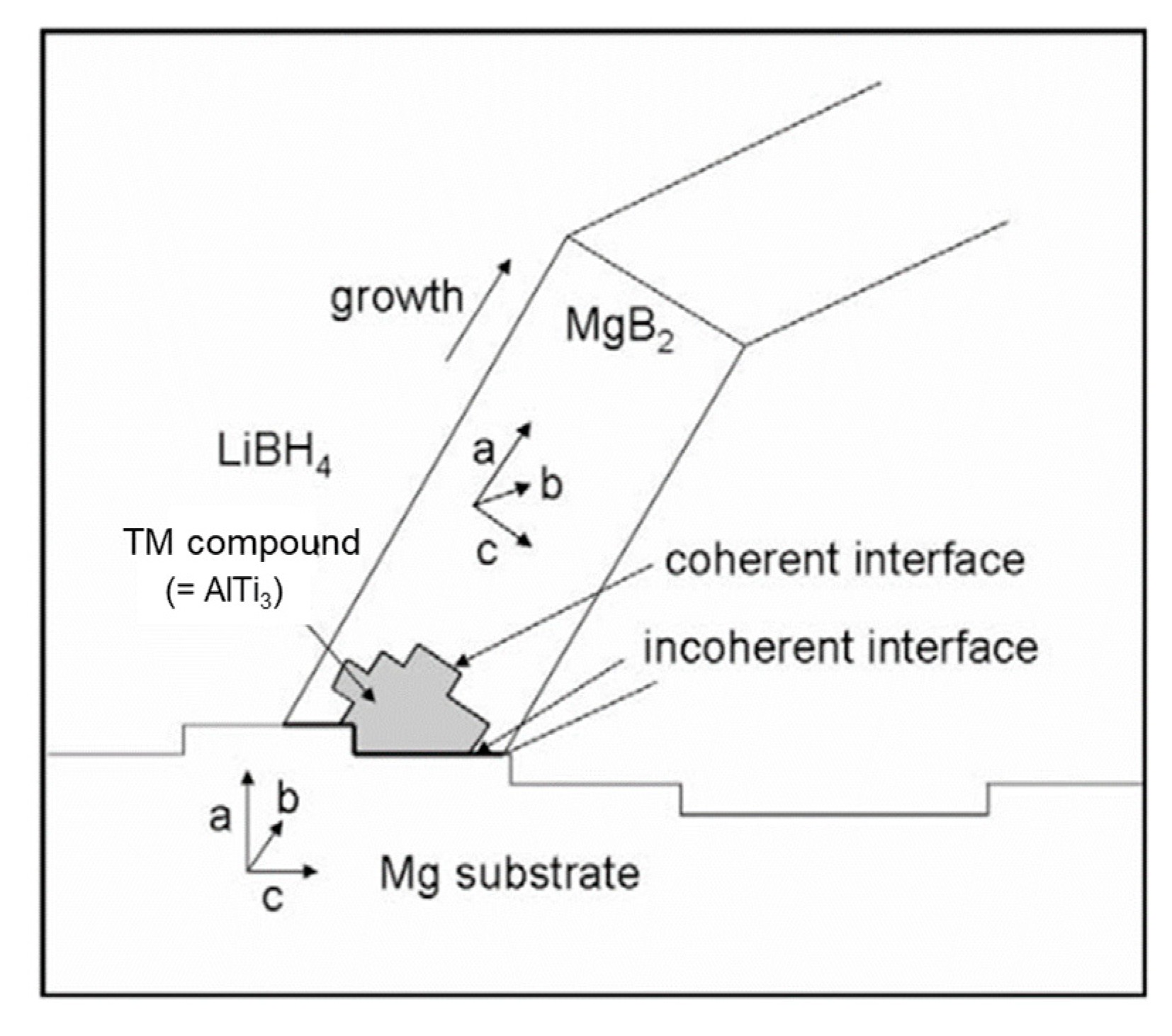{kind=link}
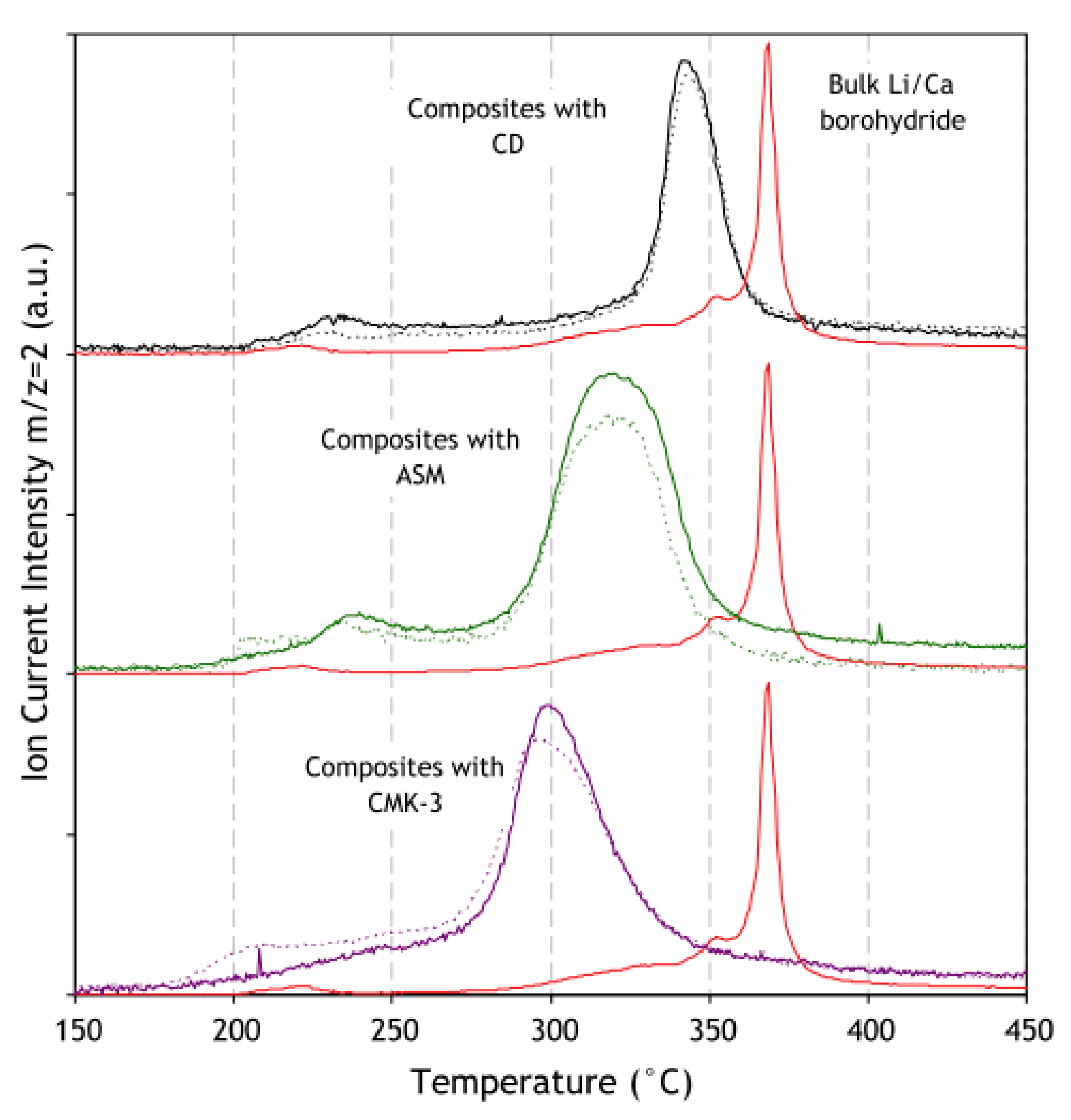{kind=link}
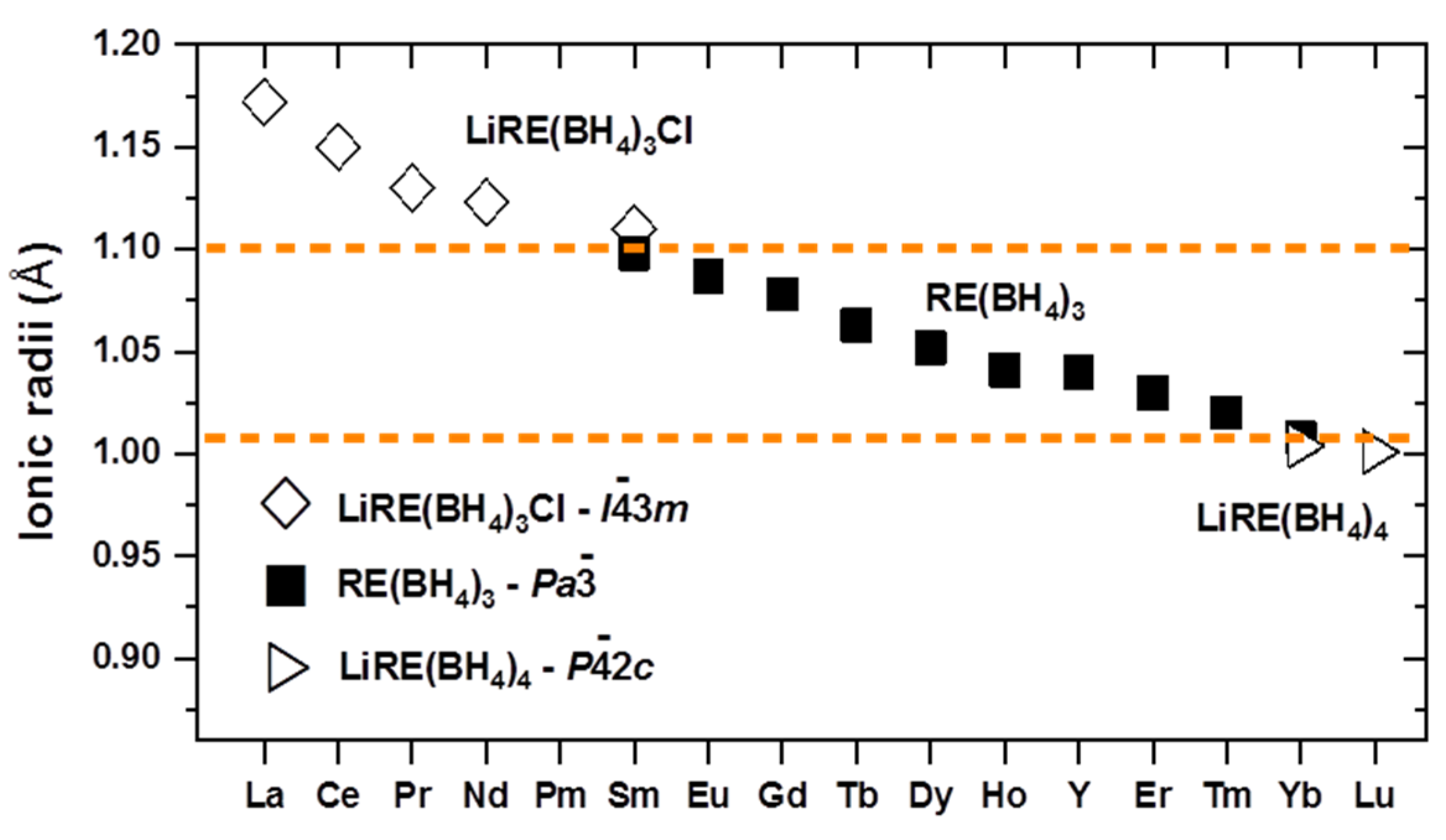{kind=link}
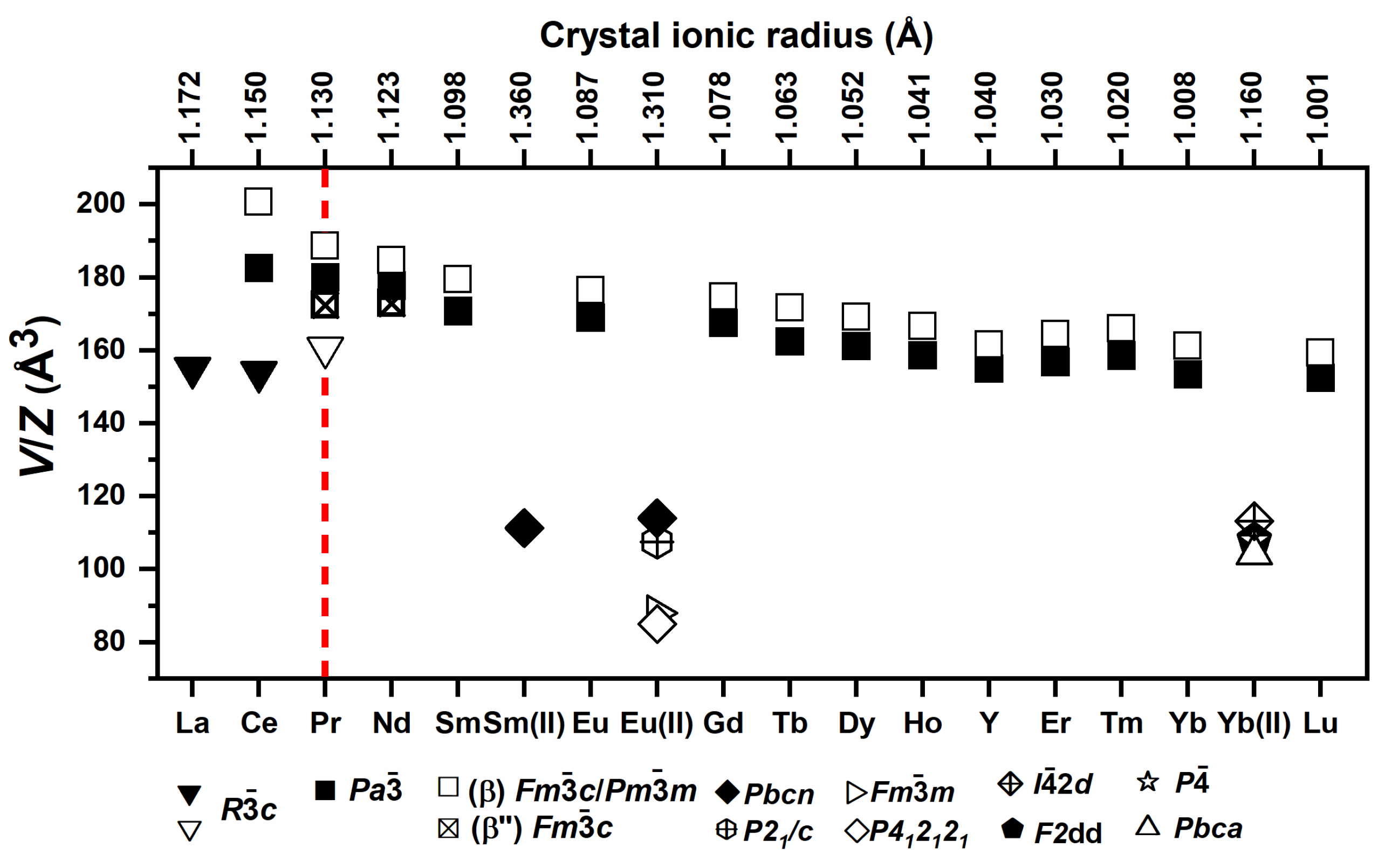{kind=link}
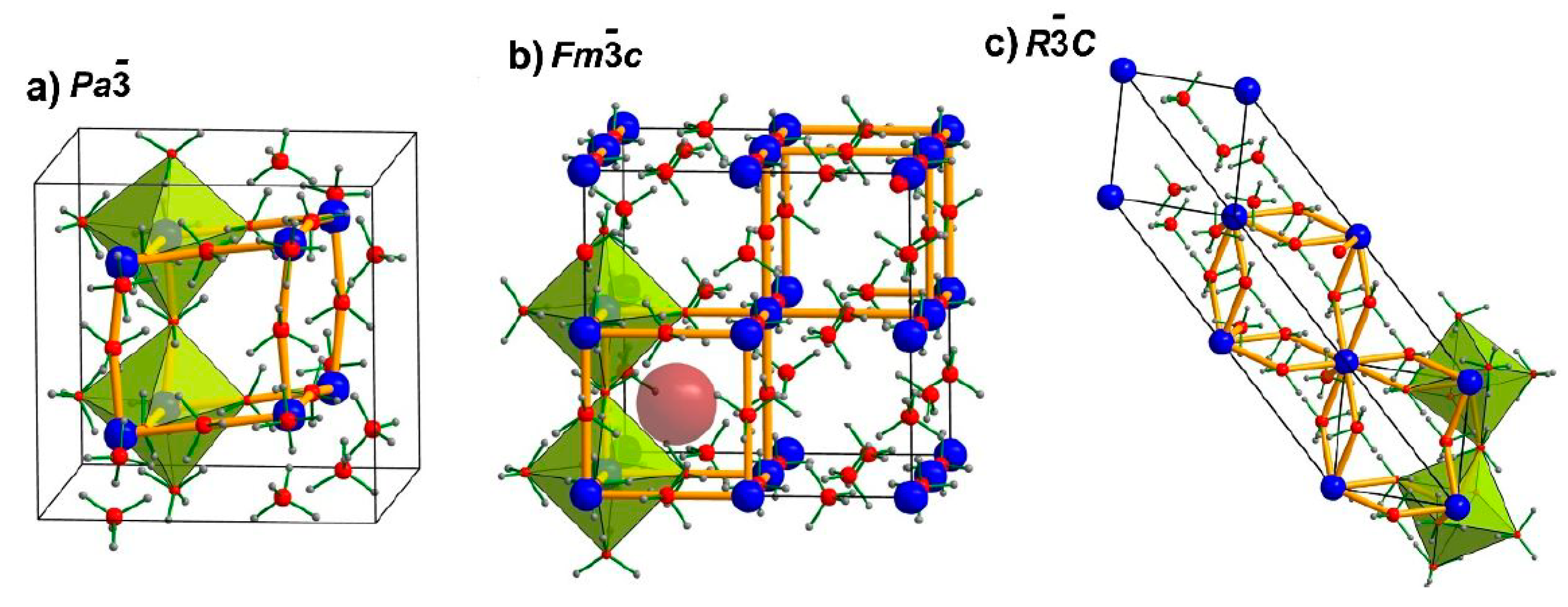{kind=link}
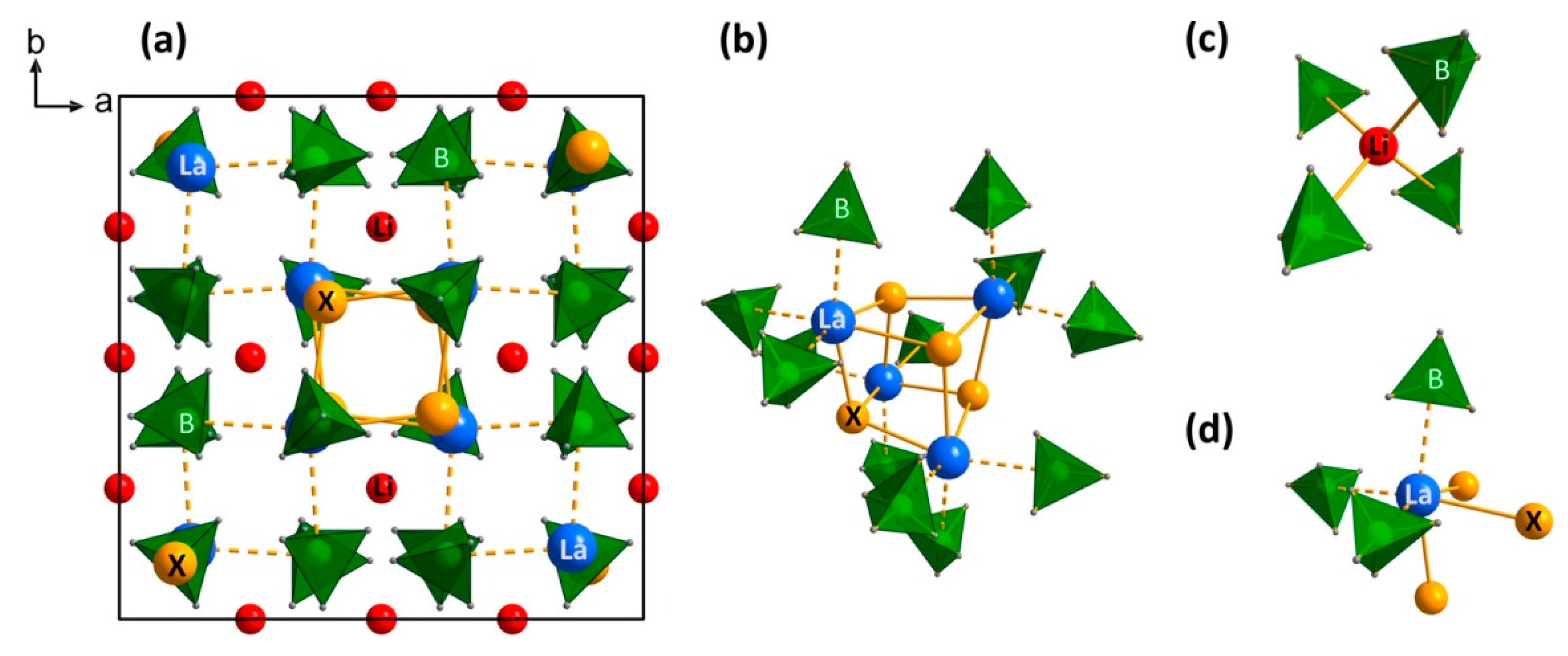{kind=link}
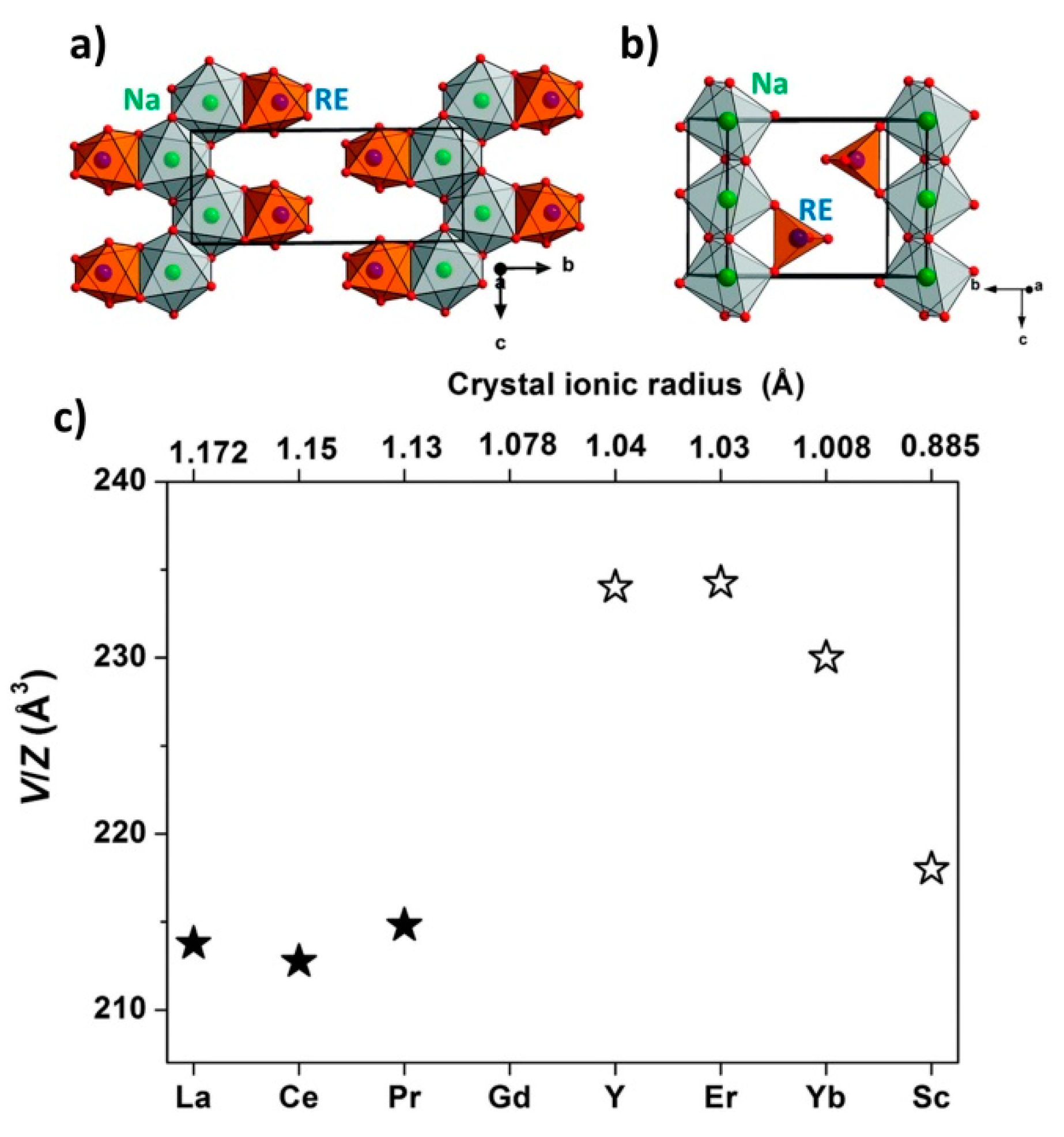{kind=link}
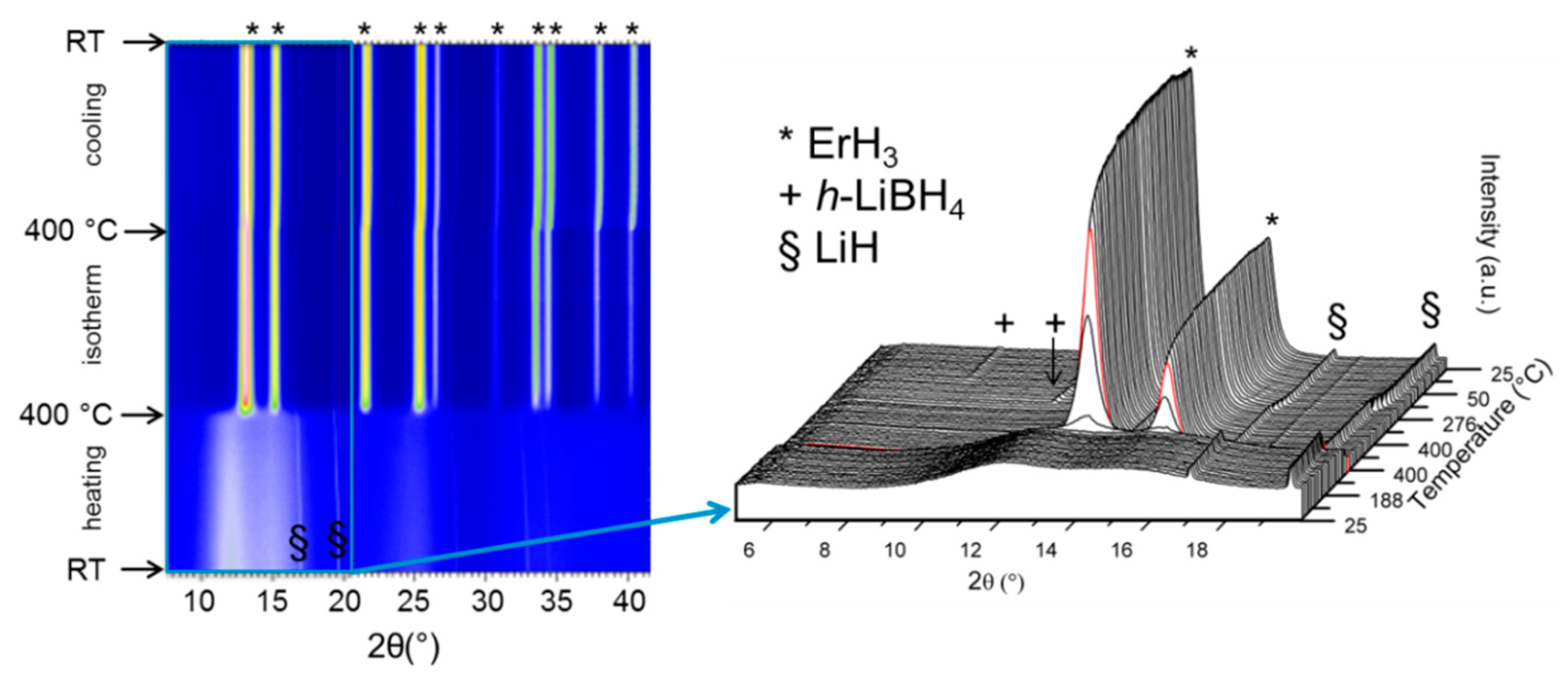{kind=link}
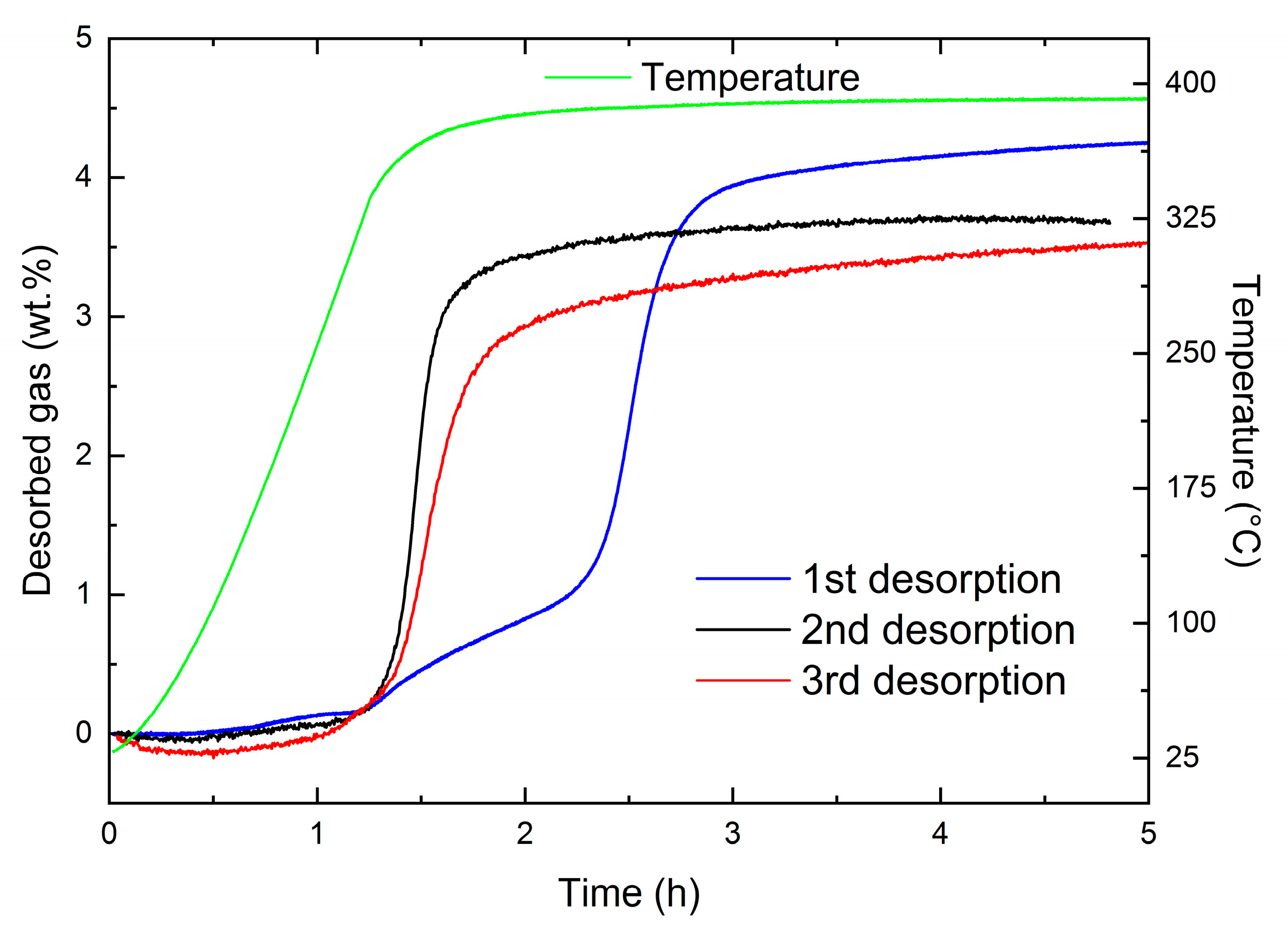{kind=link}
| Cell Configuration | Maximum Capacity, mAh g−1 | Reversible Capacity at C-Rate | Reported Cycle Number | Retention Capacity, mAh g−1 at the Last Cycle | Reference |
|---|---|---|---|---|---|
| 0.7MgH2 − 0.3TiH2│LiBH4│Li | 1950 | 90% at C/50 | 35 | 900 (51%) | [114] |
| MgH2│LiBH4│Li | 1750 | 94% at C/20 | 50 | 924 (45%) | [105] |
| TiH2│LiBH4│Li | 1052 | 86% at 2.5C | 50 | 878 (82%) | [108] |
| 25Al2O3 − 75MgH2│80Li2S − 20P2S5│Li | 3300 | 50% at | 9 | 580 (17.6%) | [106] |
| Systems | Constituents | Eutectic Mixture | Reference | ||||
|---|---|---|---|---|---|---|---|
| Tmp1 | Tmp2 | Tmp3 | Tmp | Tdec | ρg | ||
| 0.62LiBH4-0.38NaBH4 | 280 | 510 | - | 225 | 300 | 14.5 | [237,246,247] |
| 0.71LiBH4-0.29NaBH4 | 280 | 510 | - | 219 | n.a. | 15.2 | [247] |
| 0.725LiBH4-0.275KBH4 | 280 | 605 | - | 105 | 420 | 13.2 | [238,245] |
| 0.68NaBH4-0.32KBH4 | 510 | 605 | - | 460 | 465 | 9.4 | [248] |
| 0.66LiBH4-0.11NaBH4-0.23KBH4 | 280 | 510 | 605 | 102 | n.a. | 13.0 | [249] |
| 0.55LiBH4-0.45Mg(BH4)2 | 280 | 280 | - | 180 | 250 | 16.1 | [244,250,251] |
| 0.68LiBH4-0.32Ca(BH4)2 | 280 | 370 | - | 200 | 350 | 14.5 | [236,252] |
| 0.45NaBH4-0.55Mg(BH4)2 | 510 | 280 | - | 205 | 360 | 13.4 | [252,253] |
| Composition | ΔHmEXP | ΔHmASS | ΩASS | Reference |
|---|---|---|---|---|
| LiBH4 | 7.200 | [202] | ||
| NaBH4 | 16.900 | [275] | ||
| KBH4 | 19.200 | [249] | ||
| 0.70LiBH4 0.30NaBH4 | 6.990 | 6.520 | −19.604 * | [249] |
| 0.725LiBH4 0.275KBH4 | 11.025 | 9.828 | −13.016 | [249] |
| 0.682NaBH4 0.318KBH4 | 17.028 | 15.331 | 1.056 | [249] |
| M2 | M1 | M1M2(BH4)n M1M2(BH4)nClz | Structure | Structural Prototype | Investigated Application, Reference |
|---|---|---|---|---|---|
| La | Li | c-LiLa(BH4)3Cl | Spinel | Li ion conductivity, [63,391] | |
| c-LiLa(BH4)3Br | Spinel | Li ion conductivity, [411] | |||
| c-LiLa(BH4)3I | Spinel | Li ion conductivity, [411] | |||
| Na | o-NaLa(BH4)4 | Pbcn | New | Hydrogen storage, [412] | |
| K | m-K3La(BH4)6 | P21/n | rt-Na3AlF6 | Hydrogen storage, [412] | |
| Ce | Li | c-LiCe(BH4)3Cl | Spinel | Li ion conductivity, [394,413] | |
| Na | o-NaCe(BH4)4 | Pbcn | Own | Hydrogen storage, [401] | |
| K | m-K3Ce(BH4)6 | P21/n | rt-Na3AlF6 | Hydrogen storage, [414] | |
| Rb | m-Rb3Ce(BH4)6 | P21/n | rt-Na3AlF6 | not specified, [415] | |
| Pr | Li | c-LiPr(BH4)3Cl | Spinel | not specified, [391] | |
| Na | o-NaPr(BH4)4 | Pbcn | Own | Hydrogen storage, [401] | |
| Nd | Li | c-LiNd(BH4)3Cl | Spinel | not specified, [391] | |
| Sm | Li | c-LiSm(BH4)4Cl | Spinel | not specified, [391] | |
| K | o-KSm(BH4)3 | P21cn | CdTiO3 | Hydrogen storage, [416] | |
| Rb | o-RbSm(BH4)3 | Pbn21 | CdTiO3 | Hydrogen storage, [416] | |
| Cs | o-CsSm(BH4)3 | P22121 | NaNbO3 | Hydrogen storage, [416] | |
| Eu | Rb | o-RbEu(BH4)3 | Pbn21/Pna21 | CdTiO3 | not specified, [415] |
| Cs | t-CsEu(BH4)3 | P4/mbm | NaNbO3 | Luminescence, [415] | |
| Gd | Li | c-LiGd(BH4)3Cl | Spinel | Li ion conductivity, [63,391] | |
| K | m-KGd(BH4)4 | P21/c | LiMnF4 | Magneto-calorimetry, [417] | |
| m-K2Gd(BH4)5 | P21/m | Own | Magneto-calorimetry, [417] | ||
| m-K3Gd(BH4)6 | P21/n | rt-Na3AlF6 | Magneto-calorimetry, [417] | ||
| c-Cs3Gd(BH4)6 | (NH4)3AlF6 | Magneto-calorimetry, [415] | |||
| Ho | K | o-KHo(BH4)4 | Cmcm | ht-CrVO4 | not reported, [403] |
| Y | Li | t-LiY(BH4)4 | CuAlCl4 | Li ion conductivity, [418,419] | |
| Na | o-NaY(BH4)4 | C2221 | ht-CrVO4 | Na ion conductivity, [418,419] | |
| m-NaY(BH4)2Cl2 | P2/c | MgWO4 | not specified, [420] | ||
| K | o-KY(BH4)4 | Cmcm | ht-CrVO4 | not specified, [421] | |
| m-KY(BH4)4 | C2/c | rt-LaNbO4 | not specified, [422] | ||
| Rb | o-RbY(BH4)4 | Pnma | BaSO4 | not specified, [422] | |
| m-RbY(BH4)4 | P21/c | AgMnO4 (deformed BaSO4) | not specified, [423] | ||
| c-Rb3Y(BH4)6 | (NH4)3AlF6 | not specified, [415,422] | |||
| Cs | c-CsY(BH4)4 | I41/a | CaWO4-scheelite | not specified, [423] | |
| c-Cs3Y(BH4)6 | (NH4)3AlF6 | not specified, [415,422] | |||
| Er | Na | o-NaEr(BH4)4 | Cmcm | ht-CrVO4 | Hydrogen storage, [401] |
| K | o-KEr(BH4)4 | Cmcm | ht-CrVO4 | not specified, [424] | |
| Yb | Li | c-LiYb(BH4)3Cl | CuAlCl4 | not specified, [392] | |
| Na | o-NaYb(BH4)4 | Cmcm | ht-CrVO4 | not specified, [425] | |
| K | o-KYb(BH4)4 | Cmcm | ht-CrVO4 | not specified, [425] | |
| c-KYb(BH4)3-rt | CaTiO3 | Luminescence, [415] | |||
| o-KYb(BH4)3-ht | Pm21b | New | not specified, [415] | ||
| Lu | Li | t-LiLu(BH4)4 | CuAlCl4 | not specified, [391] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hadjixenophontos, E.; Dematteis, E.M.; Berti, N.; Wołczyk, A.R.; Huen, P.; Brighi, M.; Le, T.T.; Santoru, A.; Payandeh, S.; Peru, F.; et al. A Review of the MSCA ITN ECOSTORE—Novel Complex Metal Hydrides for Efficient and Compact Storage of Renewable Energy as Hydrogen and Electricity. Inorganics 2020, 8, 17. https://doi.org/10.3390/inorganics8030017
Hadjixenophontos E, Dematteis EM, Berti N, Wołczyk AR, Huen P, Brighi M, Le TT, Santoru A, Payandeh S, Peru F, et al. A Review of the MSCA ITN ECOSTORE—Novel Complex Metal Hydrides for Efficient and Compact Storage of Renewable Energy as Hydrogen and Electricity. Inorganics. 2020; 8(3):17. https://doi.org/10.3390/inorganics8030017
Chicago/Turabian StyleHadjixenophontos, Efi, Erika Michela Dematteis, Nicola Berti, Anna Roza Wołczyk, Priscilla Huen, Matteo Brighi, Thi Thu Le, Antonio Santoru, SeyedHosein Payandeh, Filippo Peru, and et al. 2020. "A Review of the MSCA ITN ECOSTORE—Novel Complex Metal Hydrides for Efficient and Compact Storage of Renewable Energy as Hydrogen and Electricity" Inorganics 8, no. 3: 17. https://doi.org/10.3390/inorganics8030017
APA StyleHadjixenophontos, E., Dematteis, E. M., Berti, N., Wołczyk, A. R., Huen, P., Brighi, M., Le, T. T., Santoru, A., Payandeh, S., Peru, F., Dao, A. H., Liu, Y., & Heere, M. (2020). A Review of the MSCA ITN ECOSTORE—Novel Complex Metal Hydrides for Efficient and Compact Storage of Renewable Energy as Hydrogen and Electricity. Inorganics, 8(3), 17. https://doi.org/10.3390/inorganics8030017