
Fluorination Effects in XPhos Gold(I) Fluorothiolates

 ,
,  , and
, and
Abstract
:1. Introduction
2. Results and Discussion
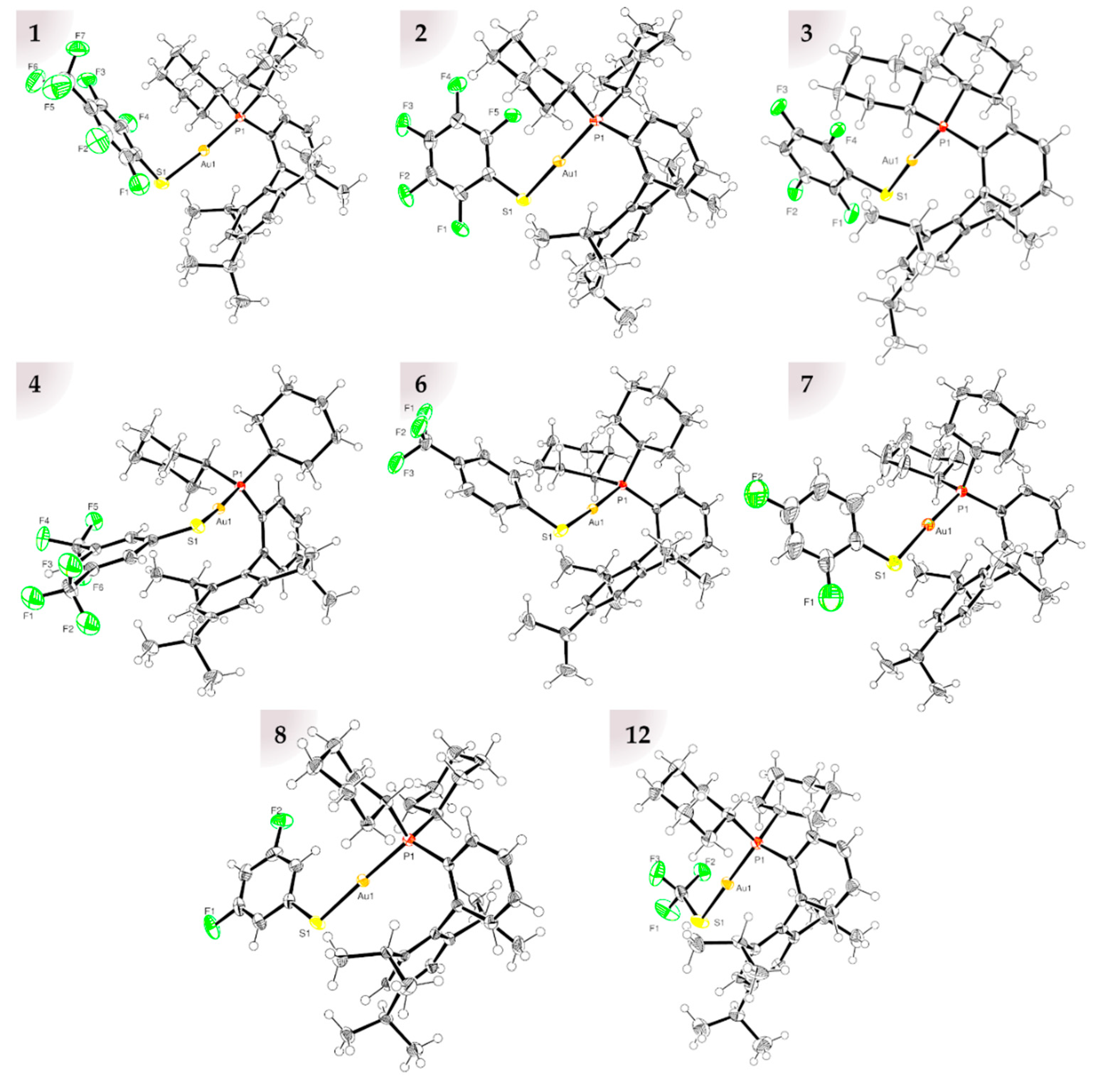
2.1. Molecular Structures
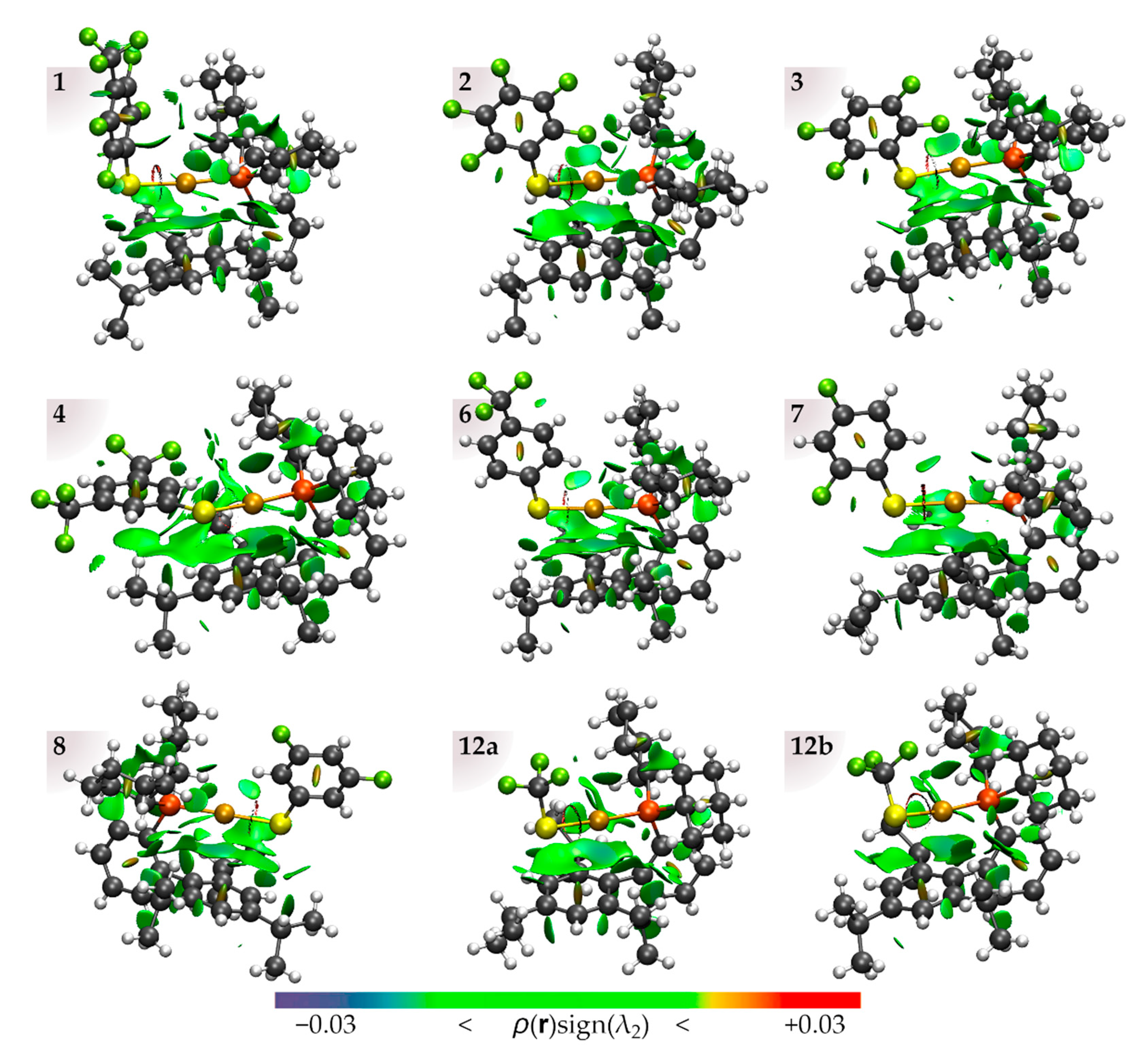
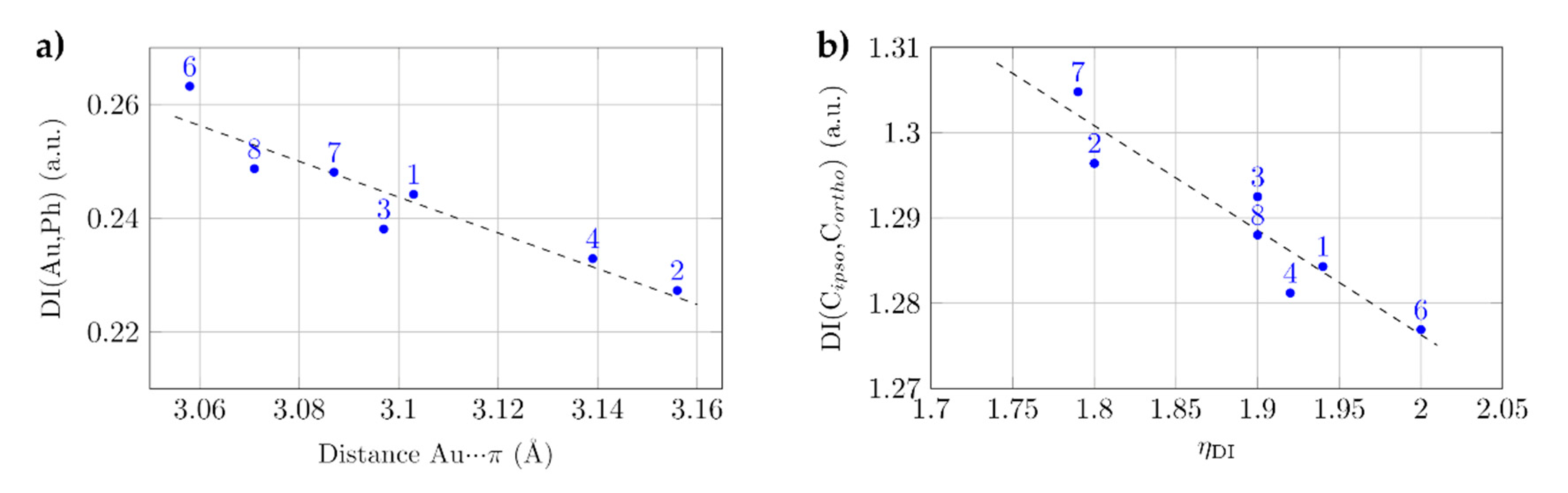
2.2. Au–Arene Contacts
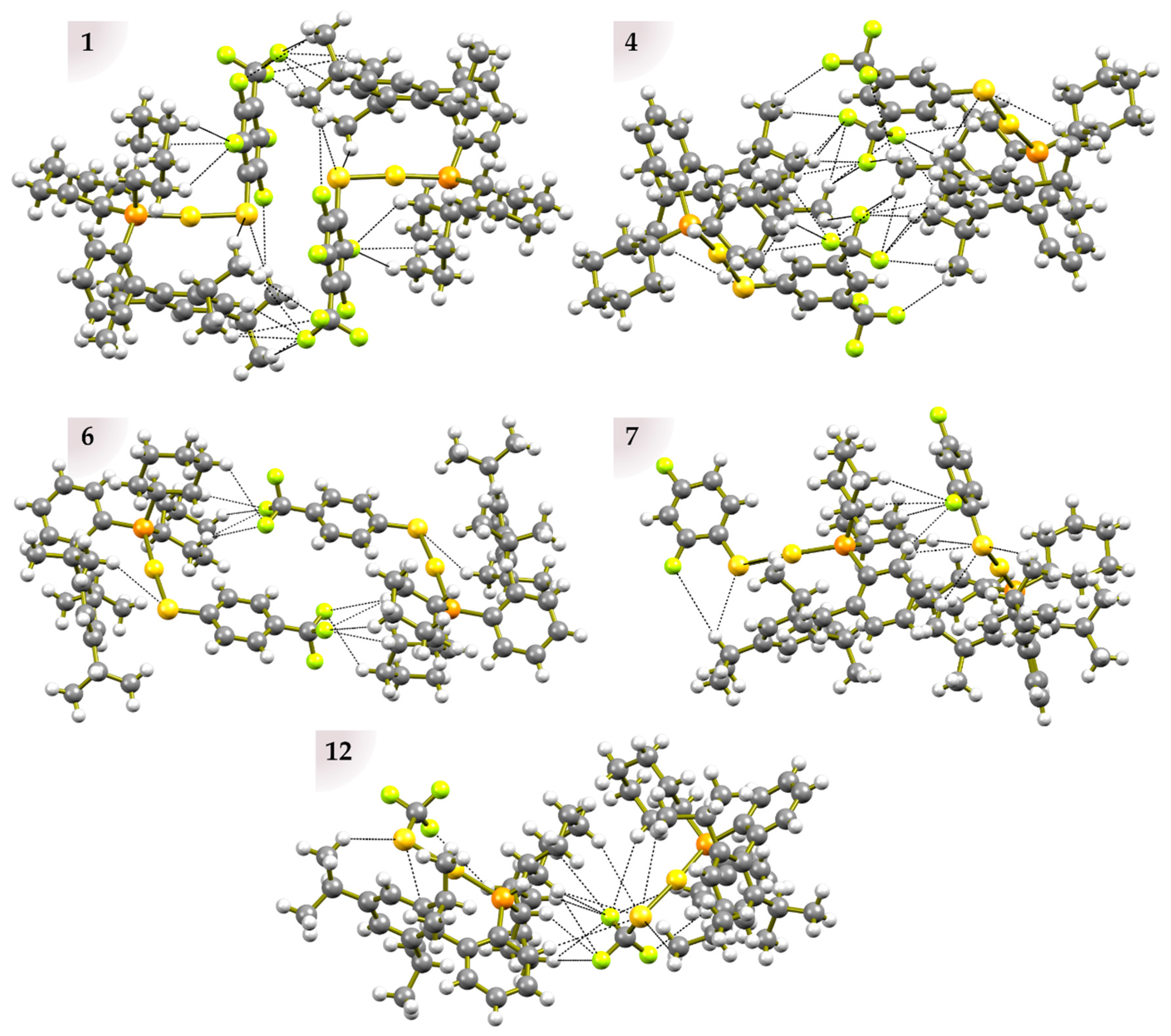
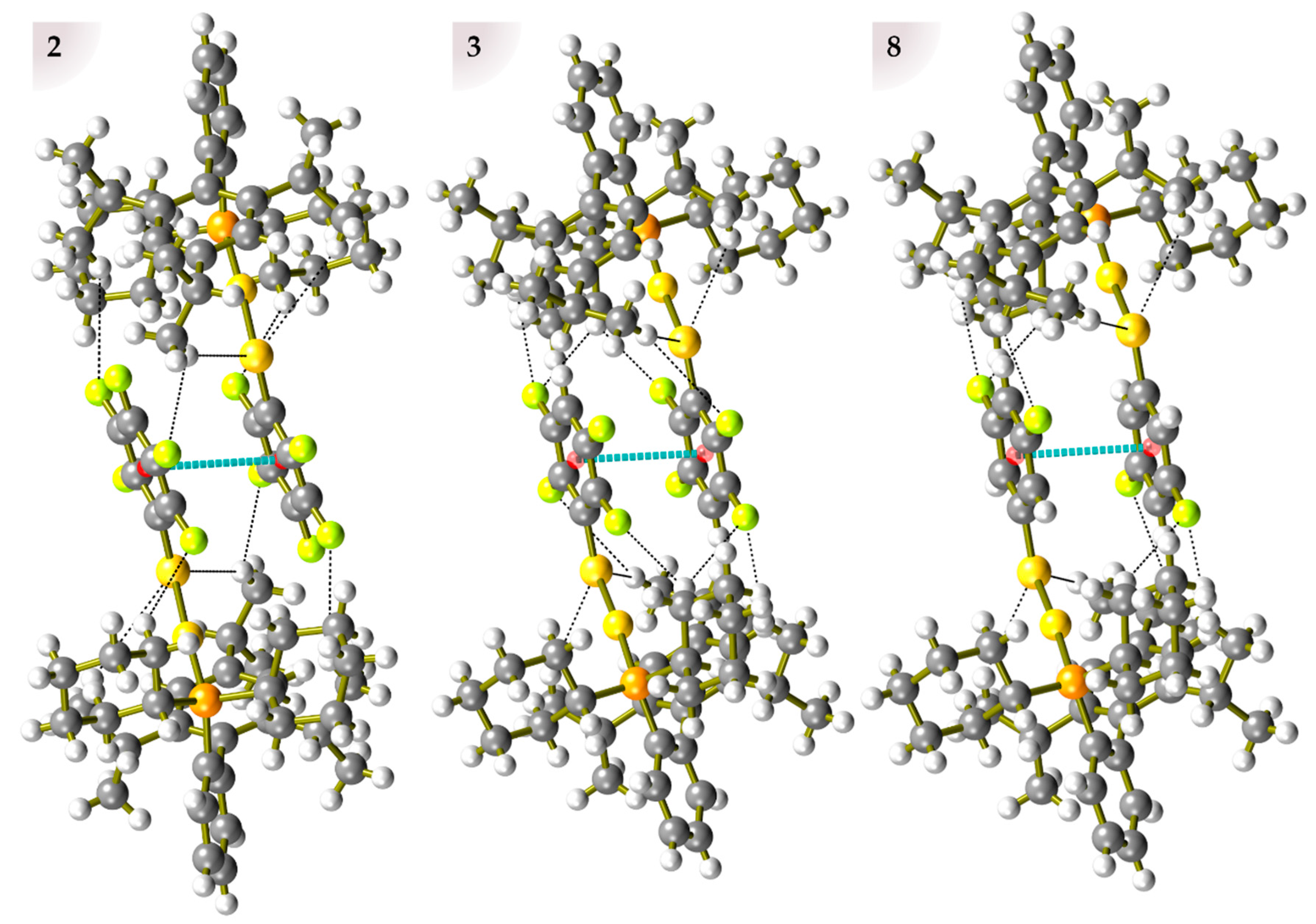
2.3. Crystal Packing
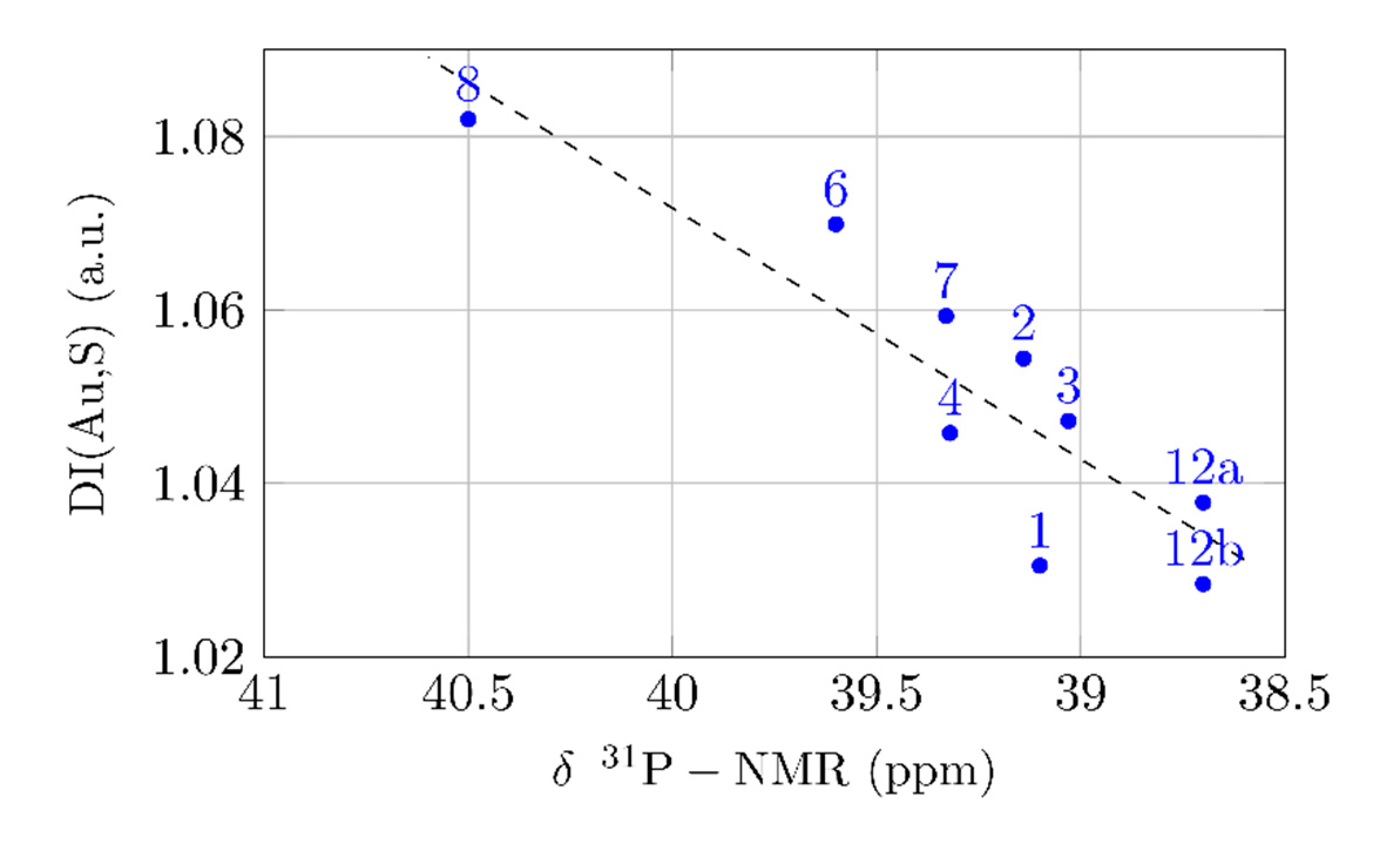
2.4. Trans-Influence
3. Materials and Methods
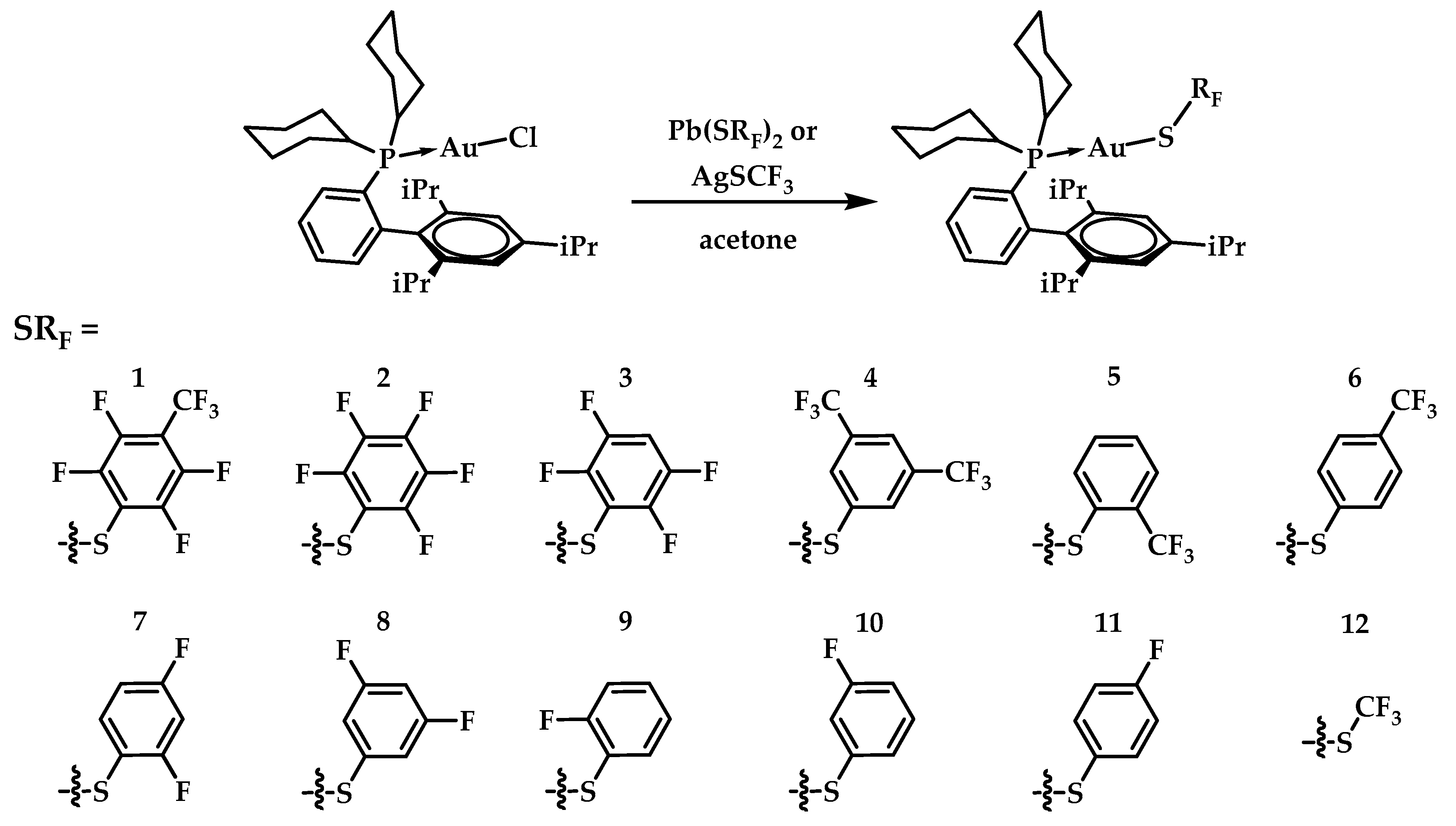
3.1. Synthesis and Characterization
3.2. Crystal Structure Determination
3.3. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Puddephatt, R.J. The Chemistry of Gold, Topics in Inorganic and General Chemistry; Elsevier Scientific Pub. Co.: Amsterdam, The Netherlands, 1978. [Google Scholar]
- Sadler, P.J. Gold chemistry. Gold Bull. 2009, 42, 220. [Google Scholar] [CrossRef] [Green Version]
- Schmidbaur, H. The fascinating implications of new results in gold chemistry. Gold Bull. 1990, 23, 11–21. [Google Scholar] [CrossRef] [Green Version]
- Schmidbaur, H.; Schier, A. Aurophilic interactions as a subject of current research: An up-date. Chem. Soc. Rev. 2012, 41, 370–412. [Google Scholar] [CrossRef] [PubMed]
- Tiekink, E.R.T. Supramolecular assembly based on “emerging” intermolecular interactions of particular interest to coordination chemists. Co-ord. Chem. Rev. 2017, 345, 209–228. [Google Scholar] [CrossRef] [Green Version]
- He, X.; Yam, V.W.-W. Luminescent gold(I) complexes for chemosensing. Coord. Chem. Rev. 2011, 255, 2111–2123. [Google Scholar] [CrossRef]
- Lima, J.C.; Rodríguez, L. Highlights on gold TADF complexes. Inorganics 2019, 7, 124. [Google Scholar] [CrossRef] [Green Version]
- Langdon-Jones, E.E.; Pope, S.J.A. Recent developments in gold(I) coordination chemistry: Luminescence properties and bioimaging opportunities. Chem. Commun. 2014, 50, 10343–10354. [Google Scholar] [CrossRef]
- Lima, J.; Rodríguez, L.; Lima, J.C.; Rodríguez, L. Supramolecular gold metallogelators: The key role of metallophilic interactions. Inorganics 2014, 3, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Blasco, D.; López-De-Luzuriaga, J.M.; Monge, M.; Olmos, M.E.; Pascual, D.; Rodríguez-Castillo, M. Cooperative Au(I)···Au(I) interactions and hydrogen bonding as origin of a luminescent adeninate hydrogel formed by ultrathin molecular nanowires. Inorg. Chem. 2018, 57, 3805–3817. [Google Scholar] [CrossRef]
- Bardají, M. Gold liquid crystals in the XXI century. Inorganics 2014, 2, 433–454. [Google Scholar] [CrossRef] [Green Version]
- Kui, S.C.F.; Huang, J.-S.; Sun, R.W.-Y.; Zhu, N.; Che, C.-M. Self-assembly of a highly stable, topologically interesting metallamacrocycle by bridging gold(I) ions with pyridyl-2,6-diphenyl2− and diphosphanes. Angew. Chem. Int. Ed. 2006, 45, 4663–4666. [Google Scholar] [CrossRef] [PubMed]
- Aguiló, E.; Moro, A.J.; Gavara, R.; Alfonso, I.; Pérez, Y.; Zaccaria, F.; Guerra, C.F.; Malfois, M.; Baucells, C.; Ferrer, M.; et al. Reversible self-assembly of water-soluble gold(I) complexes. Inorg. Chem. 2018, 57, 1017–1028. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Du, Y.; Liu, J.; Yao, Q.; Chen, T.; Cao, Y.; Zhang, H.; Xie, J. Aurophilic interactions in the self-assembly of gold nanoclusters into nanoribbons with enhanced luminescence. Angew. Chem. Int. Ed. 2019, 58, 8139–8144. [Google Scholar] [CrossRef] [PubMed]
- Shakirova, J.R.; Grachova, E.V.; Karttunen, A.J.; Gurzhiy, V.V.; Tunik, S.P.; Koshevoy, I.O. Metallophilicity-assisted assembly of phosphine-based cage molecules. Dalton Trans. 2014, 43, 6236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ott, I. On the medicinal chemistry of gold complexes as anticancer drugs. Coord. Chem. Rev. 2009, 253, 1670–1681. [Google Scholar] [CrossRef]
- Bertrand, B.; Casini, A. A golden future in medicinal inorganic chemistry: The promise of anticancer gold organometallic compounds. Dalton Trans. 2014, 43, 4209–4219. [Google Scholar] [CrossRef]
- Zou, T.; Lum, C.T.; Lok, C.-N.; Zhang, J.-J.; Che, C.-M. Chemical biology of anticancer gold(III) and gold(I) complexes. Chem. Soc. Rev. 2015, 44, 8786–8801. [Google Scholar] [CrossRef]
- Pang, B.; Yang, X.; Xia, Y. Putting gold nanocages to work for optical imaging, controlled release and cancer theranostics. Nanomedicine 2016, 11, 1715–1728. [Google Scholar] [CrossRef] [Green Version]
- Echavarren, A.M.; Hashmi, A.S.K.; Toste, F.D. Gold catalysis—steadily increasing in importance. Adv. Synth. Catal. 2016, 358, 1347. [Google Scholar] [CrossRef] [Green Version]
- Ciriminna, R.; Falletta, E.; Della Pina, C.; Teles, J.H.; Pagliaro, M. Industrial applications of gold catalysis. Angew. Chem. Int. Ed. 2016, 55, 14210–14217. [Google Scholar] [CrossRef]
- Barder, T.E.; Buchwald, S.L. Rationale behind the resistance of dialkylbiaryl phosphines toward oxidation by molecular oxygen. J. Am. Chem. Soc. 2007, 129, 5096–5101. [Google Scholar] [CrossRef] [PubMed]
- Zuccarello, G.; Zanini, M.; Echavarren, A.M. Buchwald-type ligands on gold(I) catalysis. Isr. J. Chem. 2020, 60, 360–372. [Google Scholar] [CrossRef]
- Surry, D.S.; Buchwald, S.L. Biaryl phosphane ligands in palladium-catalyzed amination. Angew. Chem. Int. Ed. 2008, 47, 6338–6361. [Google Scholar] [CrossRef] [PubMed]
- Billingsley, K.; Buchwald, S.L. Highly Efficient Monophosphine-Based Catalyst for the Palladium-Catalyzed Suzuki−Miyaura reaction of heteroaryl halides and heteroaryl boronic acids and esters. J. Am. Chem. Soc. 2007, 129, 3358–3366. [Google Scholar] [CrossRef]
- Homs, A.; Escofet, I.; Echavarren, A.M. On the silver effect and the formation of chloride-bridged digold complexes. Org. Lett. 2013, 15, 5782–5785. [Google Scholar] [CrossRef]
- Visbal, R.; Herrera, R.P.; Gimeno, M.C. Thiolate bridged gold(I)–NHC catalysts: New approach for catalyst design and its application to trapping catalytic intermediates. Chem. A Eur. J. 2019, 25, 15837–15845. [Google Scholar] [CrossRef]
- Miró, J.; Del Pozo, C. Fluorine and gold: A fruitful partnership. Chem. Rev. 2016, 116, 11924–11966. [Google Scholar] [CrossRef]
- Moreno-Alcantar, G.; Turcio-García, L.; Guevara-Vela, J.M.; Romero-Montalvo, E.; Rocha-Rinza, T.; Pendás, Á.M.; Flores-Álamo, M.; Torrens, H. Directing the crystal packing in triphenylphosphine gold(I) thiolates by ligand fluorination. Inorg. Chem. 2020, 59, 8667–8677. [Google Scholar] [CrossRef]
- Moreno-Alcantar, G.; Guevara-Vela, J.M.; Delgadillo-Ruíz, R.; Rocha-Rinza, T.; Martín Pendás, Á.; Flores-Álamo, M.; Torrens, H. Structural effects of trifluoromethylation and fluorination in gold(I) BIPHEP fluorothiolates. New J. Chem. 2017, 41, 10537–10541. [Google Scholar] [CrossRef]
- Cordón, J.; Jiménez-Osés, G.; López-De-Luzuriaga, J.M.; Monge, M. The key role of Au-substrate interactions in catalytic gold subnanoclusters. Nat. Commun. 2017, 8, 1657. [Google Scholar] [CrossRef] [Green Version]
- Partyka, D.V.; Robilotto, T.J.; Zeller, M.; Hunter, A.D.; Gray, T.G. Dialkylbiarylphosphine complexes of gold(I) halides. Gold−aryl π-interactions in the solid state. Organometallics 2008, 27, 28–32. [Google Scholar] [CrossRef]
- Nieto-Oberhuber, C.; López, S.; Echavarren, A.M. Intramolecular [4 + 2] cycloadditions of 1,3-enynes or arylalkynes with alkenes with highly reactive cationic phosphine Au(I) complexes. J. Am. Chem. Soc. 2005, 127, 6178–6179. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Galán, P.; Delpont, N.; Herrero-Gómez, E.; Maseras, F.; Echavarren, A.M. Metal-arene interactions in dialkylbiarylphosphane complexes of copper, silver, and gold. Chem. A Eur. J. 2010, 16, 5324–5332. [Google Scholar] [CrossRef] [PubMed]
- Herrero-Gómez, E.; Nieto-Oberhuber, C.; López, S.; Benet-Buchholz, J.; Echavarren, A.M. Cationic η1/η2-Gold(I) complexes of simple arenes. Angew. Chem. Int. Ed. 2006, 45, 5455–5459. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Alcántar, G.; Hess, K.; Guevara-Vela, J.M.; Rocha-Rinza, T.; Martín Pendás, Á.; Flores-Álamo, M.; Torrens, H. π-backbonding and non-covalent interactions in the JohnPhos and polyfluorothiolate complexes of gold(i). Dalton Trans. 2017, 46, 12456–12465. [Google Scholar] [CrossRef] [PubMed]
- Vasilyev, A.V.; Lindeman, S.V.; Kochi, J.K. Noncovalent binding of the halogens to aromatic donors. Discrete structures of labile Br2 complexes with benzene and toluene. Chem. Commun. 2001, 909–910. [Google Scholar] [CrossRef] [Green Version]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; Clarendon Press: Oxford, UK, 1990; ISBN 0198558651. [Google Scholar]
- Fradera, X.; Austen, M.A.; Bader, R.F.W. The Lewis Model and beyond. J. Phys. Chem. A 1999, 103, 304–314. [Google Scholar] [CrossRef]
- Johnson, E.R.; Keinan, S.; Mori-Sánchez, P.; Contreras-García, J.; Cohen, A.J.; Yang, W. Revealing noncovalent interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef] [Green Version]
- Contreras-García, J.; Johnson, E.R.; Keinan, S.; Chaudret, R.; Piquemal, J.-P.; Beratan, D.N.; Yang, W. NCIPLOT: A program for plotting noncovalent interaction regions. J. Chem. Theory Comput. 2011, 7, 625–632. [Google Scholar] [CrossRef]
- Bondi, A. van der Waals Volumes and Radii. J. Phys. Chem. 1964, 68, 441–451. [Google Scholar] [CrossRef]
- Appleton, T.; Clark, H.; Manzer, L. The trans-influence: Its measurement and significance. Coord. Chem. Rev. 1973, 10, 335–422. [Google Scholar] [CrossRef]
- Moreno-Alcantar, G.; Hernández-Toledo, H.; Guevara-Vela, J.M.; Rinza, T.R.; Pendás, Á.M.; Flores-Álamo, M.; Torrens, H. Stability and trans influence in fluorinated gold(I) coordination compounds. Eur. J. Inorg. Chem. 2018, 2018, 4413–4420. [Google Scholar] [CrossRef] [Green Version]
- Cervantes, R.; Tiburcio, J.; Torrens, H. Cis and trans influences on [Pt(SR F )(triphos)] + complexes (SR F = polyfluorobenzothiolate). New J. Chem. 2014, 39, 631–638. [Google Scholar] [CrossRef]
- Moreno-Alcantar, G.; Salazar, L.; Romo-Islas, G.; Flores-Álamo, M.; Torrens, H. Exploring the self-assembled tacticity in aurophilic polymeric arrangements of diphosphanegold(I) fluorothiolates. Molecules 2019, 24, 4422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biggs, A.I.; Baddiley, J.; Hughes, N.A.; Mehra, H.S.; Trotman-Dickenson, A.F.; Verbeke, G.J.O.; Barraclough, C.C.; Nyholm, R.S.; Beattie, I.R.; Gilson, T.; et al. Notes. J. Chem. Soc. 1961, 2572–2600. [Google Scholar] [CrossRef]
- Agilent. CrysAlis PRO; Agilent Technologies Ltd.: Yarnton, UK, 2014. [Google Scholar]
- Clark, R.C.; Reid, J.S. The analytical calculation of absorption in multifaceted crystals. Acta Crystallogr. Sect. A Found. Crystallogr. 1995, 51, 887–897. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT– Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Farrugia, L.J. WinGXandORTEP for Windows: An update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From visualization to analysis, design and prediction. J. Appl. Crystallogr. 2020, 53, 226–235. [Google Scholar] [CrossRef] [Green Version]
- Keith, T.A. AIMAll 2019, version 19.10.12; Gristmill Software: Overland Park, KS, USA, 2019. [Google Scholar]
- Neese, F. The ORCA program system. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Neese, F. Software update: The ORCA program system, version 4.0. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2017, e8, 1327. [Google Scholar] [CrossRef]
- Levy, M.; Perdew, J.P. Success of quantum mechanical approximations for molecular geometries and electron–nuclear attraction expectation values: Gift of the Coulomb potential? J. Chem. Phys. 1986, 84, 4519–4523. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Pantazis, D.A.; Chen, X.-Y.; Landis, C.R.; Neese, F. All-Electron Scalar Relativistic Basis Sets for Third-Row Transition Metal Atoms. J. Chem. Theory Comput. 2008, 4, 908–919. [Google Scholar] [CrossRef]
- Van Lenthe, E.; Baerends, E.J.; Snijders, J.G. Relativistic regular two-component Hamiltonians. J. Chem. Phys. 1993, 99, 4597–4610. [Google Scholar] [CrossRef]
- van Wüllen, C. Molecular density functional calculations in the regular relativistic approximation: Method, application to coinage metal diatomics, hydrides, fluorides and chlorides, and comparison with first-order relativistic calculations. J. Chem. Phys. 1998, 109, 392–399. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | dAu–P (Å) | dAu–S (Å) | ∠P–Au–S (°) | ∠Au–P–CPh (°) | ∠Au–S–C (°) |
|---|---|---|---|---|---|
| 1 | 2.246(1) | 2.297(2) | 174.44(5) | 115.8(2) | 101.3(2) |
| 2 | 2.261(1) | 2.307(1) | 177.62(3) | 117.5(1) | 109.0(1) |
| 3 | 2.265(1) | 2.308(1) | 175.62(3) | 116.3(1) | 111.9(1) |
| 4 | 2.259(1) | 2.305(1) | 173.25(4) | 118.3(1) | 106.7(2) |
| 6 | 2.260(1) | 2.298(1) | 175.57(3) | 117.1(1) | 106.7(1) |
| 7 | 2.264(2) | 2.313(4) | 177.60(10) | 115.4(3) | 108.2(4) |
| 2.262(2) | 2.316(3) | 176.78(9) | 115.6(3) | 109.9(4) | |
| 8 | 2.270(1) | 2.300(1) | 175.55(4) | 115.4(1) | 111.7(1) |
| 12 | 2.250(1) | 2.310(2) | 173.19(5) | 116.0(1) | 096.9(2) |
| 2.256(1) | 2.311(1) | 177.42(5) | 114.6(1) | 101.1(2) |
| Compound | Au–π (Å) | η | Au/S/C/C (°) | ∠π/ πF (°) |
|---|---|---|---|---|
| 1 | 3.103 | 1.99 | 61.7 | 83.9 |
| 2 | 3.156 | 1.75 | 18.7 | 88.1 |
| 3 | 3.097 | 2.00 | 12.9 | 67.9 |
| 4 | 3.139 | 1.98 | 5.4 | 9.1 |
| 6 | 3.058 | 1.92 | 5.5 | 69.9 |
| 7 | 3.087 | 1.81 | 15.7 | 87.6 |
| 3.099 | 1.77 | 6.2 | 85.5 | |
| 8 | 3.071 | 2.00 | 8.7 | 68.1 |
| 12 | 3.050 | 1.37 | ||
| 3.099 | 1.49 | - | - |
| Structure | p (rbcp) | DI (A,B) | ηDI | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Au–P | Au–S | Au⋯Cipso | Au, P | Au, S | Au, Cipso | Au, Cortho | Au, Ph | Cipso,Cortho | ||
| 1 | 0.125 | 0.110 | 0.016 | 1.017 | 1.031 | 0.082 | 0.077 | 0.244 | 1.284 | 1.94 |
| 2 | 0.122 | 0.109 | 0.015 | 1.009 | 1.054 | 0.080 | 0.064 | 0.227 | 1.296 | 1.80 |
| 3 | 0.121 | 0.108 | 0.016 | 1.007 | 1.047 | 0.084 | 0.076 | 0.238 | 1.293 | 1.90 |
| 4 | 0.123 | 0.110 | 0.015 | 1.006 | 1.046 | 0.076 | 0.070 | 0.233 | 1.281 | 1.92 |
| 6 | 0.122 | 0.112 | 0.017 | 0.998 | 1.070 | 0.084 | 0.084 | 0.263 | 1.277 | 2.00 |
| 7 | 0.121 | 0.107 | 0.016 | 1.022 | 1.059 | 0.091 | 0.072 | 0.248 | 1.305 | 1.79 |
| 8 | 0.120 | 0.110 | 0.017 | 0.998 | 1.082 | 0.088 | 0.080 | 0.249 | 1.288 | 1.90 |
| 12a | 0.123 | 0.108 | 0.018 | 1.025 | 1.038 | 0.105 | 0.063 | 0.271 | 1.281 | 1.60 |
| 12b | 0.124 | 0.108 | 0.016 | 1.027 | 1.028 | 0.097 | 0.058 | 0.234 | 1.298 | 1.60 |
| Compound | δ 31P-NMR (ppm) |
|---|---|
| 1 | 39.10 |
| 2 | 39.14 |
| 3 | 39.03 |
| 4 | 39.32 |
| 6 | 39.60 |
| 7 | 39.33 |
| 8 | 40.50 |
| 12 | 38.70 |
| 12b | 38.70 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moreno-Alcántar, G.; Díaz-Rosas, C.; Fernández-Alarcón, A.; Turcio-García, L.; Flores-Álamo, M.; Rocha-Rinza, T.; Torrens, H. Fluorination Effects in XPhos Gold(I) Fluorothiolates. Inorganics 2021, 9, 14. https://doi.org/10.3390/inorganics9020014
Moreno-Alcántar G, Díaz-Rosas C, Fernández-Alarcón A, Turcio-García L, Flores-Álamo M, Rocha-Rinza T, Torrens H. Fluorination Effects in XPhos Gold(I) Fluorothiolates. Inorganics. 2021; 9(2):14. https://doi.org/10.3390/inorganics9020014
Chicago/Turabian StyleMoreno-Alcántar, Guillermo, Cristian Díaz-Rosas, Alberto Fernández-Alarcón, Luis Turcio-García, Marcos Flores-Álamo, Tomás Rocha-Rinza, and Hugo Torrens. 2021. "Fluorination Effects in XPhos Gold(I) Fluorothiolates" Inorganics 9, no. 2: 14. https://doi.org/10.3390/inorganics9020014
APA StyleMoreno-Alcántar, G., Díaz-Rosas, C., Fernández-Alarcón, A., Turcio-García, L., Flores-Álamo, M., Rocha-Rinza, T., & Torrens, H. (2021). Fluorination Effects in XPhos Gold(I) Fluorothiolates. Inorganics, 9(2), 14. https://doi.org/10.3390/inorganics9020014