
Study of Anharmonicity in Zirconium Hydrides Using Inelastic Neutron Scattering and Ab-Initio Computer Modeling

 , ,
, ,  , , and
, , and
Abstract
:
1. Introduction
2. Methods
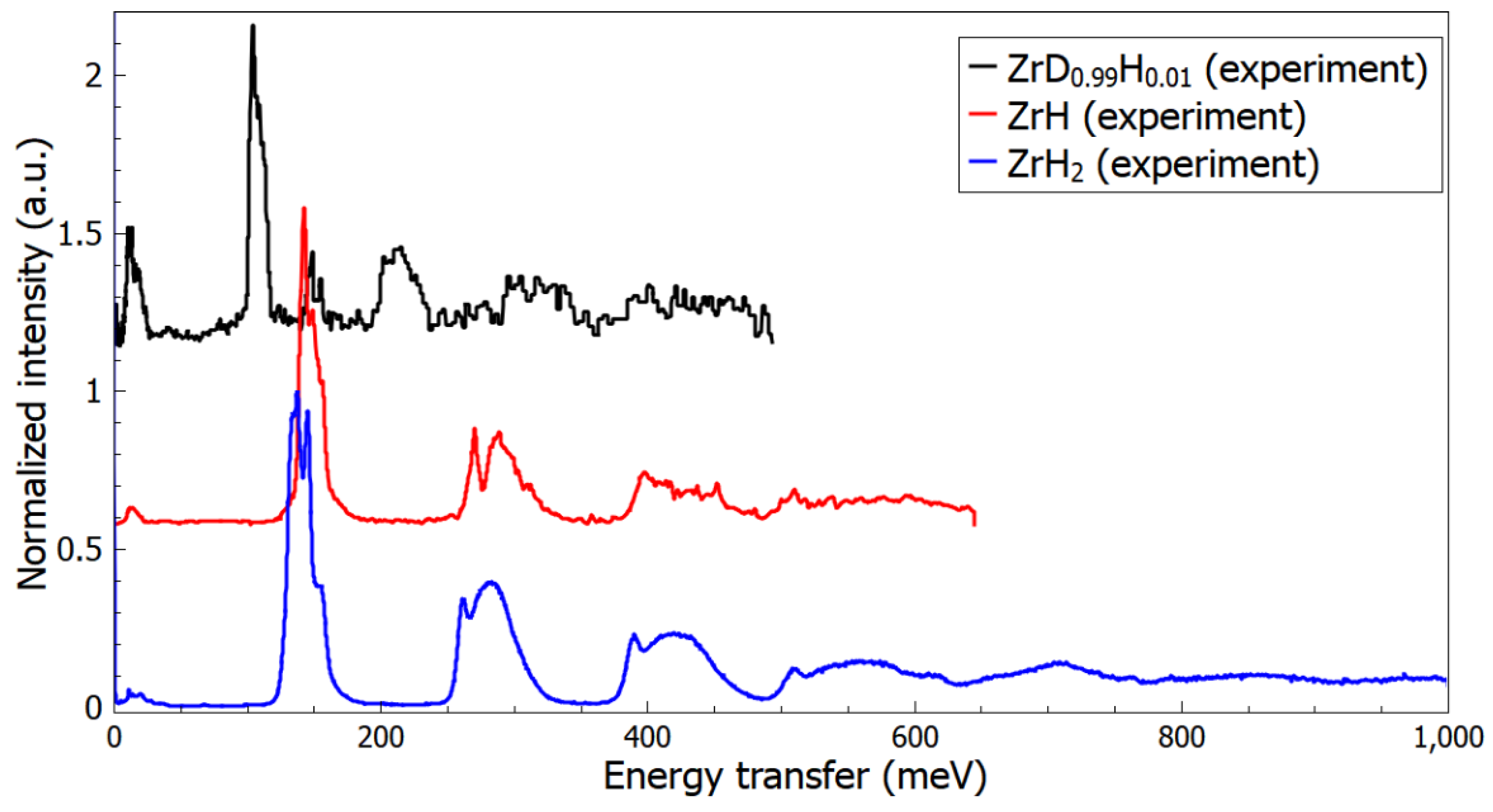
3. Results and Discussion
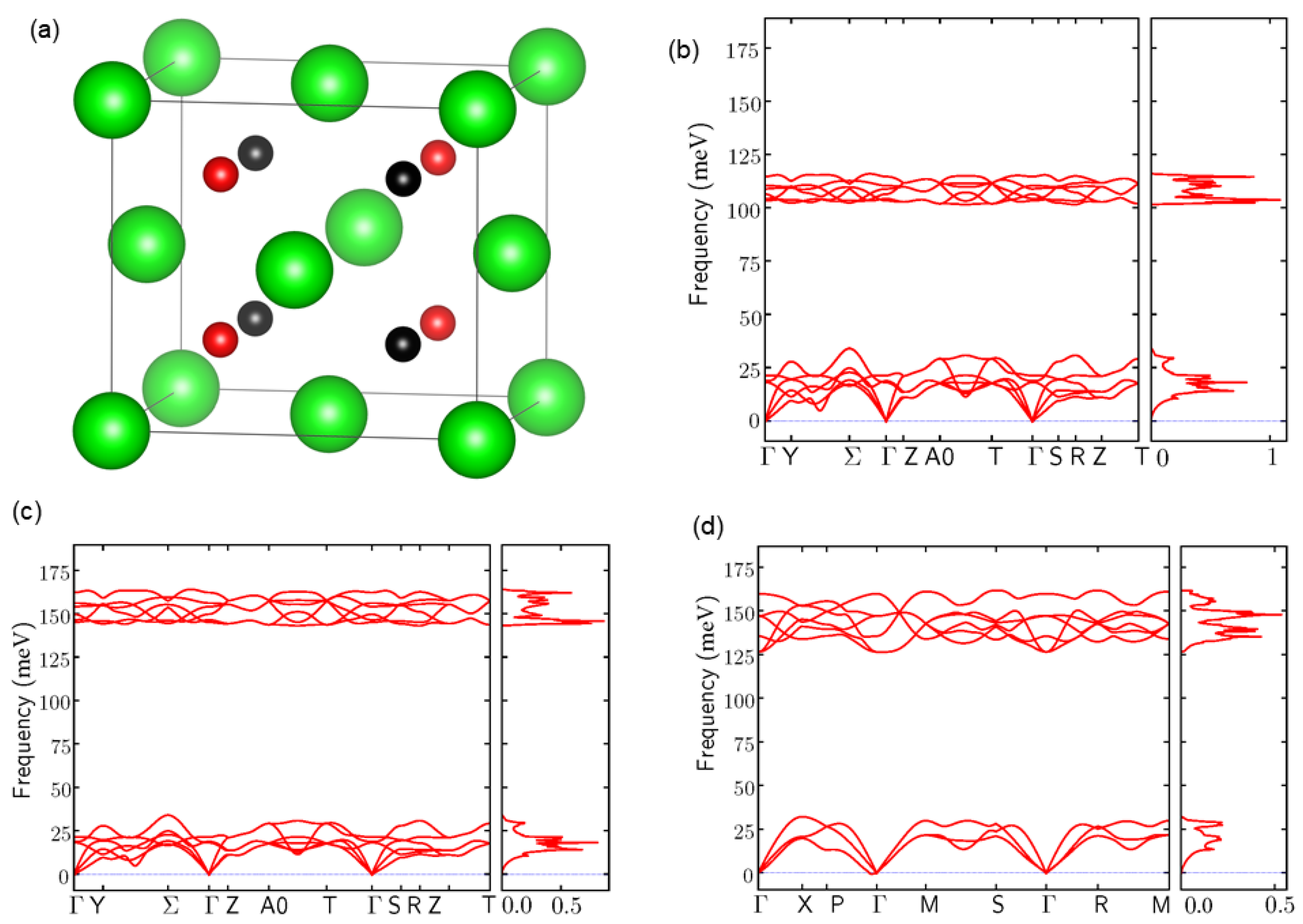
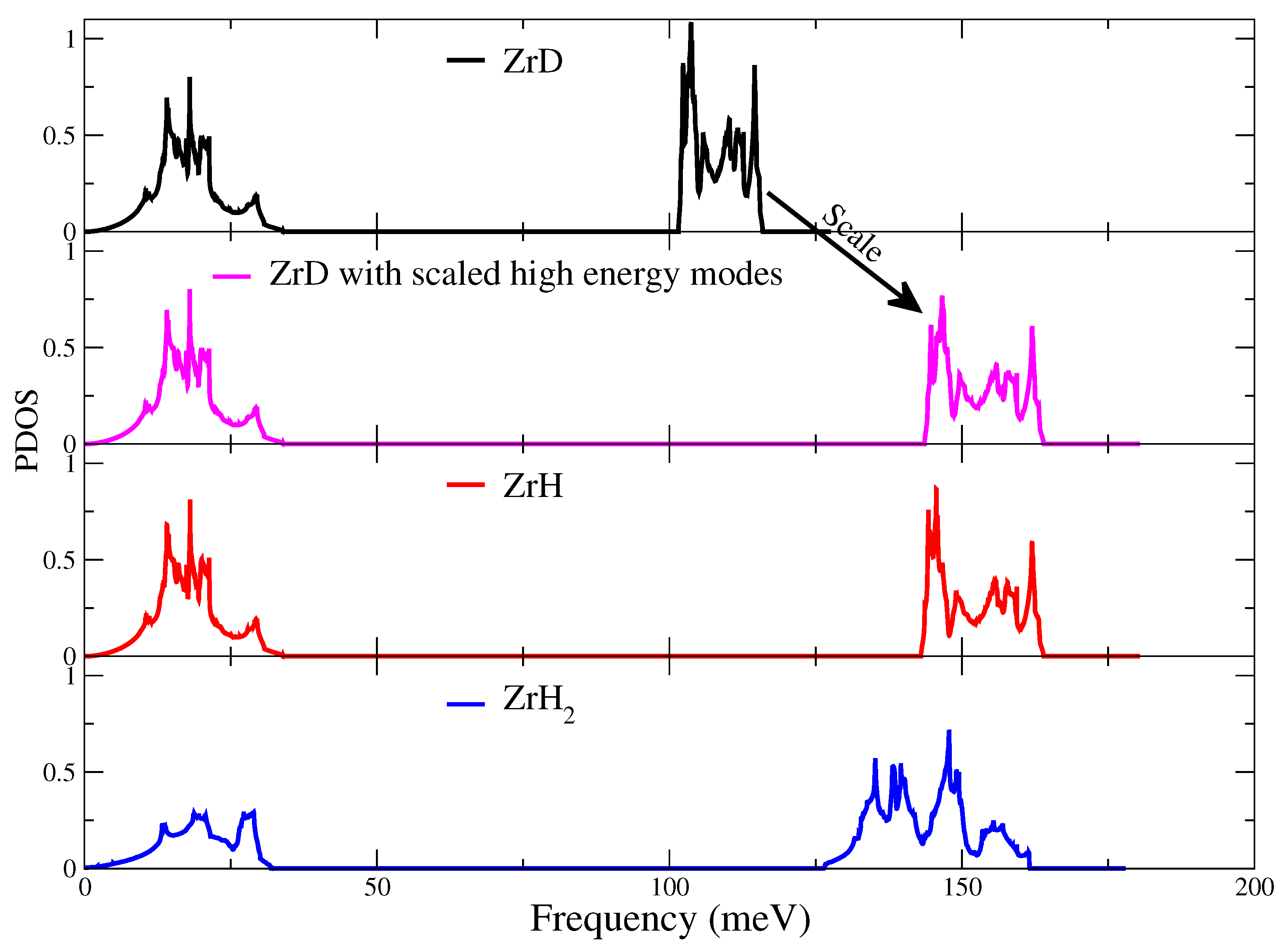
3.1. Atomic Structures and Phonon Properties of ZrHx
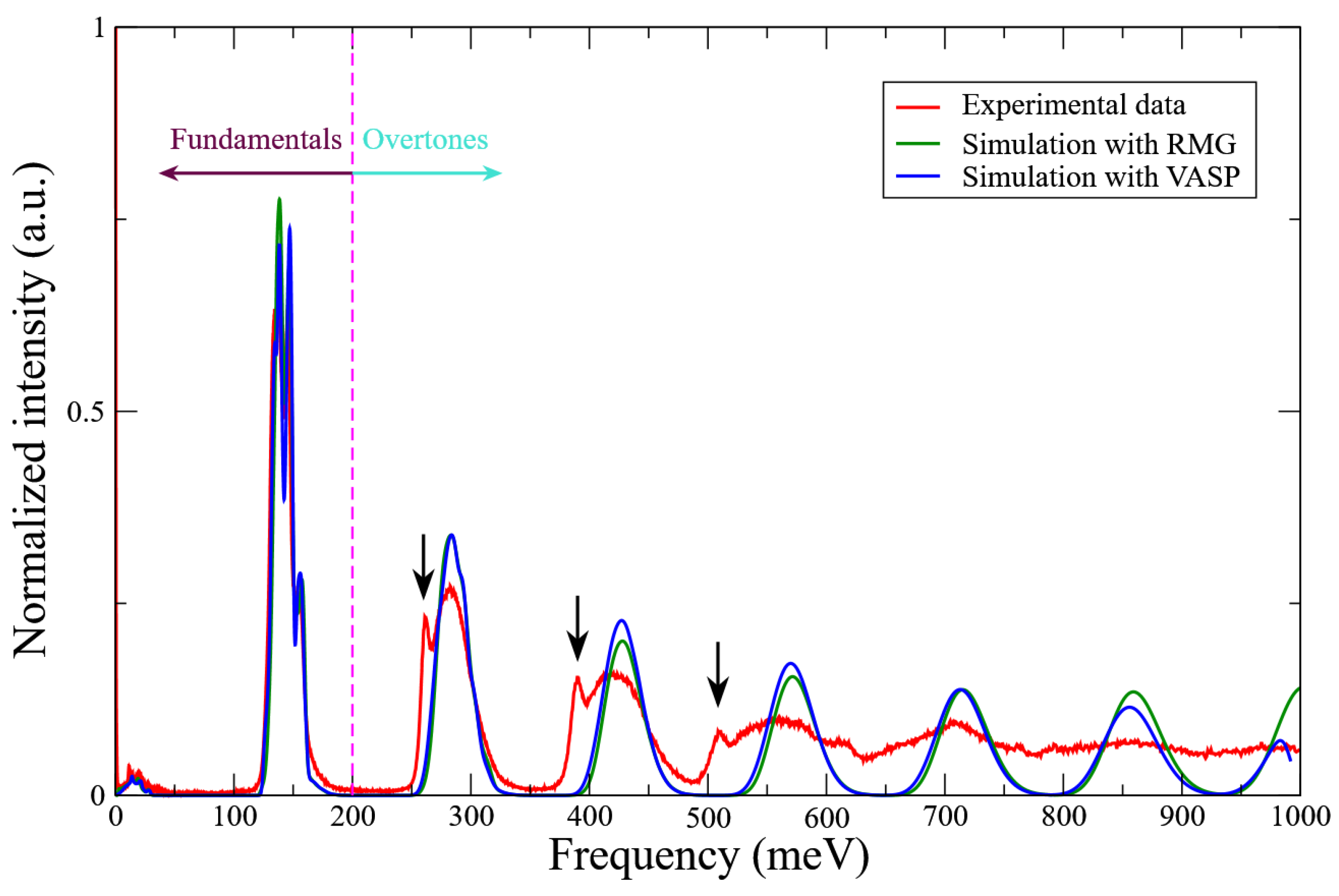
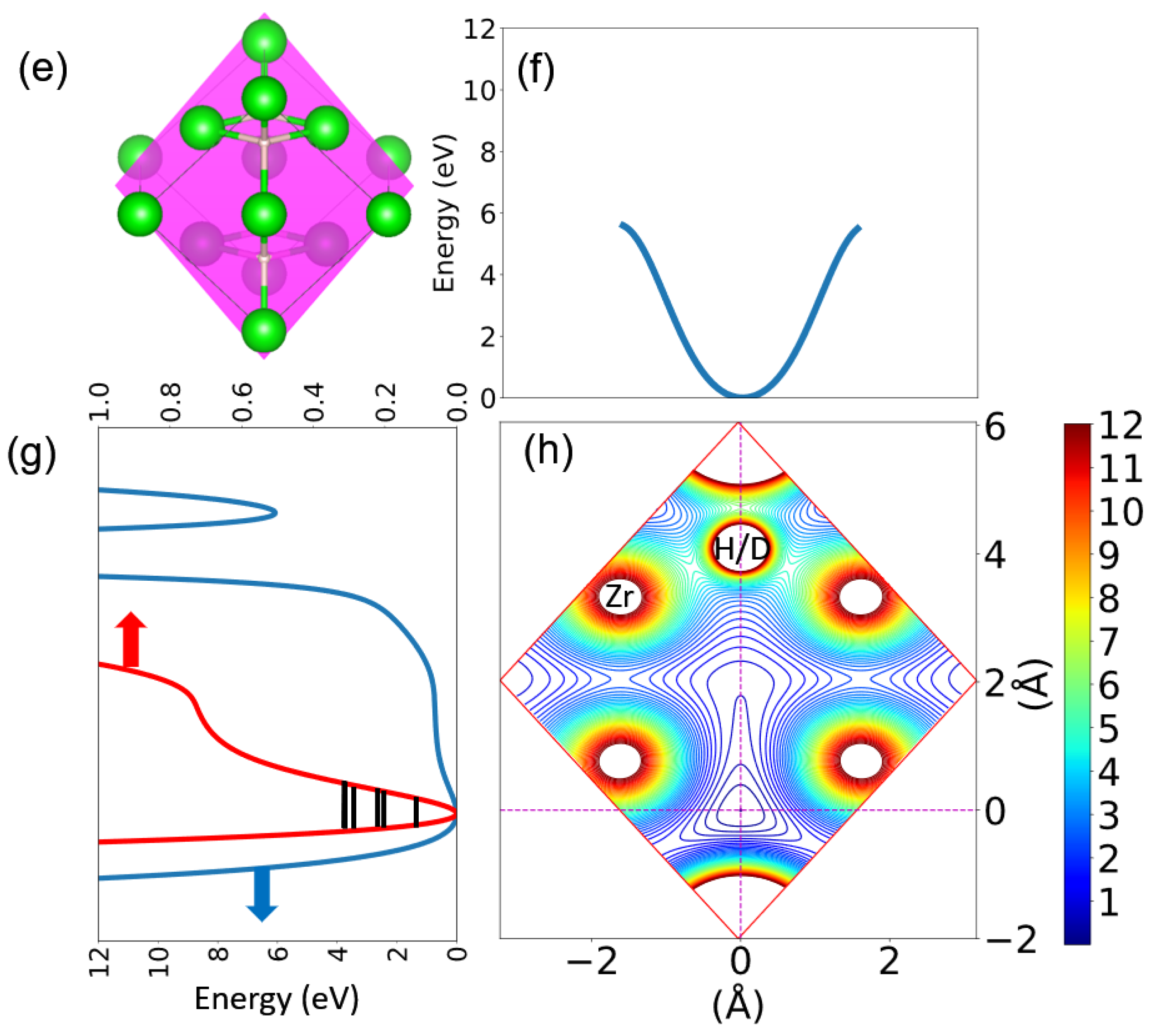
3.2. Harmonic Approximation: Frozen Phonon Method
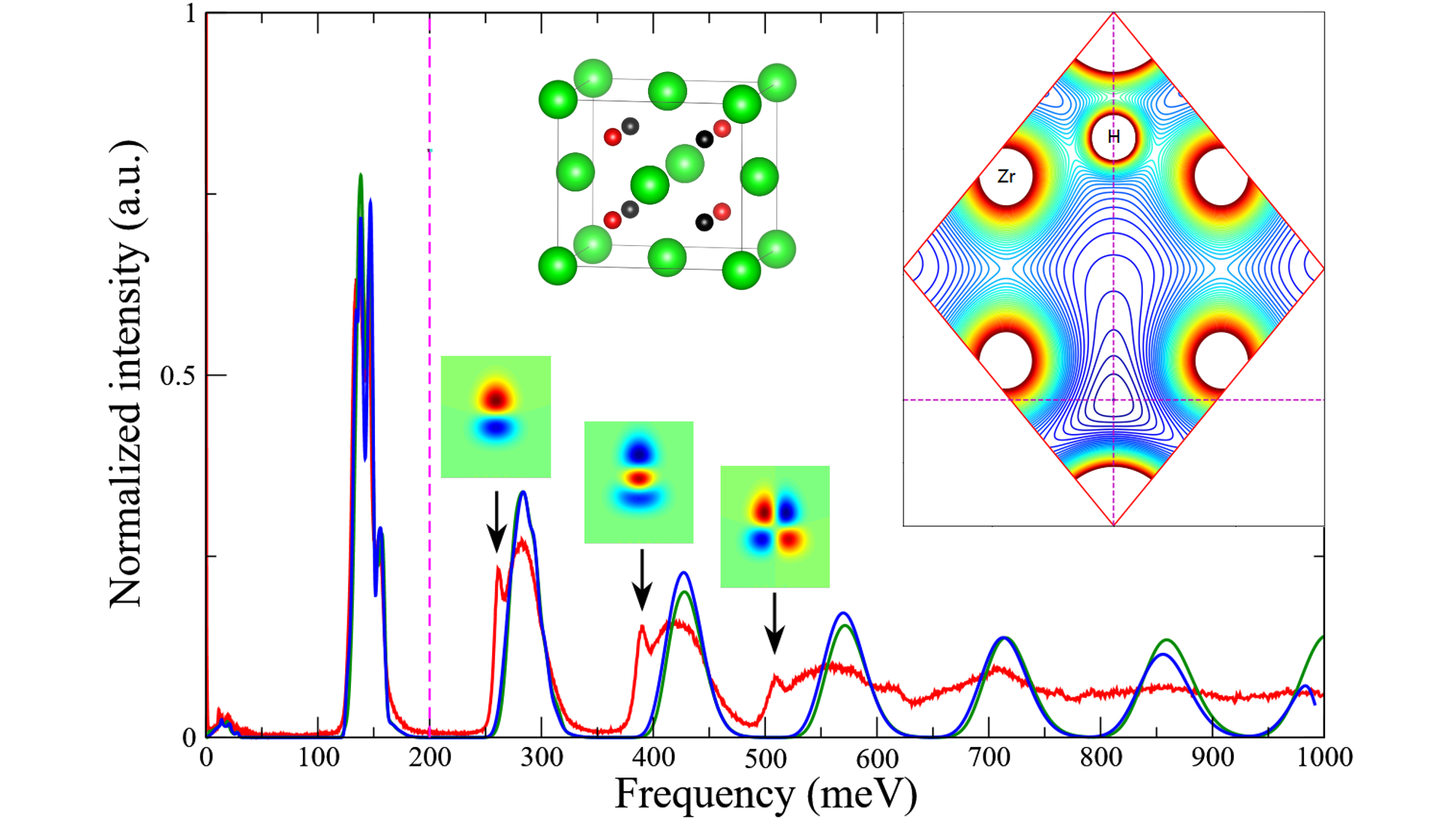
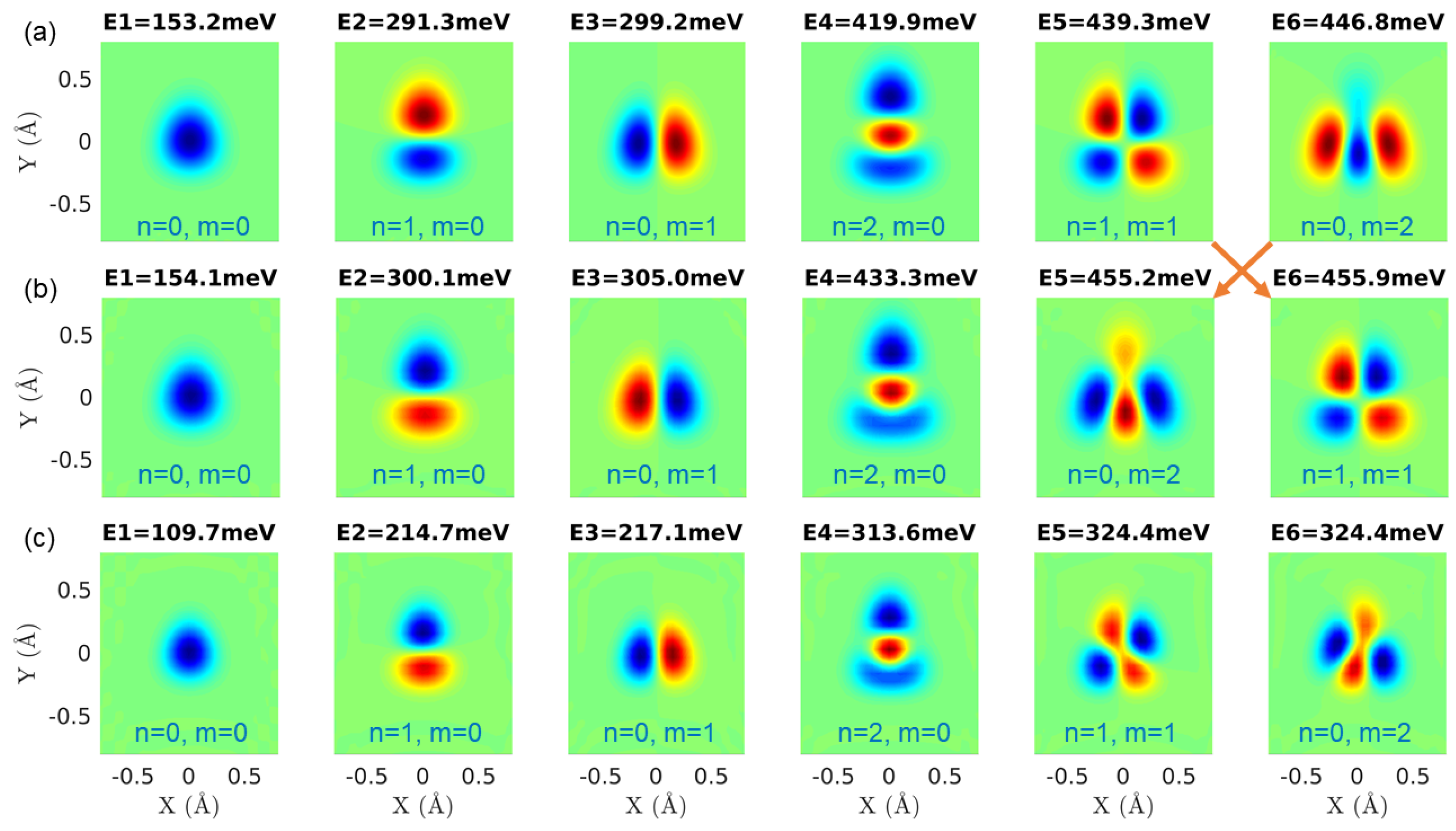
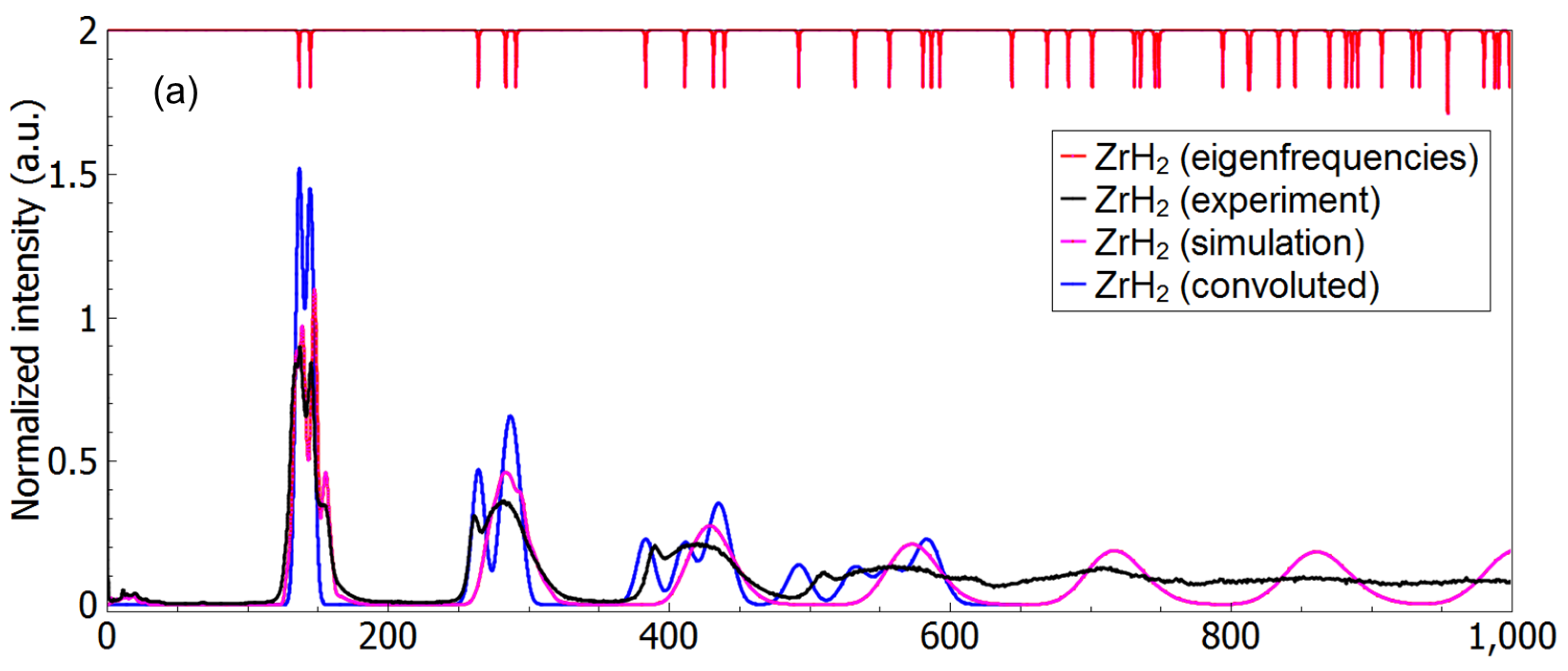
3.3. A Direct Method: Mapping Eigenfrequencies from Schrödinger Equations with the Simulated INS Spectra
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| DFT | Density functional theory |
| MD | Molecular dynamics |
| INS | Inelastic neutron scattering |
| PDOS | Phonon density of states |
| VACF | Velocity autocorrelation function |
| TDEP | Temperature-dependent effective potential |
| MSD | Mean square displacement |
| PES | Potential energy surface |
References
- Bickel, P.; Berlincourt, T. Electrical properties of hydrides and deuterides of zirconium. Phys. Rev. B 1970, 2, 4807. [Google Scholar] [CrossRef]
- Holliger, L.; Legris, A.; Besson, R. Hexagonal-based ordered phases in H-Zr. Phys. Rev. B 2009, 80, 094111. [Google Scholar] [CrossRef]
- Zuzek, E.; Abriata, J.; San-Martin, A.; Manchester, F. The H-Zr (hydrogen-zirconium) system. Bull. Alloy. Phase Diagrams 1990, 11, 385–395. [Google Scholar] [CrossRef]
- Setoyama, D.; Yamanaka, S. Phase diagram of Zr–O–H ternary system. J. Alloys Compd. 2004, 370, 144–148. [Google Scholar] [CrossRef]
- Dupin, N.; Ansara, I.; Servant, C.; Toffolon, C.; Lemaignan, C.; Brachet, J. A thermodynamic database for zirconium alloys. J. Nucl. Mater. 1999, 275, 287–295. [Google Scholar] [CrossRef]
- Zhao, Z.; Morniroli, J.P.; Legris, A.; Ambard, A.; Khin, Y.; Legras, L.; Blat-Yrieix, M. Identification and characterization of a new zirconium hydride. J. Microsc. 2008, 232, 410–421. [Google Scholar] [CrossRef]
- Weck, P.F.; Kim, E.; Tikare, V.; Mitchell, J.A. Mechanical properties of zirconium alloys and zirconium hydrides predicted from density functional perturbation theory. Dalton Trans. 2015, 44, 18769–18779. [Google Scholar] [CrossRef]
- Andersen, H.C. Molecular dynamics simulations at constant pressure and/or temperature. J. Chem. Phys. 1980, 72, 2384–2393. [Google Scholar] [CrossRef] [Green Version]
- Yamanaka, S.; Yoshioka, K.; Uno, M.; Katsura, M.; Anada, H.; Matsuda, T.; Kobayashi, S. Thermal and mechanical properties of zirconium hydride. J. Alloys Compd. 1999, 293, 23–29. [Google Scholar] [CrossRef]
- Ackland, G. Embrittlement and the bistable crystal structure of zirconium hydride. Phys. Rev. Lett. 1998, 80, 2233. [Google Scholar] [CrossRef]
- Elsässer, C.; Schweizer, S.; Fähnle, M. Hydrogen Vibration in Cubic Dihydrides MH 2 (M = Ti, Zr), and Localization in Cubic Laves Phases ZrM 2 H 1/2 (M = V, Cr, Fe, Co). MRS Online Proc. Libr. Arch. 1996, 453. [Google Scholar] [CrossRef]
- Ikeda, S.; Watanabe, N.; Kai, K. Crystal analyser TOF spectrometer (CAT). Physica B + C 1983, 120, 131–135. [Google Scholar] [CrossRef]
- Kolesnikov, A.; Bashkin, I.; Belushkin, A.; Ponyatovsky, E.; Prager, M. Inelastic neutron scattering study of ordered gamma-ZrH. J. Phys. Condens. Matter 1994, 6, 8989. [Google Scholar] [CrossRef]
- Kolesnikov, A.; Balagurov, A.; Bashkin, I.; Belushkin, A.; Ponyatovsky, E.; Prager, M. Neutron scattering studies of ordered gamma-ZrD. J. Phys. Condens. Matter 1994, 6, 8977. [Google Scholar] [CrossRef]
- Hellman, O.; Abrikosov, I.; Simak, S. Lattice dynamics of anharmonic solids from first principles. Phys. Rev. B 2011, 84, 180301. [Google Scholar] [CrossRef] [Green Version]
- Hellman, O.; Abrikosov, I.A. Temperature-dependent effective third-order interatomic force constants from first principles. Phys. Rev. B 2013, 88, 144301. [Google Scholar] [CrossRef] [Green Version]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- RMG Website. Available online: https://www.rmgdft.org/ (accessed on 10 March 2021).
- Briggs, E.; Sullivan, D.; Bernholc, J. Real-space multigrid-based approach to large-scale electronic structure calculations. Phys. Rev. B 1996, 54, 14362. [Google Scholar] [CrossRef]
- Hodak, M.; Wang, S.; Lu, W.; Bernholc, J. Implementation of ultrasoft pseudopotentials in large-scale grid-based electronic structure calculations. Phys. Rev. B 2007, 76, 085108. [Google Scholar] [CrossRef] [Green Version]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758. [Google Scholar] [CrossRef]
- Perdew, J.P.; Chevary, J.A.; Vosko, S.H.; Jackson, K.A.; Pederson, M.R.; Singh, D.J.; Fiolhais, C. Atoms, molecules, solids, and surfaces: Applications of the generalized gradient approximation for exchange and correlation. Phys. Rev. B 1992, 46, 6671. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [Green Version]
- Methfessel, M.; Paxton, A. High-precision sampling for Brillouin-zone integration in metals. Phys. Rev. B 1989, 40, 3616. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Cheng, Y.; Lu, W.; Briggs, E.; Ramirez-Cuesta, A.J.; Bernholc, J. Large-Scale Phonon Calculations Using the Real-Space Multigrid Method. J. Chem. Theory Comput. 2019, 15, 6859–6864. [Google Scholar] [CrossRef] [PubMed]
- Togo, A.; Tanaka, I. First principles phonon calculations in materials science. Scr. Mater. 2015, 108, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, P.C.H. Vibrational Spectroscopy with Neutrons: With Applications in Chemistry, Biology, Materials Science and Catalysis; World Scientific: Singapore, 2005; Volume 3. [Google Scholar]
- Ramirez-Cuesta, A. aCLIMAX 4.0. 1, The new version of the software for analyzing and interpreting INS spectra. Comput. Phys. Commun. 2004, 157, 226–238. [Google Scholar] [CrossRef]
- Cheng, Y.; Daemen, L.; Kolesnikov, A.; Ramirez-Cuesta, A. Simulation of inelastic neutron scattering spectra using OCLIMAX. J. Chem. Theory Comput. 2019, 15, 1974–1982. [Google Scholar] [CrossRef] [PubMed]
- Seeger, P.A.; Daemen, L.L.; Larese, J.Z. Resolution of VISION, a crystal-analyzer spectrometer. Nucl. Instrum. Methods Phys. Res. Sect. A 2009, 604, 719–728. [Google Scholar] [CrossRef]
- Goyal, P.S.; Penfold, J.; Tomkinson, J. Internal Rep. Technical Report, RAL-86-070; Rutherford Appleton Lab.: Chilton, UK, 1986. [Google Scholar]
- Penfold, J.; Tomkinson, J. Internal Rep. Technical Report, RAL-86-019; Rutherford Appleton Lab.: Chilton, UK, 1986. [Google Scholar]
- Rundle, R.; Shull, C.; Wollan, E.O. The crystal structure of thorium and zirconium dihydrides by X-ray and neutron diffraction. Acta Crystallogr. 1952, 5, 22–26. [Google Scholar] [CrossRef] [Green Version]
- Momma, K.; Izumi, F. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystallogr. 2011, 44, 1272–1276. [Google Scholar] [CrossRef]
- Hinuma, Y.; Pizzi, G.; Kumagai, Y.; Oba, F.; Tanaka, I. Band structure diagram paths based on crystallography. Comput. Mater. Sci. 2017, 128, 140–184. [Google Scholar] [CrossRef] [Green Version]
- Arnold, O.; Bilheux, J.C.; Borreguero, J.; Buts, A.; Campbell, S.I.; Chapon, L.; Doucet, M.; Draper, N.; Leal, R.F.; Gigg, M.; et al. Mantid—Data analysis and visualization package for neutron scattering and μ SR experiments. Nucl. Instrum. Methods Phys. Res. Sect. A 2014, 764, 156–166. [Google Scholar] [CrossRef] [Green Version]
- Ackland, G.; Warren, M.; Clark, S. Practical methods in ab initio lattice dynamics. J. Phys. Condens. Matter 1997, 9, 7861. [Google Scholar] [CrossRef] [Green Version]
- Olsson, P.; Massih, A.; Blomqvist, J.; Holston, A.M.A.; Bjerkén, C. Ab initio thermodynamics of zirconium hydrides and deuterides. Comput. Mater. Sci. 2014, 86, 211–222. [Google Scholar] [CrossRef]
- Zhang, P.; Wang, B.T.; He, C.H.; Zhang, P. First-principles study of ground state properties of ZrH2. Comput. Mater. Sci. 2011, 50, 3297–3302. [Google Scholar] [CrossRef]
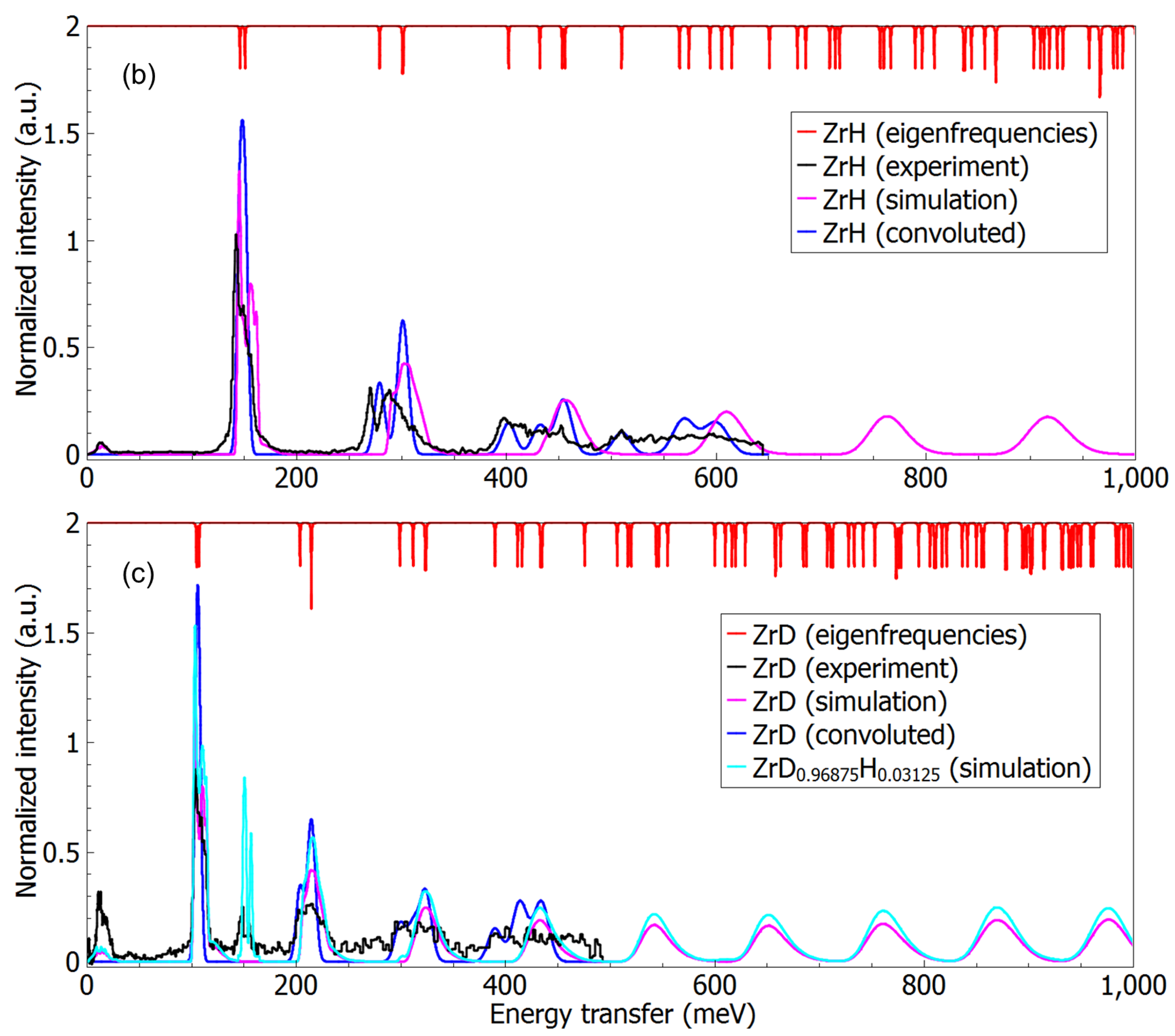
- See Supplementary Materials for details of structural parameters of ZrHx, and wavefunctions and their comparisons for H/D in selected planes.
- Nevou, L. 2D Time Indep. Schroedinger Equ. Solver. Available online: https://github.com/LaurentNevou/Q_Schrodinger2D_demo/ (accessed on 10 March 2021).











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Property | <60 meV | >60 meV |
|---|---|---|
| -ZrH | ||
| MSD(H) | 0.00063 (0.025) | 0.0144 (0.12) |
| MSD(Zr) | 0.0014 (0.037) | 0.00000132 (0.00115) |
| 0.45 (0.67) | 10,909.09 (104.45) | |
| -ZrH | ||
| MSD(H) | 0.0008 (0.0283) | 0.0136 (0.1166) |
| MSD(Zr) | 0.0015 (0.0387) | 0.00000079 (0.00089) |
| 0.53 (0.73) | 17,215.19 (131.21) | |
| -ZrD | ||
| MSD(D) | 0.0008 (0.0283) | 0.0095 (0.0976) |
| MSD(Zr) | 0.0015 (0.0387) | 0.000002245 (0.001498) |
| 0.53 (0.73) | 4231.63 (65.05) | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, J.; Cheng, Y.; Kolesnikov, A.I.; Bernholc, J.; Lu, W.; Ramirez-Cuesta, A.J. Study of Anharmonicity in Zirconium Hydrides Using Inelastic Neutron Scattering and Ab-Initio Computer Modeling. Inorganics 2021, 9, 29. https://doi.org/10.3390/inorganics9050029
Zhang J, Cheng Y, Kolesnikov AI, Bernholc J, Lu W, Ramirez-Cuesta AJ. Study of Anharmonicity in Zirconium Hydrides Using Inelastic Neutron Scattering and Ab-Initio Computer Modeling. Inorganics. 2021; 9(5):29. https://doi.org/10.3390/inorganics9050029
Chicago/Turabian StyleZhang, Jiayong, Yongqiang Cheng, Alexander I. Kolesnikov, Jerry Bernholc, Wenchang Lu, and Anibal J. Ramirez-Cuesta. 2021. "Study of Anharmonicity in Zirconium Hydrides Using Inelastic Neutron Scattering and Ab-Initio Computer Modeling" Inorganics 9, no. 5: 29. https://doi.org/10.3390/inorganics9050029
APA StyleZhang, J., Cheng, Y., Kolesnikov, A. I., Bernholc, J., Lu, W., & Ramirez-Cuesta, A. J. (2021). Study of Anharmonicity in Zirconium Hydrides Using Inelastic Neutron Scattering and Ab-Initio Computer Modeling. Inorganics, 9(5), 29. https://doi.org/10.3390/inorganics9050029