
A Rapid LC-MS/MS Method for Simultaneous Determination of Ten Flavonoid Metabolites of Naringin in Rat Urine and Its Application to an Excretion Study


Abstract
:1. Introduction
2. Materials and Methods
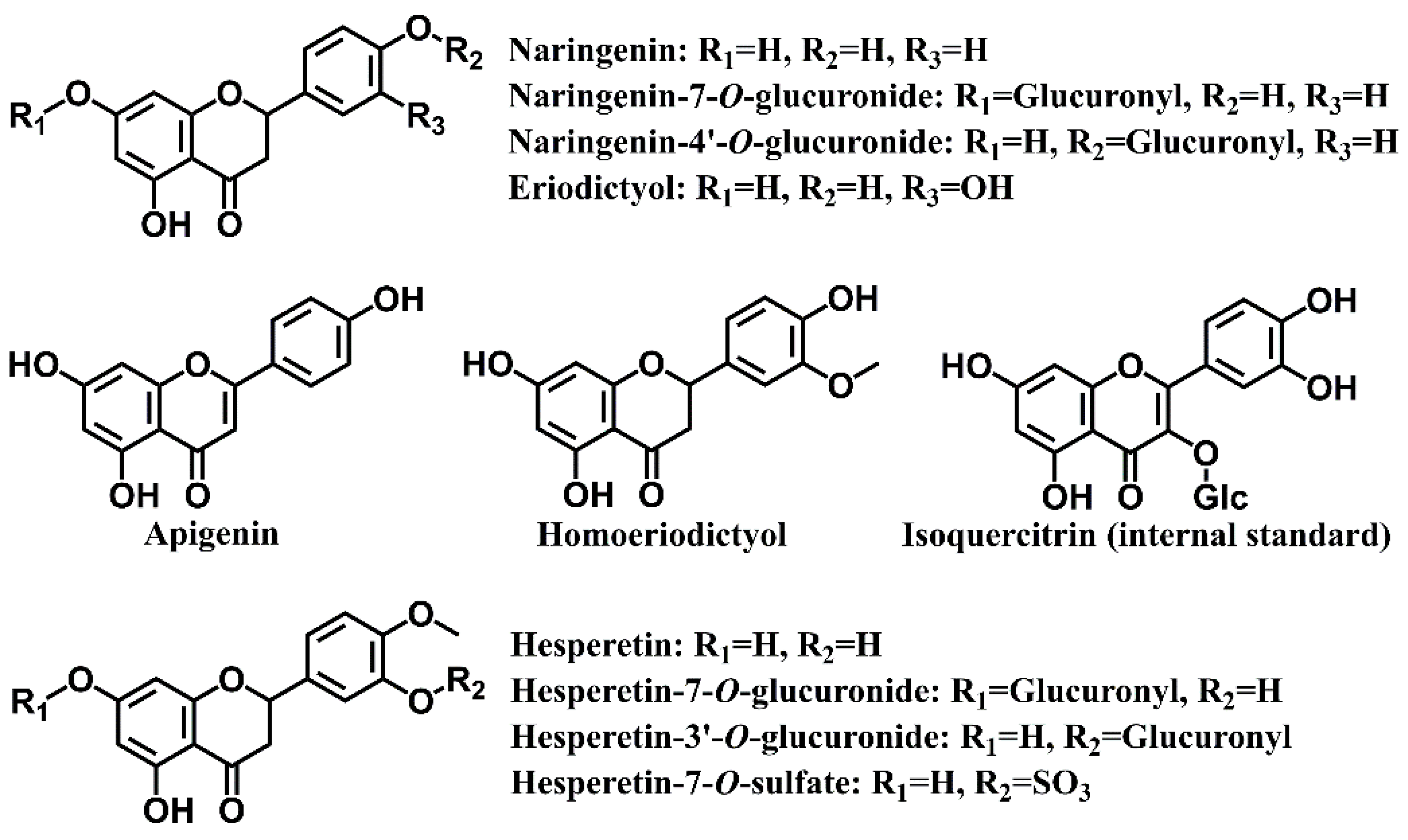
2.1. Chemicals and Reagents
2.2. Preparation of Calibration Standard and Quality Control (QC) Samples
2.3. Sample Preparation for LC-MS/MS Analysis
2.4. LC-MS/MS Conditions
2.5. Method Validation
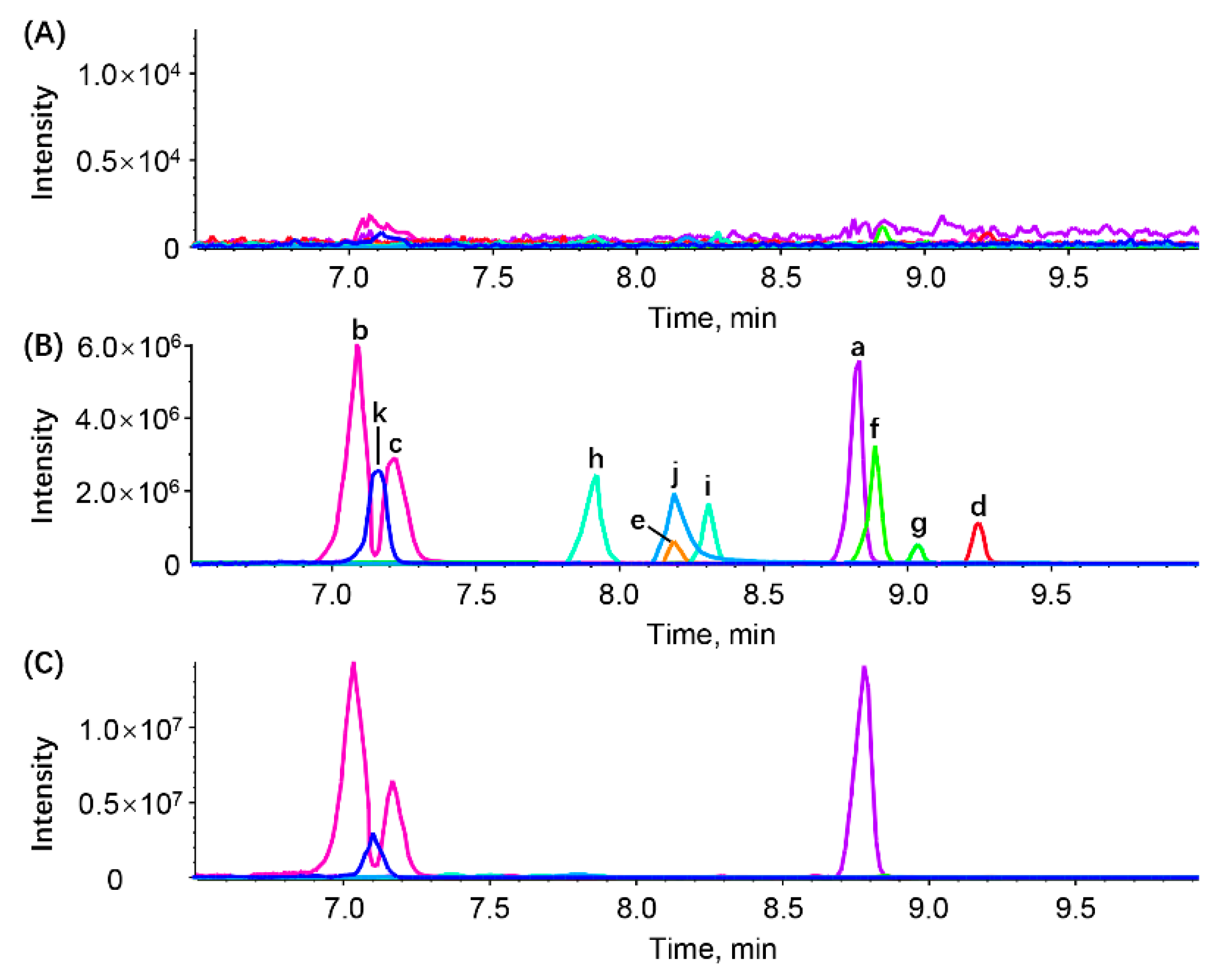
2.5.1. Selectivity
2.5.2. Carry-Over Effect
2.5.3. Calibration Curve and LLOQ
2.5.4. Precision and Accuracy
2.5.5. Matrix Effect
2.5.6. Dilution Integrity
2.5.7. Stability
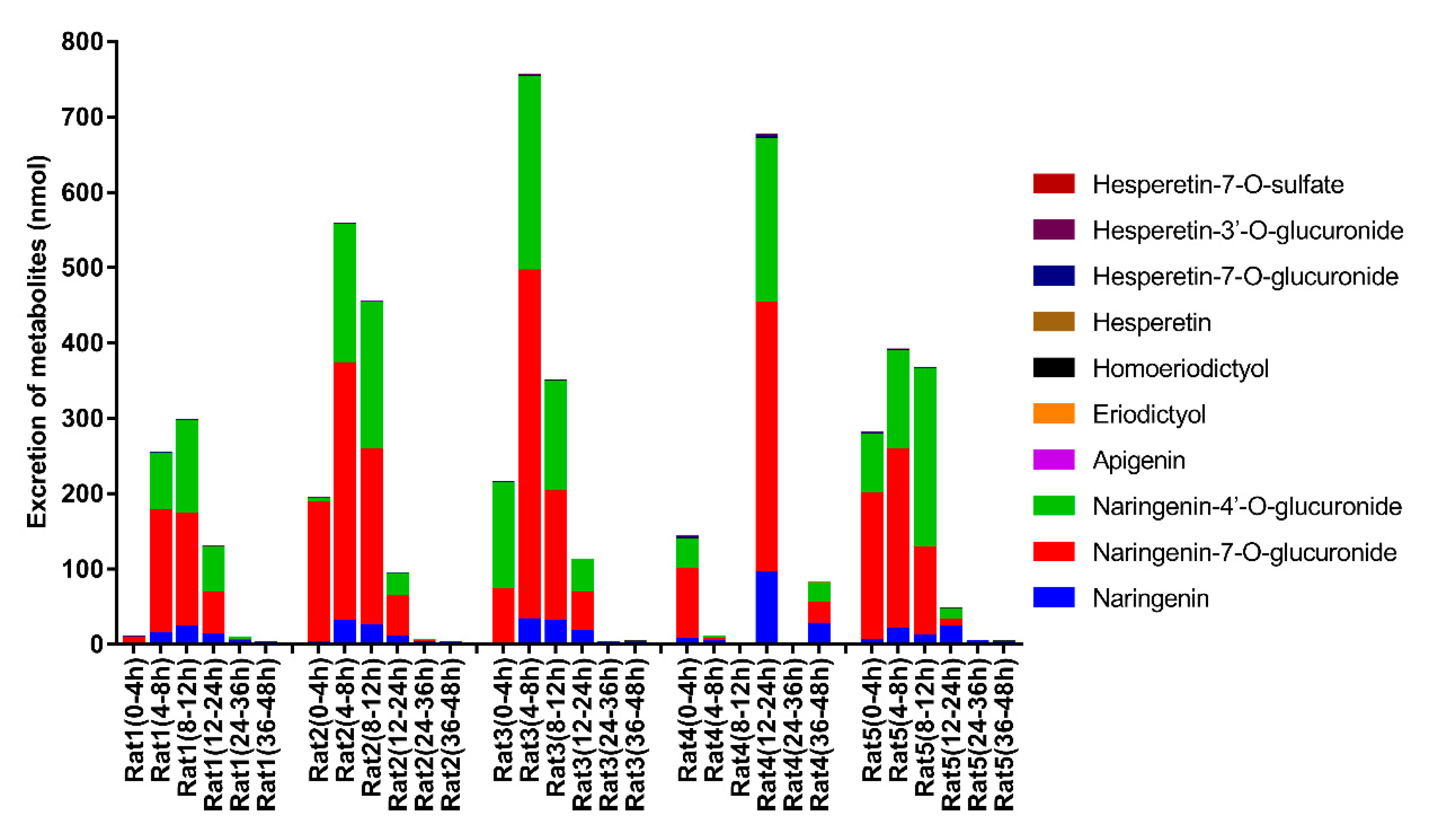
2.6. Excretion Study
2.7. Data Analysis
3. Results and Discussion
3.1. Method Development
3.2. Method Validation
3.2.1. Selectivity
3.2.2. Carry-Over Effect
3.2.3. Calibration Curve and LLOQ
3.2.4. Precision and Accuracy
3.2.5. Matrix Effect
3.2.6. Dilution Integrity
3.2.7. Stability
3.3. Excretion Study
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Singh, B.; Singh, J.; Kaur, A.; Singh, N. Phenolic composition, antioxidant potential and health benefits of citrus peel. Food Res. Int. 2020, 132, 109114. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Qi, Q.; Wang, M.; Li, Q. Therapeutic potential of naringin: An overview. Pharm. Biol. 2016, 54, 3203–3210. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.; Khan, H.; Aschner, M.; Hasan, M.; Hassan, S. Therapeutic potential of naringin in neurological disorders. Food Chem. Toxicol. 2019, 132, 110646. [Google Scholar] [CrossRef]
- Raja Kumar, S.; Mohd Ramli, E.; Abdul Nasir, N.; Ismail, N.; Mohd Fahami, N. Preventive effect of naringin on metabolic syndrome and its mechanism of action: A systematic review. Evid. Based Complement. Alternat. Med. 2019, 2019, 9752826. [Google Scholar] [CrossRef] [Green Version]
- Heidary Moghaddam, R.; Samimi, Z.; Moradi, S.; Little, P.; Xu, S.; Farzaei, M. Naringenin and naringin in cardiovascular disease prevention: A preclinical review. Eur. J. Pharmacol. 2020, 887, 173535. [Google Scholar] [CrossRef]
- Zeng, X.; Su, W.; Liu, B.; Chai, L.; Shi, R.; Yao, H. A review on the pharmacokinetic properties of naringin and its therapeutic efficacies in respiratory diseases. Mini Rev. Med. Chem. 2020, 20, 286–293. [Google Scholar] [CrossRef]
- Salehi, B.; Fokou, P.; Sharifi-Rad, M.; Zucca, P.; Pezzani, R.; Martins, N.; Sharifi-Rad, J. The therapeutic potential of naringenin: A review of clinical trials. Pharmaceuticals 2019, 12, 11. [Google Scholar] [CrossRef] [Green Version]
- Cassidy, A.; Minihane, A. The role of metabolism (and the microbiome) in defining the clinical efficacy of dietary flavonoids. Am. J. Clin. Nutr. 2017, 105, 10–22. [Google Scholar] [CrossRef] [Green Version]
- Xiao, J.; Zhou, B.; Ressom, H.-W. Metabolite identification and quantitation in LC-MS/MS-based metabolomics. Trends Analyt. Chem. 2012, 32, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Reddy, G.-N.; Laltanpuii, C.; Sonti, R. Review on in vivo profiling of drug metabolites with LC-MS/MS in the past decade. Bioanalysis 2021, 13, 1697–1722. [Google Scholar] [CrossRef]
- Orrego-Lagaron, N.; Martinez-Huelamo, M.; Vallverdu-Queralt, A.; Lamuela-Raventos, R.; Escribano-Ferrer, E. High gastrointestinal permeability and local metabolism of naringenin: Influence of antibiotic treatment on absorption and metabolism. Br. J. Nutr. 2015, 114, 169–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, X.; Jiang, J.; Duan, J.; Xie, Y.; Wang, J.; Zhai, S. Development and validation of a sensitive liquid chromatography-tandem mass spectrometry method for the determination of naringin and its metabolite, naringenin, in human plasma. J. Chromatogr. Sci. 2014, 52, 654–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, Y.; Peng, W.; Yang, C.; Zou, W.; Liu, M.; Wu, H.; Fan, L.; Li, P.; Zeng, X.; Su, W. Pharmacokinetics and metabolism of naringin and active metabolite naringenin in rats, dogs, humans, and the differences between species. Front. Pharmacol. 2020, 11, 364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joshi, R.; Kulkarni, Y.; Wairkar, S. Pharmacokinetic, pharmacodynamic and formulations aspects of naringenin: An update. Life Sci. 2018, 215, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Zou, W.; Yang, C.; Peng, W.; Su, W. Metabolism and excretion studies of oral administered naringin, a putative antitussive, in rats and dogs. Biopharm. Drug Dispos. 2012, 33, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Su, W.; Zheng, Y.; He, Y.D.; He, Y.; Rao, H.; Peng, W.; Yao, H. Pharmacokinetics, tissue distribution, metabolism, and excretion of naringin in aged rats. Front. Pharmacol. 2019, 10, 34. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Yao, H.; Zheng, Y.; He, Y.D.; He, Y.; Rao, H.; Li, P.; Su, W. Tissue distribution of naringin and derived metabolites in rats after a single oral administration. J. Chromatogr. B Analyt. Technol. Biomed Life Sci. 2020, 1136, 121846. [Google Scholar] [CrossRef]
- Pharmacopoeia Commission of the People’s Republic of China. Guidance for bioanalytical method validation. In Pharmacopoeia of the People’s Republic of China; China Medical Science Press: Beijing, China, 2020; pp. 466–472. [Google Scholar]
- Najmanová, I.; Vopršalová, M.; Saso, L.; Mladěnka, P. The pharmacokinetics of flavanones. Crit. Rev. Food Sci. Nutr. 2020, 60, 3155–3171. [Google Scholar] [CrossRef]
- Anguita-Ruiz, A.; Aguilera, C.; Gil, Á. Genetics of lactose intolerance: An updated review and online interactive world maps of phenotype and genotype frequencies. Nutrients 2020, 12, 2689. [Google Scholar] [CrossRef]
- Yang, N.; Sun, R.; Liao, X.; Aa, J.; Wang, G. UDP-glucuronosyltransferases (UGTs) and their related metabolic cross-talk with internal homeostasis: A systematic review of UGT isoforms for precision medicine. Pharmacol. Res. 2017, 121, 169–183. [Google Scholar] [CrossRef]
- Chen, B.; Wang, C.; Hou, Y.; Mao, Y.; Yang, Y. Mechanism of sulfotransferase pharmacogenetics in altered xenobiotic metabolism. Expert Opin. Drug Metab. Toxicol. 2015, 11, 1053–1071. [Google Scholar] [CrossRef] [PubMed]
- Kwon, E.-Y.; Choi, M.-S. Chapter 44—Polyphenols and Nutrigenetic/Nutrigenomic Associations With Obesity-Related Metabolic Diseases. In Principles of Nutrigenetics and Nutrigenomics; Caterina, R.D.E., Martinez, J.A., Kohlmeier, M., Eds.; American Academic Press: Salt Lake City, UT, USA, 2020; pp. 327–334. [Google Scholar]
- Chen, T.; Su, W.; Yan, Z.; Wu, H.; Zeng, X.; Peng, W.; Gan, L.; Zhang, Y.; Yao, H. Identification of naringin metabolites mediated by human intestinal microbes with stable isotope-labeling method and UFLC-Q-TOF-MS/MS. J. Pharm. Biomed Anal. 2018, 161, 262–272. [Google Scholar] [CrossRef] [PubMed]
- Vandenplas, Y.; Carnielli, V.; Ksiazyk, J.; Luna, M.; Migacheva, N.; Mosselmans, J.; Picaud, J.; Possner, M.; Singhal, A.; Wabitsch, M. Factors affecting early-life intestinal microbiota development. Nutrition 2020, 78, 110812. [Google Scholar] [CrossRef]
- Rowland, I.; Gibson, G.; Heinken, A.; Scott, K.; Swann, J.; Thiele, I.; Tuohy, K. Gut microbiota functions: Metabolism of nutrients and other food components. Eur. J. Nutr. 2018, 57, 1–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira-Caro, G.; Ludwig, I.; Polyviou, T.; Malkova, D.; García, A.; Moreno-Rojas, J.; Crozier, A. Identification of plasma and urinary metabolites and catabolites derived from orange juice (poly)phenols: Analysis by high-performance liquid chromatography-high-resolution mass spectrometry. J. Agric. Food Chem. 2016, 64, 5724–5735. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.; Wu, H.; He, Y.; Pan, W.; Yan, Z.; Liao, Y.; Peng, W.; Gan, L.; Zhang, Y.; Su, W.; et al. Simultaneously quantitative analysis of naringin and its major human gut microbial metabolites naringenin and 3-(4′-hydroxyphenyl) propanoic acid via stable isotope deuterium-labeling coupled with RRLC-MS/MS method. Molecules 2019, 24, 4287. [Google Scholar] [CrossRef] [Green Version]
- Luca, S.; Macovei, I.; Bujor, A.; Miron, A.; Skalicka-Woźniak, K.; Aprotosoaie, A.; Trifan, A. Bioactivity of dietary polyphenols: The role of metabolites. Crit. Rev. Food Sci. Nutr. 2020, 60, 626–659. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| No. | Analytes | Q1 Mass (Da) | Q3 Mass (Da) | DP (eV) | CE (eV) |
|---|---|---|---|---|---|
| 1 | Naringenin | 270.9 | 150.9 | −24 | −44 |
| 2 | Naringenin-7-O-glucuronide, Naringenin-4′-O-glucuronide | 447.0 | 271.1 | −33 | −72 |
| 3 | Apigenin | 269.0 | 151.0 | −33 | −48 |
| 4 | Eriodictyol | 286.9 | 135.0 | −38 | −56 |
| 5 | Homoeriodictyol | 301.0 | 150.9 | −26 | −50 |
| 6 | Hesperetin | 301.0 | 163.9 | −32 | −134 |
| 7 | Hesperetin-7-O-glucuronide, Hesperetin-3′-O-glucuronide | 477.1 | 301.1 | −31 | −59 |
| 8 | Hesperetin-7-O-sulfate | 381.0 | 301.1 | −28 | −53 |
| 9 | Isoquercitrin (IS) | 463.1 | 299.9 | −36 | −32 |
| No. | Analytes | r | Linear Ranges (ng/mL) |
|---|---|---|---|
| 1 | Naringenin | 0.99072 | 2.140~1070 |
| 2 | Naringenin-7-O-glucuronide | 0.99350 | 20.00~10,000 |
| 3 | Naringenin-4′-O-glucuronide | 0.99006 | 20.00~10,000 |
| 4 | Apigenin | 0.99320 | 0.9430~471.5 |
| 5 | Eriodictyol | 0.99748 | 0.4464~223.2 |
| 6 | Homoeriodictyol | 0.99574 | 0.9900~495.0 |
| 7 | Hesperetin | 0.99004 | 0.5510~275.5 |
| 8 | Hesperetin-7-O-glucuronide | 0.99681 | 4.200~2100 |
| 9 | Hesperetin-3′-O-glucuronide | 0.99316 | 3.600~1800 |
| 10 | Hesperetin-7-O-sulfate | 0.99252 | 0.9700~485.0 |
| Analytes | Conc. (ng/mL) | Intra-Batch (n = 6) | Inter-Batch (n = 6 × 3) | IS-MF (n = 3 × 6) | |||
|---|---|---|---|---|---|---|---|
| RSD% | RE% | RSD% | RE% | Mean | RSD% | ||
| Naringenin | 2.140 | 3.9 | 5.7 | 8.5 | −1.1 | - | - |
| 6.420 | 8.1 | −2.5 | 7.2 | −4.5 | 1.04 | 3.7 | |
| 64.20 | 6.3 | −8.0 | 5.7 | −5.6 | - | - | |
| 802.5 | 2.8 | −1.1 | 6.6 | −3.6 | 0.96 | 2.7 | |
| Naringenin-7-O-glucuronide | 20.00 | 6.8 | 10.7 | 8.9 | 3.8 | - | - |
| 60.00 | 4.2 | 2.6 | 6.1 | −0.1 | 1.05 | 4.5 | |
| 600.0 | 7.0 | −2.5 | 7.3 | 1.5 | - | - | |
| 7500 | 5.9 | 0.7 | 6.4 | −0.2 | 0.93 | 2.5 | |
| Naringenin-4′-O-glucuronide | 20.00 | 8.9 | −0.3 | 9.2 | 2.5 | - | - |
| 60.00 | 5.0 | −4.8 | 6.7 | −0.8 | 1.05 | 3.8 | |
| 600.0 | 3.4 | −6.1 | 5.5 | −1.8 | - | - | |
| 7500 | 4.0 | 0.7 | 6.4 | −0.7 | 0.95 | 3.9 | |
| Apigenin | 0.9430 | 5.8 | 4.0 | 8.7 | 0.5 | - | - |
| 2.829 | 7.9 | −6.5 | 6.6 | −6.0 | 1.05 | 4.4 | |
| 28.29 | 6.5 | −10.2 | 4.8 | −7.6 | - | - | |
| 353.6 | 3.4 | −2.5 | 6.3 | −3.3 | 0.98 | 3.5 | |
| Eriodictyol | 0.4464 | 8.5 | 2.8 | 7.4 | 4.0 | - | - |
| 1.339 | 5.5 | −2.6 | 5.6 | −3.2 | 1.12 | 2.2 | |
| 13.39 | 4.1 | −5.8 | 6.1 | −1.4 | - | - | |
| 167.4 | 6.5 | −2.7 | 6.6 | −0.9 | 0.98 | 1.7 | |
| Homoeriodictyol | 0.9900 | 4.6 | 6.8 | 7.5 | 1.9 | - | - |
| 2.970 | 7.2 | 4.6 | 7.7 | −2.9 | 1.01 | 5.8 | |
| 29.70 | 5.1 | −8.0 | 5.3 | −5.9 | - | - | |
| 371.3 | 3.0 | 0.1 | 5.8 | −2.4 | 0.98 | 3.9 | |
| Hesperetin | 0.551 | 3.8 | 7.6 | 4.0 | 8.6 | - | - |
| 1.653 | 6.2 | −4.2 | 6.1 | −2.8 | 1.09 | 4.7 | |
| 16.53 | 5.4 | −6.3 | 6.3 | −2.8 | - | - | |
| 206.6 | 2.6 | 0.2 | 5.5 | −1.1 | 1.08 | 2.2 | |
| Hesperetin-7-O-glucuronide | 4.200 | 6.3 | 4.3 | 7.7 | 3.3 | - | - |
| 12.60 | 5.0 | −4.9 | 6.4 | 0.4 | 1.00 | 7.4 | |
| 126.0 | 6.0 | −9.1 | 6.8 | −2.4 | - | - | |
| 1575 | 4.6 | −1.1 | 5.2 | 1.7 | 0.95 | 3.2 | |
| Hesperetin-3′-O-glucuronide | 3.600 | 3.2 | 7.5 | 7.5 | 1.5 | - | - |
| 10.80 | 6.0 | 0.6 | 6.5 | −0.2 | 1.06 | 4.0 | |
| 108.0 | 6.7 | −5.5 | 7.4 | −2.9 | - | - | |
| 1350 | 2.5 | 3.0 | 5.2 | 3.8 | 1.00 | 2.7 | |
| Hesperetin-7-O-sulfate | 0.9700 | 9.8 | 2.3 | 8.3 | 2.2 | - | - |
| 2.910 | 8.8 | 3.4 | 8.6 | −0.3 | 1.14 | 2.1 | |
| 29.10 | 8.0 | −4.2 | 7.5 | −1.5 | - | - | |
| 363.8 | 6.4 | 5.6 | 5.2 | 3.9 | 0.93 | 5.1 | |
| Analytes | Conc. (ng/mL) | Long Term (−70 °C, 1 Month) | Long Term (−70 °C, 3 Months) | Freeze–Thaw (1 Cycle) | Freeze–Thaw (2 Cycles) | Placement in Autosampler (15 °C, 24 h) | Dilution Integrity (5 Times) | Dilution Integrity (10 Times) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| RSD% | RE% | RSD% | RE% | RSD% | RE% | RSD% | RE% | RSD% | RE% | RSD% | RE% | RSD% | RE% | ||
| Naringenin | 6.420 | 5.0 | 2.3 | 4.0 | −2.0 | 4.0 | 2.9 | 12.0 | −1.9 | 4.0 | 8.2 | - | - | - | - |
| 802.5 | 1.8 | 0.3 | 7.1 | −1.1 | 7.1 | −4.7 | 4.2 | 0.6 | 1.8 | 3.1 | 5.0 | 0.3 | 0.9 | −0.2 | |
| Naringenin-7-O-glucuronide | 60.00 | 5.2 | 6.9 | 6.4 | 4.3 | 4.9 | −1.5 | 6.9 | 4.1 | 3.8 | 10.5 | - | - | - | - |
| 7500 | 7.0 | −5.5 | 6.7 | −2.2 | 3.9 | −4.4 | 6.7 | −9.3 | 7.5 | −4.6 | 2.8 | −7.0 | 5.9 | −9.9 | |
| Naringenin-4′-O-glucuronide | 60.00 | 6.3 | 0.4 | 5.1 | 2.3 | 11.0 | 3.8 | 3.0 | 8.0 | 7.1 | 5.4 | - | - | - | - |
| 7500 | 8.2 | −3.7 | 6.8 | 1.4 | 6.5 | −3.7 | 4.5 | 2.1 | 4.4 | 6.1 | 1.6 | 0.1 | 5.7 | 0.7 | |
| Apigenin | 2.829 | 3.6 | −1.1 | 6.7 | 1.4 | 9.4 | −0.1 | 8.1 | 7.6 | 3.4 | 9.3 | - | - | - | - |
| 353.6 | 1.8 | 0.6 | 6.5 | 1.2 | 6.3 | −7.0 | 5.4 | 2.3 | 2.6 | 1.3 | 5.8 | −1.1 | 4.3 | −3.1 | |
| Eriodictyol | 1.339 | 5.5 | 0.1 | 9.1 | 0.9 | 7.5 | 1.6 | 11.3 | −3.9 | 5.9 | −2.3 | - | - | - | - |
| 167.4 | 3.4 | −2.3 | 6.1 | −1.8 | 7.1 | −6.4 | 4.2 | −3.2 | 6.4 | −3.9 | 5.6 | −0.7 | 5.6 | −0.4 | |
| Homoeriodictyol | 2.970 | 10.7 | −3.7 | 7.7 | 4.1 | 3.8 | 10.7 | 9.8 | 5.8 | 9.4 | 2.3 | - | - | - | - |
| 371.3 | 3.3 | 3.5 | 9.0 | −1.8 | 8.0 | −6.0 | 2.1 | 4.1 | 1.3 | 1.4 | 6.6 | −0.7 | 5.0 | 1.1 | |
| Hesperetin | 1.653 | 10.4 | −1.1 | 6.9 | −2.4 | 3.3 | 6.6 | 9.7 | 4.3 | 6.2 | 7.5 | - | - | - | - |
| 206.6 | 4.3 | 1.1 | 5.4 | 0.4 | 7.8 | −4.7 | 1.2 | −1.0 | 5.6 | −1.4 | 7.5 | 0.7 | 5.2 | −0.9 | |
| Hesperetin-7-O-glucuronide | 12.60 | 5.2 | −6.0 | 7.9 | −7.4 | 10.0 | −1.3 | 3.6 | −0.5 | 5.1 | 4.5 | - | - | - | - |
| 1575 | 4.7 | 1.1 | 6.7 | −2.4 | 7.3 | −6.4 | 2.9 | −1.6 | 4.0 | 2.3 | 2.9 | 0.0 | 4.4 | −3.1 | |
| Hesperetin-3′-O-glucuronide | 10.80 | 2.1 | 2.9 | 11.3 | 0.5 | 4.6 | 7.2 | 9.1 | 3.8 | 6.8 | −3.9 | - | - | - | - |
| 1350 | 1.8 | 4.0 | 8.2 | 0.0 | 7.2 | −0.6 | 3.4 | 6.1 | 5.6 | 1.1 | 2.0 | −1.5 | 2.7 | 5.6 | |
| Hesperetin-7-O-sulfate | 2.910 | 2.7 | 3.8 | 10.4 | 2.4 | 10.2 | 2.0 | 8.2 | 4.7 | 8.6 | 0.8 | - | - | - | - |
| 363.8 | 4.3 | −2.5 | 7.3 | −1.5 | 8.9 | −4.2 | 1.6 | 8.3 | 8.8 | −4.9 | 6.5 | 5.9 | 7.3 | −0.1 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zeng, X.; Zheng, Y.; He, Y.; Peng, W.; Su, W. A Rapid LC-MS/MS Method for Simultaneous Determination of Ten Flavonoid Metabolites of Naringin in Rat Urine and Its Application to an Excretion Study. Foods 2022, 11, 316. https://doi.org/10.3390/foods11030316
Zeng X, Zheng Y, He Y, Peng W, Su W. A Rapid LC-MS/MS Method for Simultaneous Determination of Ten Flavonoid Metabolites of Naringin in Rat Urine and Its Application to an Excretion Study. Foods. 2022; 11(3):316. https://doi.org/10.3390/foods11030316
Chicago/Turabian StyleZeng, Xuan, Yuying Zheng, Yan He, Wei Peng, and Weiwei Su. 2022. "A Rapid LC-MS/MS Method for Simultaneous Determination of Ten Flavonoid Metabolites of Naringin in Rat Urine and Its Application to an Excretion Study" Foods 11, no. 3: 316. https://doi.org/10.3390/foods11030316
APA StyleZeng, X., Zheng, Y., He, Y., Peng, W., & Su, W. (2022). A Rapid LC-MS/MS Method for Simultaneous Determination of Ten Flavonoid Metabolites of Naringin in Rat Urine and Its Application to an Excretion Study. Foods, 11(3), 316. https://doi.org/10.3390/foods11030316