1. Introduction
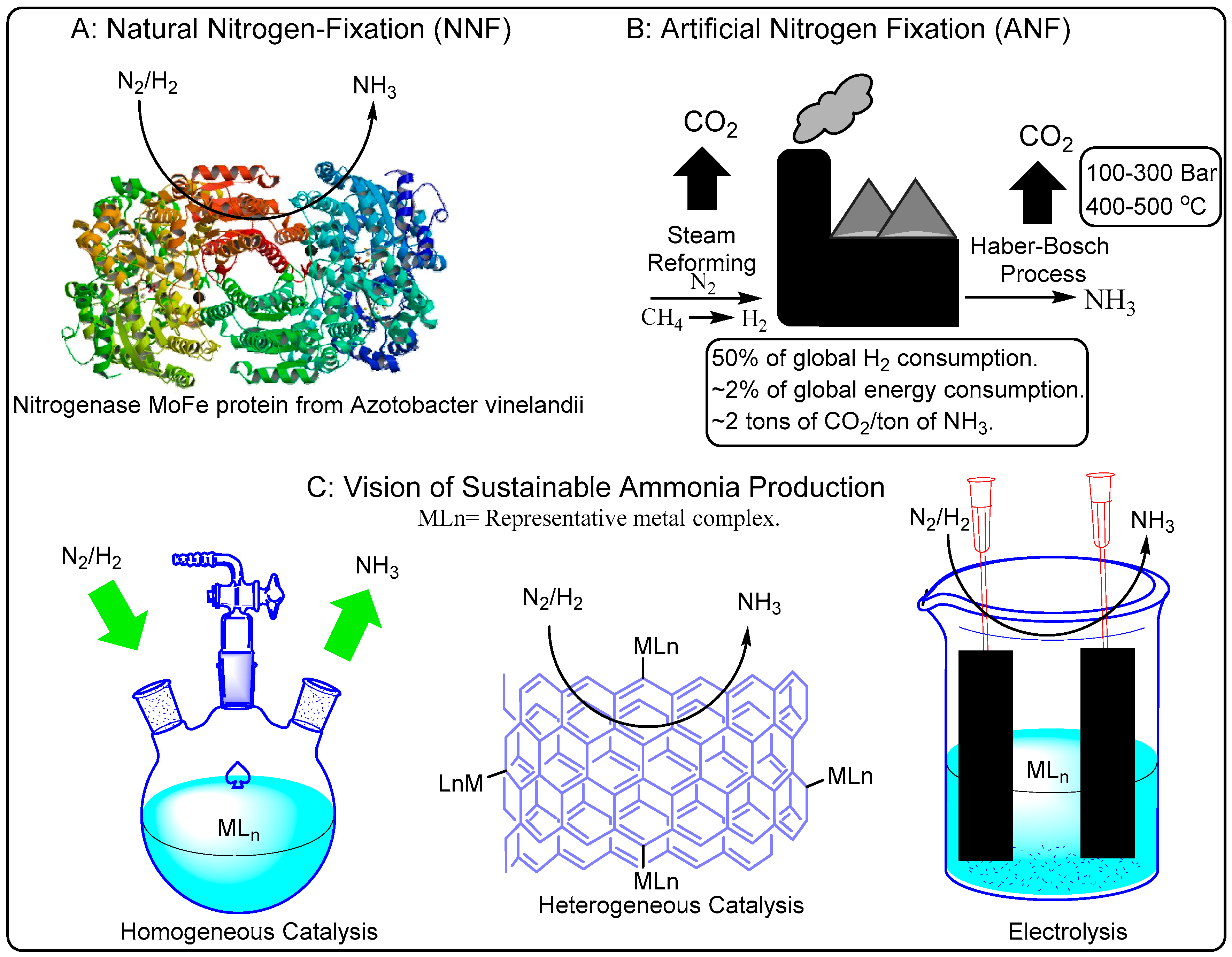
Nitrogen is one of the most important elements present in essential molecules (amino acids, proteins, enzymes and nucleic acids and medicines) for life [
1]. The bond strength of dinitrogen limits its chemical reactivity which is fortunate given its abundance in the atmosphere (78%) [
2]. The biological transformation of dinitrogen into ammonia for conversion into more complicated biomolecules, is called natural nitrogen fixation (NNF). On the other hand, conversion of dinitrogen into useful molecules unaided by nature is called artificial nitrogen fixation (ANF). Recently, ANF surpassed NNF in terms of the total amount [
1]. The challenges of ANF are enshrined in the Haber–Bosch (H–B) process (1913), which has been called the most important invention of the 20th century [
1,
2,
3]. The H–B process converts nitrogen purified from the atmosphere and externally supplied hydrogen into gaseous ammonia at a very high temperature (650–750 K) and extreme pressure (50–200 bar) in the presence of a heterogeneous catalyst [
4,
5,
6,
7,
8]. The growth of the human population has been mapped directly to the growth of ammonia production via the H–B process to supply artificial fertilizers post World War II [
8,
9]. A whopping 40% of the total human population depends on ANF for an adequate food supply [
10,
11]. The ever-expanding need for food grown with artificial fertilizers generates 450 million metric tons of carbon dioxide (~1.9 metric tons of CO
2 per metric ton of ammonia produced [
12]), consumes 2% of the world’s total natural gas output, results in 1% of all human emissions and 2% of the world’s total energy consumption on an annual basis [
13,
14,
15,
16]. Ammonia produced through ANF is utilized in multiple large-scale industries apart from fertilizers; examples include: pharmaceuticals, explosives, plastic manufacturing, mining and metallurgy, production of nitric acid, et cetera [
8]. Recently there is an increasing interest in using ammonia as the energy carrier of the future [
17,
18]. Further increase in conventional ANF will be ecologically undesirable in terms of carbon footprint, therefore new environmentally-friendly methods for ANF are required [
19].
Improving the efficiency of ANF will lead to huge benefits on a world-wide scale. Multiple approaches (
Figure 1) have been undertaken to achieve this including: improvement of the H–B process conditions by exploring biological-mimics, homogeneous or heterogeneous catalysts designed to operate at milder conditions, semiconductor-based photocatalysis for nitrogen fixation, greener microwave plasma methods to synthesize ammonia and NO
x, and hybrid systems between NNF and ANF such as development of synthetic rhizospheres (SRS), which helps intensify the NNF in plants via the development of a synthetic media [
1,
17,
18,
20,
21,
22,
23].
Binding of dinitrogen to a metal center depends on the formation of a stable reduced metal center, which can in turn reduce a dinitrogen molecule. Organometallic chemists all over the world have attempted this feat with different levels of success [
24]. Schrock and Yandulov synthesized the first successful nitrogenase model system that evolved ammonia from a dinitrogen species using (LutH)BArF
4 and CrCp*
2 as protonating and reducing agents, respectively [
25]. The Peters group followed with an organometallic complex with first row transition metal (Fe) with a maximum of 93 equivalents of ammonia evolution per iron center [
3,
26,
27]. Additional interest in first row transition metal PNP pincer complexes as ANF agents began in 2015 when the Lu group published results involving a cobalt dinitrogen complex [
26]. In 2016, the Nishibayashi group also developed Fe complexes based on a PNP pincer ligand with a pyrrolidine unit at the center and a terminal di-tert-butylphosphine to effectively catalyze ANF [
27]. Reduction of (FeCl(PNP)) with KC
8 yielded the iron(I)-N
2 complex [
27]. The Nishibayashi group developed another first-row pincer complex (Co-PNP type) with a central pyrrolide moiety, where the Co-dinitrogen complexes were synthesized via reduction of paramagnetic precursors (CoCl(
tBuPNP)) and (CoCl(
cyPNP)) with KC
8 under dinitrogen and were examined in detail with regard to the conversion to ammonia under ambient reaction conditions. Recently, the Nishibayashi group also developed a low-cost and environment-friendly Mo-based PNP pincer ligand to synthesize ammonia (up to 4350 equivalents, turnover frequency of around 117/min) under ambient conditions at a rate double that previously reported [
28,
29].
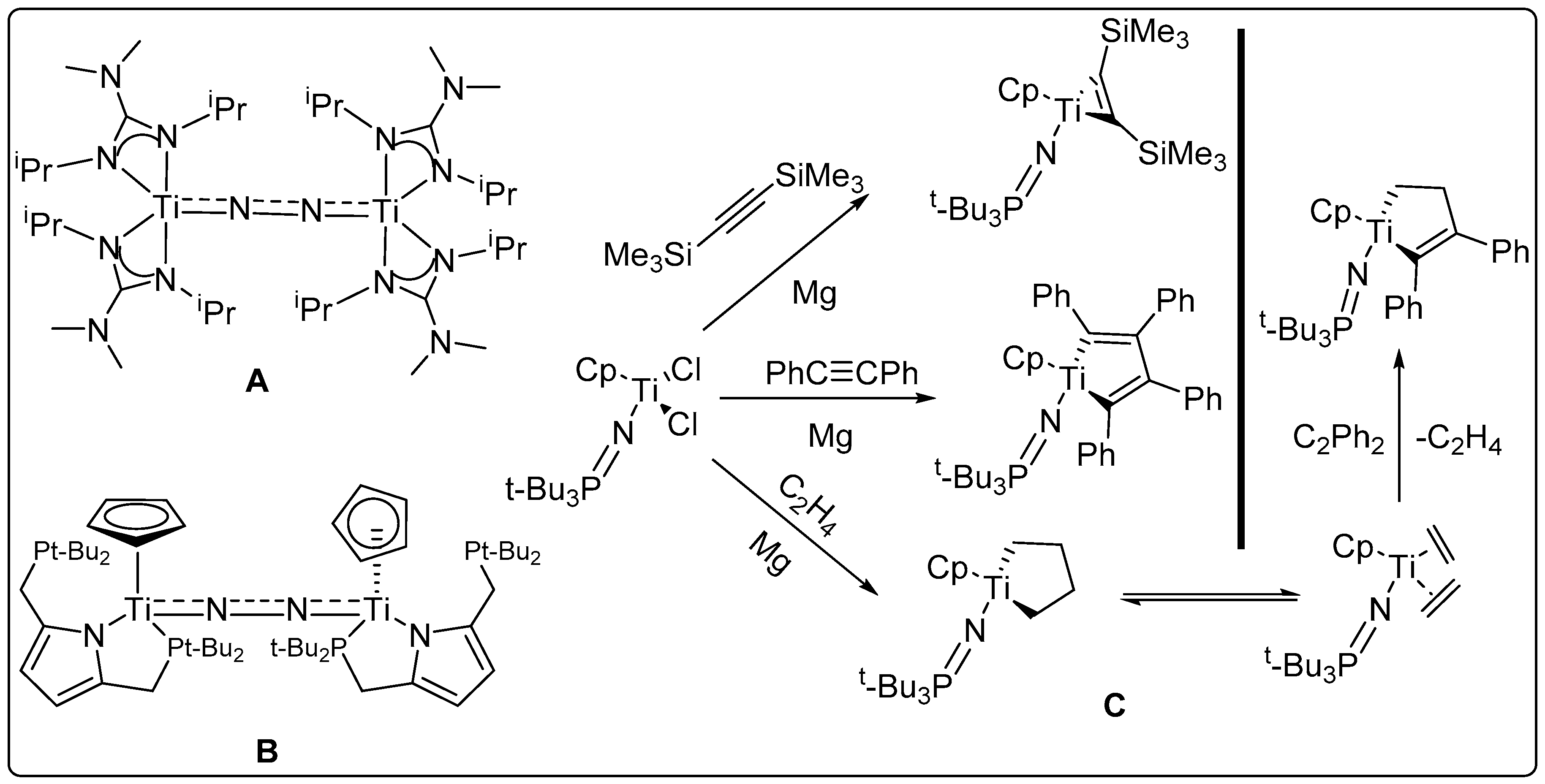
Titanium is an earth-abundant first row metal [
30]. “Ti(II)”-dinitrogen complexes are well known (
Figure 2) [
28,
29,
30,
31,
32]. Many other valuable reaction pathways for low-valent Ti(II), such as reductive coupling of alkynes and catalytic Pauson–Khand reactions are known (
Figure 2) [
33].
Pincer ligands containing
N-heterocyclic carbenes (NHC) have emerged as an important contribution for homogeneous catalysis [
34,
35,
36,
37]. Strong electron donating ability makes them an alternative to the tertiary phosphine containing ligands well known to bind dinitrogen [
38,
39,
40,
41,
42]. Recent reports of CCC-NHC pincer complexes of first row transition metal complexes have been reported to bind dinitrogen effectively [
43]. Evidence of such reactivity in an electron rich environment suggested the opportunity to pursue strong electron donating ligands first reported by Hollis et al. [
44,
45]. In light of the background, previously reported, robust and easy to synthesize first row transition metal complexes of the CCC-NHC pincer ligand were chosen as a stable starting material to access low valent (CCC-NHC)Ti pincer complexes to study their ability to bind dinitrogen [
46]. In this contribution, initial investigations aimed at reducing (CCC-NHC)Ti(IV) pincer complexes directed toward binding and reducing N
2 and studies on redox behavior of similar compounds are reported for the first time. Reductions were conducted with Mg or KC
8 and the reactivity of the reduced species were investigated. The reactivity of the reduced species with alkynes, phosphines, CO and PhICl
2 are reported.
2. Results and Discussion
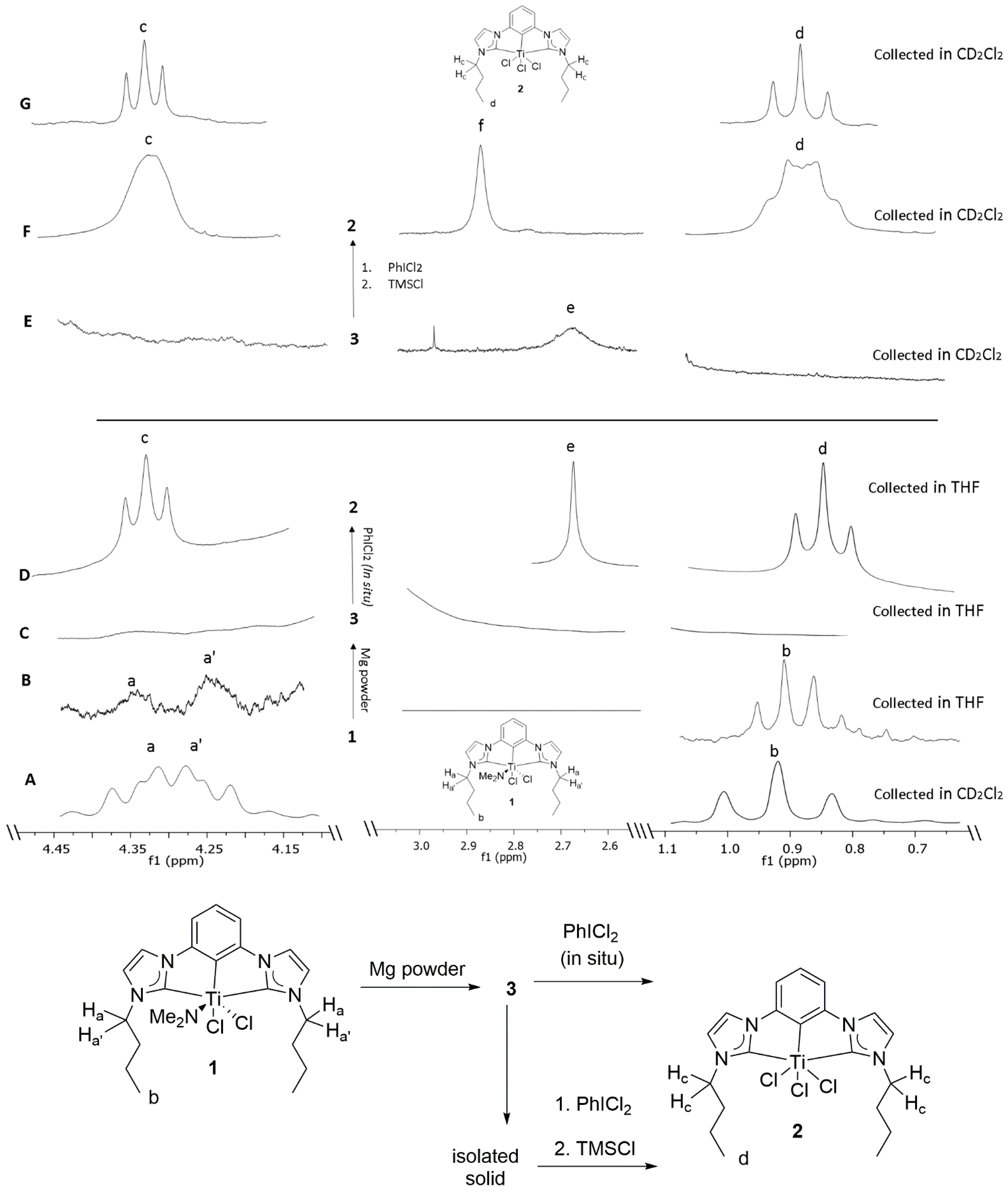
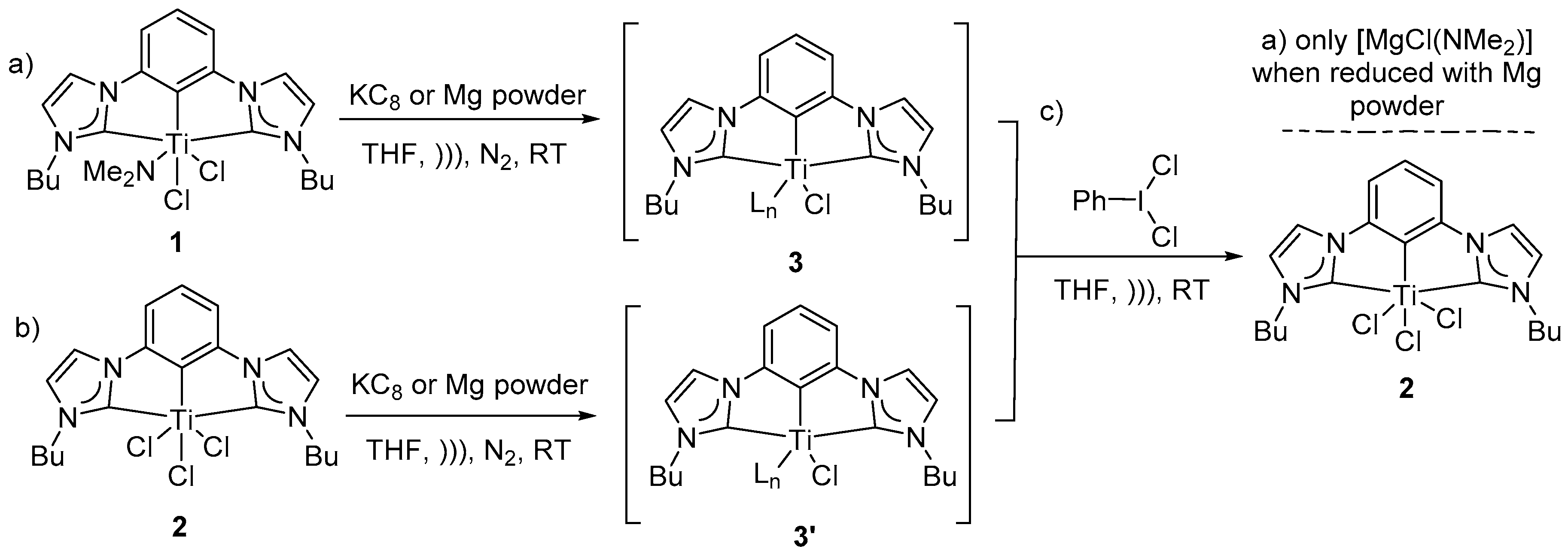
To study dinitrogen binding to low valent (CCC-NHC)Ti pincer complexes, well known, previously reported (CCC-NHC)dichloro mono-amido Ti pincer complex
1 was chosen as a starting material for initial studies (
Scheme 1a,
Figure 3A–D). All reactions were monitored by no-D NMR techniques in THF with a few drops of C
6D
6 [
47]. Illustrated in
Figure 3A (CD
2Cl
2) and 3B (no-D THF) are the characteristic signals for identifying amido complex
1 by its diastereotopic
N-CH
2-Pr (H
a and H
a’) signals at δ~4.3 and a methyl triplet (H
b) at δ~0.9. Very slight shifts due to solvent effects were observed. Complex
1 had limited solubility in THF. Complex
1 in THF was sonicated with excess Mg-powder in THF under nitrogen atmosphere at room-temperature to attempt dinitrogen binding via low valent (CCC-NHC)Ti in situ (
Scheme 1a,
Figure 3C). Upon sonication of the reaction mixture, a thick light-yellow precipitate under a faint yellow supernatant was observed (
Scheme 1). Upon addition of the reductant, the diamagnetic NMR spectrum (
Figure 3B) became paramagnetically broadened and the spectrum was featureless (
Scheme 1a,
Figure 3C). The broadened spectrum for the Mg reduction reaction is illustrated in
Figure 3C. No peaks are discernable.
This observation was consistent with reduction of complex
1 to intermediate species
3 (
Scheme 1a,
Figure 3C,
Figures S2–S4). This faint yellow supernatant was then subjected to a multitude of crystallization conditions to identify the structure of the intermediate
3 or the target nitrogen adduct via the determination of the crystal structure. Despite our best efforts, intermediate
3 remained intractable spectroscopically and no crystalline material has been isolated thus far. With no crystalline material in hand, reoxidation reaction on reduced intermediate was sought to provide insight into the structure of the intermediate. Iodobenzene dichloride is well known to oxidatively halogenate group 4 organometallic compounds [
49,
50]. Freshly prepared iodobenzene dichloride was combined with the mixture containing reduced intermediate
3 in THF [
51] (
Scheme 1c,
Figure 3D). Oxidative halogenation of intermediate
3 dissolved the resulting faint yellow precipitate and resulted in a homogeneous yellow colored solution, which upon analysis with no-D
1H NMR spectrum was surprisingly consistent with previously reported trichloride
2 (triplets H
c and H
d at δ 4.33 and at δ 0.84) and not the amido complex
1 (diastereotopic protons H
a, H
a’ δ~4.3 and triplet H
b δ~0.9) (
Scheme 1c,
Figure 3A,D,G,
Figures S2–S4) [
46]. Additionally, a singlet was observed at δ 2.64 (THF) as illustrated in
Figure 3D, e, which was consistent with the signal for Mg-dimethyl amido species as previously reported in literature (C
6D
6) [
48]. In a separate experiment, the faint yellow supernatant was decanted to separate the light-yellow precipitate and understand its nature. The precipitate was then dried under reduced pressure and dissolved in CD
2Cl
2 for spectroscopic data collection purposes (
Figure 3E and
Figure S1). The resulting solution was subjected to a multitude of crystallization conditions to crystallize and therefore characterize the intermediate
3 or a target nitrogen adduct. Despite our best efforts, the intermediate
3 remained intractable spectroscopically and no crystalline material has been isolated thus far. The broad singlet at δ 2.64 was consistent with the signal for Mg-dimethyl amido species as previously reported in literature (δ 2.64, C
6D
6) (
Figure 3E) [
48]. The broadening of the
1H NMR peak of the Mg-dimethyl amido species was consistent with its interaction with the metal core of the reduced intermediate. With no crystal of the desired nitrogen adduct or an intermediate, reoxidation reaction on reduced intermediate was sought to provide insight into the structure of the intermediate. A faint yellow homogeneous solution of the resulting precipitate in dichloromethane was allowed to react with iodobenzene dichloride. The resulting yellow solution was observed to contain a mixture of multiple compounds via
1H NMR spectroscopy. This solution was then allowed to react with TMSCl to convert any (CCC-NHC)dimethylamido Ti species into a (CCC-NHC) TiCl
3 as previously reported [
46]. The resulting yellow solution was evaporated to dryness yielding a yellow solid. It was then precipitated with hexane from a homogeneous solution of dichloromethane. The supernatant was decanted, and the residual yellow precipitate was then dried under reduced pressure. The
1H NMR analysis of the homogeneous, yellow solution of the resulting precipitate in dichloromethane was also contrary to expectation, consistent with previously reported trichloride
2 and not the amido complex
1 (
Figure 3A,F,G and
Figure S1) [
46]. Additionally, a broad singlet was observed at δ 2.89 (THF) as illustrated in
Figure 3F, f which was attributed to the signal for (Me
2N(SiMe
3)
2)
+ as cited in literature in a similar chemical environment [
52]. The broader than usual peaks in the resulting
1H NMR spectra (
Figure 3F) were attributed to the dynamic exchange with excess chloride anions present in the solution (
Figure 3F and
Figure S1). The aromatic peaks around δ 9.5 in the resulting
1H NMR spectra were attributed to partially decomposed (CCC-NHC)Ti pincer complex present in the solution as previously reported in literature [
44] (
Figure S1). The minimal shift in the position of
1H NMR peaks observed in the resulting
1H NMR spectra was attributed to the difference in the chemical environment in the resulting solution (
Figure S1). The fact that the singlet from the resulting Mg-NMe
2 species’ NMR was also broadened in reducing conditions (
Figure 3C) is consistent with an interaction with the reduced Ti-species, which likely inhibits the coordination of weak ligands such as N
2. However, results clearly demonstrated that the CCC-NHC ligand and Ti are still bonded in the low valent state.
Since the Mg-amido species (vide supra) were likely impeding the formation of N
2-adducts, KC
8 was chosen as an alternative reducing agent. A light red colored homogeneous solution of CCC-NHC)Ti(IV)Cl
2NMe
2 1 was sonicated with two equivalents of freshly prepared KC
8 in THF at room-temperature to attempt nitrogen binding in situ in an NMR tube (
Scheme 1). Paramagnetic broadening of all (overlapping diastereotopic peaks (H
a, H
a’) δ~4.3 and triplet peaks (H
b) δ~0.9) major peaks in a no-D
1H NMR experiment of the homogeneous reaction mixture over the residual graphite was consistent with reduction to species
3 (
Figures S5 and S6). The resulting homogeneous solution was deep yellow-green in color after the removal of heterogeneous reducing agent via filtration through a celite plug. The resulting solution was subjected to a multitude of crystallization conditions to crystallize and therefore characterize the intermediate or a target nitrogen adduct. Despite our best efforts, the intermediate remained intractable spectroscopically and no crystalline material has been isolated thus far. With no crystal available; reoxidation was pursued to provide insight into the structure of the intermediate. Freshly prepared iodobenzene dichloride was combined with the homogeneous deep yellow-green colored reaction mixture containing reduced intermediate
3 at room-temperature in THF [
51] (
Scheme 1). Oxidative halogenation on
3 also resulted in a homogeneous yellow colored solution which upon analysis with
1H NMR was consistent with previously reported CCC-NHC)Ti(IV)Cl
3 2 (triplets H
c and H
d at δ 4.33 and at δ 0.84) and not the amido complex
1 (overlapping diastereotopic peaks (H
a, H
a’) δ~4.3 and triplet peaks (H
b) δ~0.9) (
Scheme 1,
SI,
Figure 3A,G) [
46]. Reoxidation of intractable intermediate
3 to complex
2 repeatedly, led to the decision to choose trichloride
2 for further evaluation (
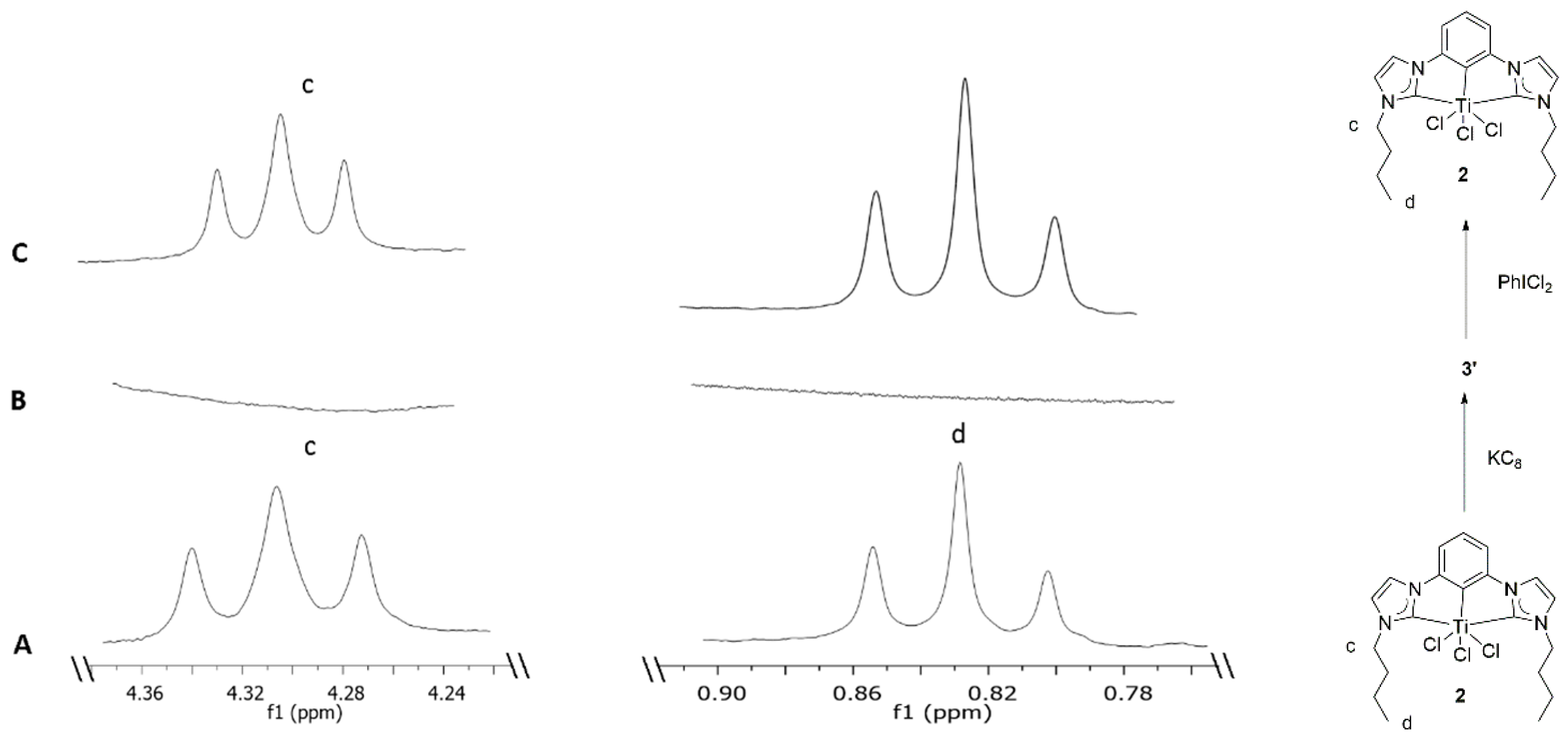
Scheme 1b,
Figure 4A–C). Illustrated in
Figure 4A (no-D THF) are the characteristic signals for identifying trichloro complex
2 by its triplets H
c and H
d at δ 4.33 and at δ 0.84. To attempt nitrogen fixation by low oxidation states of Ti exclusively, stoichiometric reduction reactions of complex
2 were executed with one- and two-fold molar excesses of freshly prepared KC
8 as follows [
53]. A homogeneous solution of compound
2 in an NMR tube, was sonicated with equimolar ratio of KC
8 in THF at ambient temperature under inert conditions (
Figure 4B). The resulting homogeneous solution looked orange-yellow upon removal of the heterogeneous reducing agent via filtration through a celite plug. Paramagnetic broadening of all (triplets H
c and H
d at δ 4.33 and at δ 0.84) major peaks in a no-D
1H NMR analysis of the solution was consistent with reduction in both the cases (
Figure 4B,
Figures S7 and S9).
The homogeneous solution of intermediate
3′ was subjected to a multitude of crystallization conditions to isolate and crystallize the intermediate. Despite our best efforts, no crystalline material has been isolated so far. Unable to isolate the target dinitrogen adduct or the intermediate, freshly prepared iodobenzene dichloride was allowed to react with a homogeneous solution containing reduced intermediate
3′ in situ in THF, at room-temperature under inert conditions. Upon sonication in an NMR tube, the reaction mixture restored its initial yellow color (
Scheme 1c,
Figure 4C). The re-appearance of the characteristic triplets (H
c and H
d) at δ 4.33 and at δ 0.84 in the no-D
1H NMR experiment in THF was consistent with formation of compound
2 via oxidative halogenation (
Scheme 1c,
Figure 4C,
Figures S7 and S8). A homogeneous solution of compound
2 in an NMR tube was also sonicated with two equivalents of freshly prepared KC
8 in THF in order to attempt nitrogen fixation at ambient temperature under inert conditions. The resulting solution was light yellow in color after the removal of resulting heterogeneous reducing agent via filtration through a celite plug. Upon addition of the reducing agent, diamagnetic NMR spectrum becomes paramagnetically broadened and the resulting spectrum becomes featureless. The resulting NMR spectrum was consistent with the formation of reduced species
3′. The homogeneous solution of intermediate
3′ was subjected to a multitude of crystallization conditions to isolate and crystallize the intermediate, despite our best efforts, no crystalline material has been isolated so far. Unable to isolate the target dinitrogen adduct or the intermediate, freshly prepared iodobenzene dichloride was allowed to react with a homogeneous solution containing reduced intermediate
3′ in situ in THF, at room-temperature under inert conditions. Upon sonication in an NMR tube, the reaction mixture restored its initial yellow color (
Scheme 1c,
Figure 4C). The re-appearance of the characteristic triplets (H
c and H
d) at δ 4.33 and at δ 0.84 in the no-D
1H NMR experiment in THF was consistent with formation of compound
2 via oxidative halogenation (
Scheme 1c,
Figure 4C and
SI). Since quantitative reoxidation reactions on the reduced species with one and two equivalents of reducing agent resulted in the same final compound, compound
2 was sonicated with excess of KC
8 to ensure quantitative reduction. The resulting homogeneous solution looked wine-red upon removal of the resulting heterogeneous reducing agent via filtration through a celite plug. Paramagnetic broadening of all major peaks (triplets H
c and H
d at δ 4.33 and at δ 0.84) in the no-D
1H NMR experiment was consistent with reduction. The resulting homogeneous solution containing intermediate
3′ in THF was also subjected to a multitude of crystallization conditions to isolate and crystallize the intermediate, despite our best efforts, no crystalline material has been isolated thus far. Unable to isolate the target dinitrogen adduct or the intermediate, freshly prepared iodobenzene dichloride was allowed to react with a homogeneous solution of reduced intermediate
3′ at room-temperature in THF in situ under inert conditions. The resulting homogeneous reaction mixture restored its initial yellow color. The re-appearance of the characteristic peaks (triplets H
c and H
d at δ 4.33 and at δ 0.84) in the no-D
1H NMR experiment in THF was consistent with formation of compound
2 via oxidative halogenation (
Scheme 1,
Figure 4). A homogeneous solution of compound
2 was subsequently sonicated with excess Mg-powder at ambient temperature and under inert reaction conditions. A fluffy yellow precipitate was observed (
Scheme 1b,
Figure S8). Paramagnetic broadening of all peaks (triplets H
c and H
d at δ 4.33 and at δ 0.84 of
2) in the no-D
1H NMR spectrum of the supernatant was consistent with reduction to species
3′(
Figure S8). A homogeneous solution of the resulting precipitate was collected and subjected to a multitude of crystallization conditions to crystallize and therefore characterize the intermediate or a target nitrogen adduct. Despite our best efforts, the intermediate remained intractable spectroscopically and no crystalline material has been isolated so far. With no crystal of desired nitrogen adduct or an intermediate available, in situ reoxidation reaction on intermediate was pursued to provide insight into the structure of the intermediate (
Scheme 1c). Freshly prepared iodobenzene dichloride was allowed to react with a homogeneous solution of reduced intermediate
3′ in THF at room-temperature under inert atmosphere. Upon sonication, the resulting homogeneous reaction mixture restored its initial yellow color. The re-appearance of the characteristic triplets (H
c and H
d) at δ 4.33 and at δ 0.84 in the no-D
1H NMR experiment in THF was consistent with formation of compound
2 (
Scheme 1) via oxidative halogenation (
Figure S8).
Hence, it was interesting to observe that both intermediates
3 and
3′ gave rise to the same symmetric characteristic signals of compound
2 after the redox cycling was complete (
Scheme 1,
Figure 3,
Figure 4 and
supplementary). This phenomenon was consistent with a robust, redox stable metal binding to the CCC-NHC core and a common reduced intermediate. Since the reactions with stoichiometric as well as excess reducing agents resulted in complex
2 upon reoxidation, only excess reductants were used from here onwards to ensure complete reduction. Evidence of (MgClNMe
2) and (Me
2N(SiMe
3)
2)
+ complexes after reoxidation of reduced complex
1 with Mg was consistent with the facile elimination of electron donating dimethylamine leading to a stable reduced intermediate (
Figure 3 and
SI). Resurgence of complex
2 after the oxidation of both reduced intermediates
3 and
3′ hints at their similarity in structure and robustness of the metal-backbone binding (
Scheme 1,
Figure 3,
Figure 4 and
SI). Reduction followed by re-oxidation with iodobenzene dichloride followed similar trends in KC
8 and Mg powder (
Scheme 1,
Figure 3,
Figure 4 and
SI). Unable to isolate any dinitrogen bound coordination compound or the reduced intermediates, recourse to reactivity of the intermediates were sought.
Attempted reactions to trap reduced intermediate 3 or 3′: Low-valent Ti intermediates have well-known reactivity patterns as illustrated before. Any resulting low-valent Ti adducts could be utilized to determine the nature of the reactive intermediate.
A homogeneous solution of reduced species
3 or
3′ in THF were subjected to various phosphines (PPh
3, P(
o-tol)
3) and alkyne (diphenyl acetylene) at temperatures ambient to 100 °C and under inert atmosphere, to synthesize any (CCC-NHC)Ti-phosphine adduct as previously cited (
Scheme 2) [
54]. Highly reactive PEt
3 was reacted with quantitatively reduced (CCC-NHC)Ti complex
2 with 1, 2 and 4 equivalents of freshly prepared KC
8 to accommodate any difference in reactivity between quantitative reductants used (
Scheme 2). For all the reaction conditions illustrated below, the color of the solution remained unchanged after the addition of the phosphines. The
31P NMR spectra showed no complex bound phosphines. The observations listed above were consistent with no reaction between the reduced intermediates
3 or
3′ and phosphine. Therefore, the results were suggestive of an alternative reactivity pattern in the present (CCC-NHC)Ti complex system compared to the well-established Ti-chemistry reported in literature [
54].
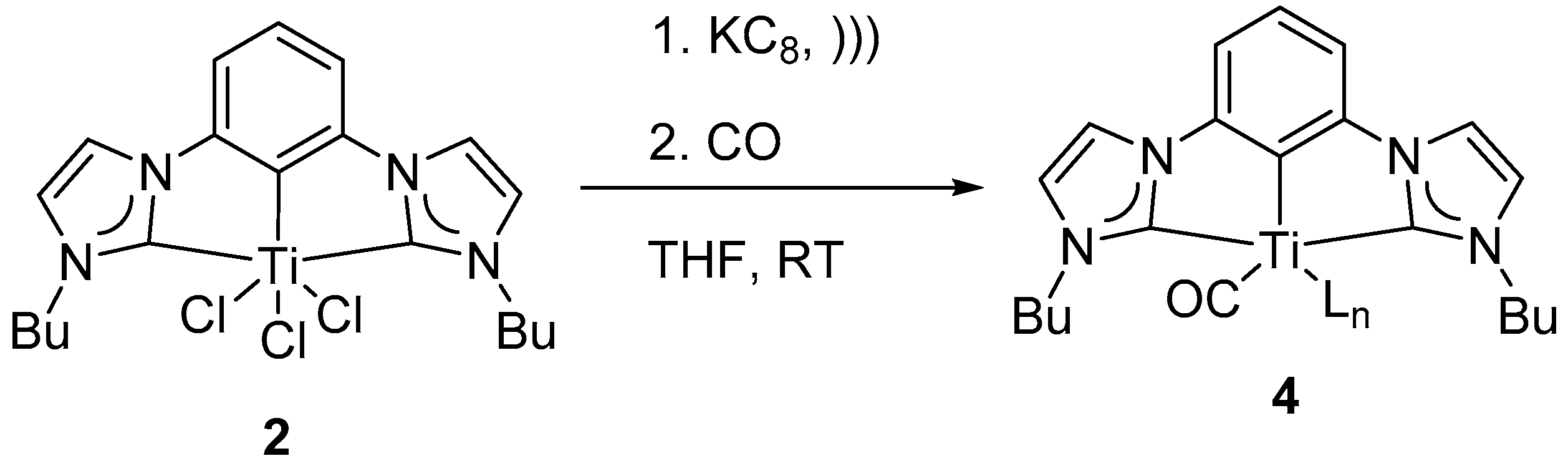
Carbon monoxide is isoelectronic to dinitrogen molecule. Stable metal carbonyl complexes are abundant in literature [
55]. Identification of any discreet CO-bound (CCC-NHC) could be utilized to determine the nature of the reactive intermediate. Reduced homogeneous wine-red colored solution of
3′ in THF was bubbled with carbon monoxide (
Scheme 3). Upon reaction, the wine-red color of the solution changed to pale yellow. The resulting no-D
1H NMR spectrum in THF changed to a diamagnetic spectrum with a new multiplet at δ 0.77 instead of the characteristic triplet peaks (triplets H
c and H
d at δ 4.33 and at δ 0.84 of
2) of complex
2 at δ 4.33 and δ 0.84 (
Figure S13). The diamagnetic nature of the resulting spectrum along with the observed color change of the solution were consistent with the formation of a new species
4 (
Scheme 3 and
Figure S13). Further attempts at characterizing the newly formed pincer-CO adduct was intractable, seemingly due to loss of CO when CO-atmosphere was removed.
Since the original target was the nitrogen adducts of the low valent Ti-pincer complex, extensive effort was expended to obtain the crystalline material. No crystals with or without N2-binding were obtained. It was reasoned that the (CCC-NHC)Ti system has an interestingly alternative reactivity pattern compared to literature precedence and the observed Mg-amido complex may have been complicating the equilibria preventing the N2-adduct and crystal formation.
3. Materials and Methods
3.1. General
Standard inert-atmosphere techniques were used unless stated otherwise. Solvents were dried using a standalone solvent purification system. (1,3-bis(3-butylimidazol-1-yl-2-idene)-2-phenylene)(dimethylamido)dichlorotitanium
1, (1,3-bis(3-butylimidazol-1-yl-2-idene)-2-phenylene)trichlorotitanium
2 were synthesized according to the literature procedure [
46]. Benzene-d
6 was purchased and passed through a column of activated basic alumina. KC
8 was freshly prepared following the procedure reported in literature [
53]. Iodobenzene dichloride was freshly prepared using literature procedure [
51].
1H NMR spectra were collected on a 300 MHz, or a 500 MHz NMR spectrometer at ambient temperature. No-D
1H NMR experiments in THF were conducted with two drops of C
6D
6 [
47].
3.2. Reaction of Complex 1 with Mg Powder
((1,3-Bis(3-butylimidazol-1-yl-2-idene)-2-phenylene)(dimethylamido)dichlorotitanium(IV)) 1 (11 mg, 0.02 mmol) was dissolved in 2.5 mL of dry protic-THF (2 drops of C6D6 was added to lock) in an NMR tube. To this solution, Mg-powder (10 mg, 0.42 mmol) was added. The resulting heterogeneous mixture was sonicated for 3 h. The mixture was left in the glovebox for 48 h for the completion of the reaction; a thick light-yellow precipitate of 3 was observed. Spectroscopic data for the supernatant was collected.
1H NMR (300 MHz, THF no-D, locking solvent: C6D6): para-magnetic broadening caused relevant peaks to disappear.
3.3. Oxidation of Intermediate 3 with PhICl2 Followed by Addition of TMSCl
((1,3-Bis(3-butylimidazol-1-yl-2-idene)-2-phenylene)(dimethylamido)dichlorotitanium(IV)) 1 (13.7 mg, 0.03 mmol) was dissolved in 2.5 mL of dry protic-THF in an NMR tube. To this solution, Mg-metal powder (11 mg, 0.45 mmol) was added. The resulting heterogeneous mixture was sonicated for 3 h with occasional manual shaking. The mixture was left in the glovebox for 48 h for the completion of the reaction; a thick faint yellow precipitate of intermediate 3 was observed. The supernatant was decanted followed by collecting the resulting solid. The solid was dried under reduced pressure. The residue was dissolved in CH2Cl2 and freshly prepared PhICl2 (18.0 mg, 0.07 mmol) followed by TMSCl (25.0 mg, 0.23 mmol) were added to the mixture and the reaction mixture was left in the inert atmosphere overnight. The reaction mixture was evaporated to dryness. The residue was dissolved in CH2Cl2. To the solution dry hexane was added to precipitate the resulting complex. Supernatant was decanted and the resulting precipitate was dried under reduced pressure. Spectroscopic data was collected.
1H NMR (500 MHz, CD2Cl2): δ 7.99 (br, 4H), 7.77 (br, 1H), 7.52 (br, 2H), 4.38 (br, 4H), 2.88 (s, 1H), 2.02 (br, 4H), 1.49 (br, 4H), 1.03 (br, 6H).
3.4. Reduction with Mg Followed by Reoxidation of 1 with PhICl2
((1,3-Bis(3-butylimidazol-1-yl-2-idene)-2-phenylene)(dimethylamido)dichlorotitanium(IV)) 1 (11 mg, 0.02 mmol) was dissolved in 2.5 mL of dry protic-THF in an NMR tube. To this solution, Mg-metal powder (10 mg, 0.42 mmol) was added. The resulting heterogeneous mixture was sonicated for 3 h with occasional manual shaking. The mixture was left in the glovebox for 48 h for the completion of the reaction; a thick yellow precipitate was observed. Iodobenzene dichloride (18 mg, 0.07 mmol) was added in situ. Upon mixing, the yellow precipitate dissolved; forming a clear yellow solution. 1H NMR (300 MHz, THF no-D, locking solvent: C6D6): δ 4.33 (t), 0.84 (t).
3.5. Reaction of Complex 1 with KC8
((1,3-Bis(3-butylimidazol-1-yl-2-idene)-2-phenylene)(dimethylamido)dichlorotitanium(IV)) 1 (5 mg, 0.01 mmol) was dissolved in 2.5 mL of dry protic-THF in an NMR tube. To this solution KC8 (4.5 mg, 0.03 mmol) was added. The resulting heterogeneous mixture was sonicated for 3 min with occasional manual shaking. The reaction mixture turned deep yellow-green. The resulting solution was passed through a celite plug to remove the heterogeneous reducing agent. The solvent was removed from the solid under reduced pressure. 1H NMR (300 MHz, THF no-D, locking solvent: C6D6): para-magnetic broadening caused relevant peaks to disappear.
3.6. Reduction with KC8 Followed by Reoxidation of 1 with PhICl2
((1,3-Bis(3-butylimidazol-1-yl-2-idene)-2-phenylene)(dimethylamido)dichlorotitanium(IV)) 1 (5 mg, 0.01 mmol) was dissolved in 2.5 mL of dry protic-THF in an NMR tube. To this solution, KC8 (4.5 mg, 0.03 mmol) was added. The resulting heterogeneous mixture was sonicated for 3 min with occasional manual shaking. Iodobenzene dichloride (10 mg, 0.04 mmol) was added in situ. Upon mixing, the yellow precipitate dissolved forming a clear yellow solution. 1H NMR (300 MHz, THF no-D, locking solvent: C6D6): δ 4.33 (t), 0.84 (t).
3.7. Reduction of Complex 2 with KC8
((1,3-Bis(3-butylimidazol-1-yl-2-idene)-2-phenylene)trichlorotitanium(IV)) 2 (5.1 mg, 0.01 mmol) was dissolved in 2.42 mL of dry protic-THF (2 drops of C6D6 was added to lock) in an NMR tube. To this solution, KC8 (5.2 mg, 0.04 mmol) was added. The resulting heterogeneous mixture was sonicated for 0.11 h with occasional manual shaking. A yellow-green solution was observed. The resulting solution was passed through a celite plug to remove the heterogeneous reducing agent. The solvent was removed from the solid under reduced pressure. 1H NMR (300 MHz, THF no-D, locking solvent: C6D6): para-magnetic broadening caused relevant peaks to disappear.
3.8. Reduction of Complex 2 with KC8 Followed by Reoxidation with PhICl2
((1,3-Bis(3-butylimidazol-1-yl-2-idene)-2-phenylene)trichlorotitanium(IV)) 2 (5.1 mg, 0.01 mmol) was dissolved in 2.42 mL of 30% C6D6 in dry protic-THF in an NMR tube. To this solution, KC8 (5.2 mg, 0.04 mmol) was added. The resulting heterogeneous mixture was sonicated for 0.11 h with occasional manual shaking. A yellow-green solution was observed. To the decanted supernatant, iodobenzene dichloride (16 mg, 0.06 mmol) was added in situ. Upon mixing, a clear yellow solution was observed. 1H NMR (300 MHz, THF no-D, locking solvent: C6D6): δ 4.33 (t), 0.84 (t).
3.9. Reduction of Complex 2 with Mg
((1,3-Bis(3-butylimidazol-1-yl-2-idene)-2-phenylene)trichlorotitanium(IV)) 2 (10 mg, 0.02 mmol) was dissolved in 2.5 mL of dry protic-THF (2 drops of C6D6 was added to lock) in an NMR tube. To this solution, Mg-powder (12 mg, 0.49 mmol) was added. The resulting heterogeneous mixture was sonicated for 2h with occasional manual shaking. A yellow precipitate was observed. Spectroscopic data was collected for the supernatant. The supernatant was then decanted and the resulting solid was dried under reduced pressure. 1H NMR (300 MHz, THF no-D, locking solvent: C6D6): para-magnetic broadening caused relevant peaks to disappear.
3.10. Reduction of Complex 2 with Mg Followed by Reoxidation with PhICl2
((1,3-Bis(3-butylimidazol-1-yl-2-idene)-2-phenylene)trichlorotitanium(IV)) 2 (10 mg, 0.02 mmol) was dissolved in 2.5 mL of dry protic-THF in an NMR tube. To this solution, Mg-metal powder (12 mg, 0.49 mmol) was added. The resulting heterogeneous mixture was sonicated for 2 h with occasional manual shaking. A yellow precipitate was observed. Iodobenzene dichloride (17 mg, 0.06 mmol) was added in situ. Upon mixing, the yellow precipitate dissolved; forming a clear yellow solution. 1H NMR (300 MHz, THF no-D, locking solvent: C6D6): δ 4.33 (t), 0.84 (t).
3.11. Reduction of Complex 2 with KC8 Followed by Reaction with PPh3
((1,3-Bis(3-butylimidazol-1-yl-2-idene)-2-phenylene)trichlorotitanium(IV)) 2 (25 mg, 0.05 mmol) was dissolved in 2.5 mL of dry protic-THF in an NMR tube. To this solution, excess KC8 (28.3 mg, 0.21 mmol) was added. The resulting heterogeneous mixture was sonicated for 0.08 h with occasional manual shaking. Due to para-magnetic broadening; all the characteristic 1H NMR peaks disappeared; broad solvent peaks persisted. The wine-red colored heterogeneous solution was filtered through celite plug to get rid of the resulting graphite. To this clear, wine-red colored solution, PPh3 (22.86 mg, 0.09 mmol) was added. The mixture was sonicated for 2 h with occasional manual shaking. No change in color was observed. 1H NMR (300 MHz, THF no-D, locking solvent: C6D6): para-magnetic broadening caused relevant peaks to disappear. The characteristic region in 31P NMR remained unchanged.
3.12. Reduction of Complex 2 with KC8 Followed by Reaction with P(o-tol)3
((1,3-Bis(3-butylimidazol-1-yl-2-idene)-2-phenylene)trichlorotitanium(IV)) 2 (25 mg, 0.05 mmol) was dissolved in 2.4 mL of dry protic-THF (2 drops of C6D6 was added to lock) in an NMR tube. To this solution, excess KC8 (28.3 mg, 0.21 mmol) was added. The resulting heterogeneous mixture was sonicated for 0.08 h with occasional manual shaking. Due to para-magnetic broadening; all the characteristic 1H NMR peaks disappeared; broad solvent peaks persisted. The wine-red colored heterogeneous solution was filtered through a celite plug to get rid of the resulting graphite. To this clear, wine-red colored solution, P(o-tol)3 (26.51 mg, 0.09 mmol) was added. The mixture was sonicated for 2 h with occasional manual shaking. No change in color was observed. 1H NMR (300 MHz, THF no-D, locking solvent: C6D6): para-magnetic broadening caused relevant peaks to disappear. The characteristic region in 1H NMR and 31P NMR remained unchanged.
3.13. Reduction of Complex 2 with One Equivalent of KC8 Followed by Reaction with PEt3
((1,3-Bis(3-butylimidazol-1-yl-2-idene)-2-phenylene)trichlorotitanium(IV)) 2 (10 mg, 0.02 mmol) was dissolved in 2.5 mL of dry protic-THF (2 drops of C6D6 was added to lock) in an NMR tube. To this solution, KC8 (2.8 mg, 0.21 mmol) was added. The resulting heterogeneous mixture was sonicated for 2 h with occasional manual shaking. Due to para-magnetic broadening; all the characteristic 1H NMR peaks disappeared; broad solvent peaks persist. The orange-red colored heterogeneous solution was filtered through a celite plug to get rid of the resulting graphite. To this clear, orange-red colored solution, PEt3 (8.85 µL, 0.06 mmol) was added. The mixture was sonicated for 2 h with occasional manual shaking. No change in color was observed. 1H NMR (300 MHz, THF no-D, locking solvent: C6D6): para-magnetic broadening caused relevant peaks to disappear. The characteristic region in 1H NMR and 31P NMR remained unchanged.
3.14. Reduction of Complex 2 with Two Equivalents of KC8 Followed by Reaction with PEt3
((1,3-Bis(3-butylimidazol-1-yl-2-idene)-2-phenylene)trichlorotitanium(IV)) 2 (10 mg, 0.02 mmol) was dissolved in 2.5 mL of dry protic-THF (2 drops of C6D6 was added to lock) in an NMR tube. To this solution, KC8 (5.4 mg, 0.04 mmol) was added. The resulting heterogeneous mixture was sonicated for 2 h with occasional manual shaking. Due to para-magnetic broadening; all the characteristic 1H NMR peaks disappeared; broad solvent peaks persisted. The wine-red colored heterogeneous solution was filtered through a celite plug to get rid of the resulting graphite. To this clear, wine-red colored solution, PEt3 (8.85 µL, 0.06 mmol) was added. The mixture was sonicated for 2 h with occasional manual shaking. No change in color was observed. The characteristic region in 1H NMR and 31P NMR remained unchanged. 1H NMR (300 MHz, THF no-D, locking solvent: C6D6): para-magnetic broadening caused relevant peaks to disappear. The characteristic region in 1H NMR and 31P NMR remained unchanged.
3.15. Reduction of Complex 2 with KC8 Followed by Reaction with Diphenyl Acetylene
((1,3-Bis(3-butylimidazol-1-yl-2-idene)-2-phenylene)trichlorotitanium(IV)) 2 (10 mg, 0.02 mmol) was dissolved in 1.5 mL of dry protic-THF (2 drops of C6D6 was added to lock) in an NMR tube. To this solution, excess KC8 (14.1 mg, 0.01 mmol) was added. The resulting heterogeneous mixture was sonicated for 0.08 h with occasional manual shaking. Due to para-magnetic broadening; all the characteristic 1H NMR peaks disappeared; broad solvent peaks persisted. The wine-red colored heterogeneous solution was filtered to get rid of the resulting graphite. To this clear, wine-red colored solution, diphenyl acetylene (7.81 mg, 0.04 mmol) was added. The mixture was sonicated for 2 h with occasional manual shaking. The color of the resulting solution and the characteristic region in 31P NMR remained unchanged. 1H NMR (300 MHz, THF no-D, locking solvent: C6D6): para-magnetic broadening caused relevant peaks to disappear.
3.16. Reduction of Complex 2 with KC8 Followed by Reaction with CO
((1,3-Bis(3-butylimidazol-1-yl-2-idene)-2-phenylene)trichlorotitanium(IV)) 2 (10 mg, 0.02 mmol) was dissolved in 2.5 mL of dry protic-THF (2 drops of C6D6 was added to lock) in an NMR tube. To this solution, KC8 (5.4 mg, 0.04 mmol) was added. The resulting heterogeneous mixture was sonicated for 2 h with occasional manual shaking. Due to para-magnetic broadening; all the characteristic 1H NMR peaks disappeared; broad solvent peaks persisted. The wine-red colored heterogeneous solution was filtered through a celite plug to get rid of the resulting graphite. To this clear, wine-red colored solution, CO was bubbled for 0.03 h with occasional manual shaking. The resulting mixture was sonicated for 2 h with occasional manual shaking. The color of the resulting solution changed from wine-red to dirty-yellow. 1H NMR (300 MHz, THF no-D, locking solvent: C6D6): δ 0.77 (t).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
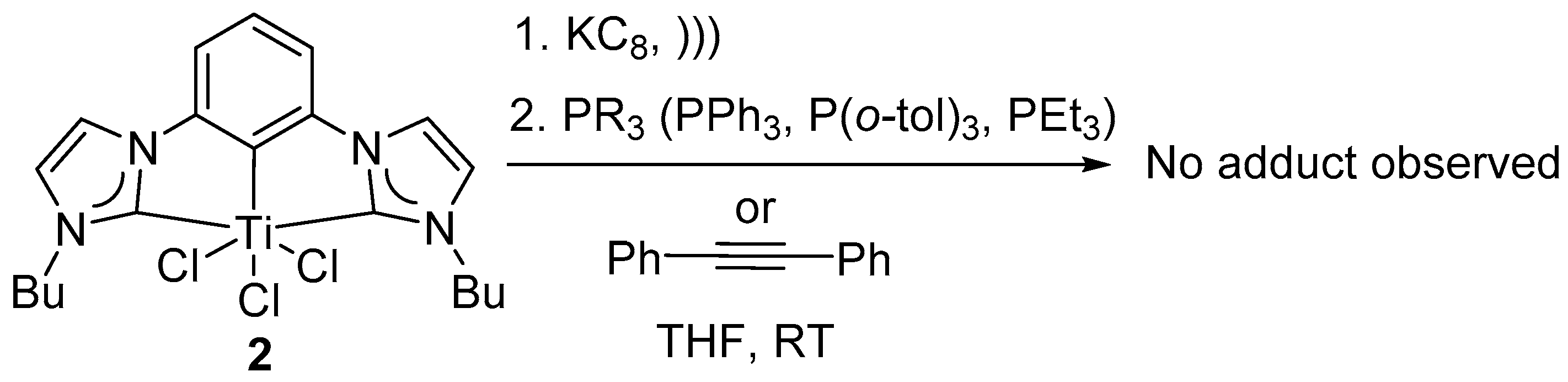{kind=link}
{kind=link}






