

Improved LC/MS/MS Quantification Using Dual Deuterated Isomers as the Surrogates: A Case Analysis of Enrofloxacin Residue in Aquatic Products
 , ,
, , 
Abstract
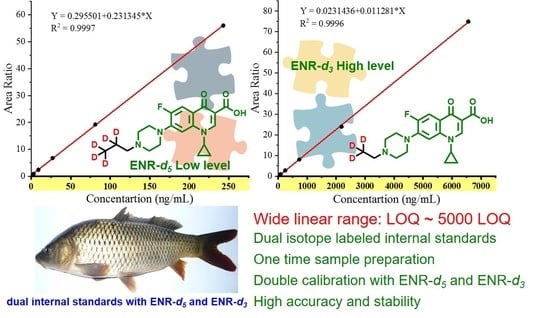
:
1. Introduction
2. Materials and Methods
2.1. Experimental Materials
2.2. Synthesis of ENR-d3
2.3. Sample Preparation
2.4. LC–MS/MS Analysis
2.5. Method Validation
3. Results and Discussion
3.1. 1HNMR Spectra of ENR-d3
3.2. Calibration Range and Deviation
3.3. Method Validation
3.4. Application of Dual Isotope Surrogates to Aquatic Products
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dai, J.; Wang, Y.; Lin, H.; Sun, Y.; Pan, Y.; Qiao, J.Q.; Lian, H.Z.; Xu, C.X. Residue screening and analysis of enrofloxacin and its metabolites in real aquatic products based on ultrahigh-performance liquid chromatography coupled with high resolution mass spectrometry. Food Chem. 2023, 404, 134757. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Bayliss, S.C.; Feil, E.J.; Liu, Y.; Wang, C.; Zhang, G.; Zhou, D.; Wei, D.; Tang, N.; Leclercq, S.O.; et al. Real time monitoring of Aeromonas salmonicida evolution in response to successive antibiotic therapies in a commercial fish farm. Environ. Microbiol. 2019, 21, 1113–1123. [Google Scholar] [CrossRef] [PubMed]
- Soto-Rodriguez, S.A.; Lozano-Olvera, R.; Abad-Rosales, S.M.; Martinez-Brown, J.M.; Ibarra-Castro, L. Susceptibility of Pacific white snook Centropomus viridis to Vibrio species. Dis. Aquat. Org. 2019, 134, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Delghandi, M.R.; El-Matbouli, M.; Menanteau-Ledouble, S. Renibacterium salmoninarum-The causative agent of bacterial kidney disease in salmonid fish. Pathogens 2020, 9, 845. [Google Scholar] [CrossRef] [PubMed]
- Danyi, S.; Widart, J.; Douny, C.; Dang, P.K.; Baiwir, D.; Wang, N.; Tu, H.T.; Tung, V.T.; Phuong, N.T.; Kestemont, P.; et al. Determination and kinetics of enrofloxacin and ciprofloxacin in Tra catfish (Pangasianodon hypophthalmus) and giant freshwater prawn (Macrobrachium rosenbergii) using a liquid chromatography/mass spectrometry method. J. Vet. Pharmacol. Ther. 2011, 34, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Flores-Miranda, B.M.; Espinosa-Plascencia, A.; Gómez-Jiménez, S.; López-Zavala, A.A.; González-Carrillo, H.H.; Bermúdez-Almada, M.d.C. Accumulation and elimination of enrofloxacin and ciprofloxacin in tissues of shrimp Litopenaeus vannamei under laboratory and farm conditions. ISRN Pharmaceutics 2012, 2012, 374212. [Google Scholar] [CrossRef] [Green Version]
- Morales-Gutiérrez, F.J.; Barbosa, J.; Barrón, D. Metabolic study of enrofloxacin and metabolic profile modifications in broiler chicken tissues after drug administration. Food Chem. 2015, 172, 30–39. [Google Scholar] [CrossRef]
- Kang, H.S.; Lee, S.B.; Shin, D.; Jeong, J.; Hong, J.H.; Rhee, G.S. Occurrence of veterinary drug residues in farmed fishery products in South Korea. Food Control 2018, 85, 57–65. [Google Scholar] [CrossRef]
- Hua, Y.Y.; Yao, Q.H.; Lin, J.; Li, X.; Yang, Y. Comprehensive survey and health risk assessment of antibiotic residues in freshwater fish in southeast China. J. Food Compos. Anal. 2022, 114, 104821. [Google Scholar] [CrossRef]
- Cao, L.; Naylor, R.; Henriksson, P.; Leadbitter, D.; Metian, M.; Troell, M.; Zhang, W.B. China’s aquaculture and the world’s wild fisheries. Science 2015, 347, 133–135. [Google Scholar] [CrossRef]
- Santos, L.; Ramos, F. Analytical strategies for the detection and quantification of antibiotic residues in aquaculture fishes: A review. Trends Food Sci. Technol. 2016, 52, 16–30. [Google Scholar] [CrossRef]
- Quesada, S.P.; Paschoal, J.A.R.; Reyes, F.G.R. Considerations on the aquaculture development and on the use of veterinary drugs: Special issue for fluoroquinolones—A review. J. Food Sci. 2013, 78, 1321–1333. [Google Scholar] [CrossRef] [PubMed]
- Griboff, J.; Carrizo, J.C.; Bonansea, R.I.; Valdés, M.E.; Wunderlin, D.A.; Amé, M.V. Multiantibiotic residues in commercial fish from Argentina. The presence of mixtures of antibiotics in edible fish, achallenge to health risk assessment. Food Chem. 2020, 332, 127380. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Zhang, R.; Zhu, L.; Deng, H.; Li, F.; Xu, L.; Huan, J.; Sun, X.; Xu, Z. Establishment of a peptide-based enzyme-linked immunosorbent assay for detecting antibodies against PRRSV M protein. BMC Vet. Res. 2021, 17, 355. [Google Scholar] [CrossRef]
- Yan, C.; Teng, J.; Liu, F.Y.; Yao, B.B.; Xu, Z.L.; Yao, L.; Chen, W. Signal amplified enzyme-linked immunosorbent assay with gold nanoparticles for sensitive detection of trace furaltadone metabolite. Microchem. J. 2020, 159, 105414. [Google Scholar] [CrossRef]
- Zhang, W.W.J.; Zheng, G.; Yin, Y.; Zhu, X.; Shan, Q.; Yang, Y.; Ma, L.; Li, L.; Liu, S. Pharmacokinetics, tissue distribution, and depletion of enrofloxacin and its metabolite ciprofloxacin in the northern snake-head (Channa argus) following multiple oral administration. Aquaculture 2021, 533, 736183. [Google Scholar] [CrossRef]
- Liu, Y.Z.G.; Sun, R.; Zhou, S.; Dong, J.; Yang, Y.; Yang, Q.; Xu, N.; Ai, X. Determination of pharmacokinetic parameters and tissue distribution characters of enrofloxacin and its metabolite ciprof-loxacin in Procambarus clarkii after two routes of administration. Aquacult. Rep. 2022, 22, 100939. [Google Scholar] [CrossRef]
- Dickson, L.C. Performance characterization of a quantitative liquid chromatography–tandem mass spectrometric method for 12 macro-lide and lincosamide antibiotics in salmon, shrimp and tilapia. J. Chromatogr. B 2014, 967, 203–210. [Google Scholar] [CrossRef]
- Monteiro, S.H.; Francisco, J.G.; Campion, T.F.; Pimpinato, R.F.; Andrade, G.C.R.M.; Garcia, F.; Tornisielo, V.L. Multiresidue antimicrobial determination in Nile tilapia (Oreochromis niloticus) cage farming by liquid chromatography tandem mass spectrometry. Aquaculture 2015, 447, 37–43. [Google Scholar] [CrossRef]
- Yu, R.; Chen, L.; Shen, R.; Li, P.; Shi, N.B. Quantification of ultratrace levels of fluoroquinolones in wastewater by molecularly imprinted solid phase extraction and liquid chromatography triple quadrupole mass. Environ. Technol. Innov. 2020, 19, 100919. [Google Scholar] [CrossRef]
- Jadhav, M.R.; Pudale, A.; Raut, P.; Utture, S.; Ahammed Shabeer, T.P.; Banerjee, K. A unified approach for high-throughput quantitative analysis of the residues of multi-class veterinary drugs and pesticides in bovine milk using LC-MS/MS and GC-MS/MS. Food Chem. 2019, 272, 292–305. [Google Scholar] [CrossRef] [PubMed]
- Hoff, R.B.M.L.; Deolindo, C.T.P.; Vargas, M.O.; Kleemann, C.R.; Daguer, H. Determination of 62 veterinary drugs in feeding stuffs by novel pressurized liquid extraction methods and LC-MS/MS. J. Chromatogr. B 2020, 1152, 122232. [Google Scholar] [CrossRef] [PubMed]
- Geddes, E.J.; Li, Z.; Hergenrother, P.J. An LC-MS/MS assay and complementary web-based tool to quantify and predict compound accumulation in E. coli. Nat. Protoc. 2021, 16, 4833–4854. [Google Scholar] [CrossRef] [PubMed]
- Wilkes, E.H.; Whitlock, M.J.; Williams, E.L. A data-driven approach for the detection of internal standard outliers in targeted LC-MS/MS assays. J. Mass Spectrom. Adv. Clin. Lab 2021, 20, 42–47. [Google Scholar] [CrossRef] [PubMed]
- Burns, D.T.; Walker, M.J. Origins of the method of standard additions and of the use of an internal standard in quantitative instrumental chemical analyses. Anal. Bioanal. Chem. 2019, 411, 2749–2753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleigrewe, K.; Niehaus, E.M.; Wiemann, P.; Tudzynski, B.; Humpf, H.U. New approach via gene knockout and single-step chemical reaction for the synthesis of isotopically labeled fusarin C as an internal standard for the analysis of this fusarium mycotoxin in food and feed samples. J. Agric. Food Chem. 2012, 60, 8350–8355. [Google Scholar] [CrossRef]
- Wang, H.X.; Ren, L.S.; Yu, X.; Hu, J.; Chen, Y.; He, G.S.; Jiang, Q.W. Antibiotic residues in meat, milk and aquatic products in Shanghai and human exposure assessment. Food Control 2017, 80, 217–225. [Google Scholar] [CrossRef]
- Chen, H.; Liu, S.; Xu, X.R.; Diao, Z.H.; Sun, K.F.; Hao, Q.W.; Liu, S.S.; Ying, G.G. Tissue distribution, bioaccumulation characteristics and health risk of antibiotics in cultured fish from a typical aquaculture area. J. Hazard. Mater. 2018, 343, 140–148. [Google Scholar] [CrossRef]
- Guidi, L.R.; Santos, F.A.; Ribeiro, A.C.; Fernandes, C.; Silva, L.H.; Gloria, M.B. A simple, fast and sensitive screening LC-ESI-MS/MS method for antibiotics in fish. Talanta 2017, 163, 85–93. [Google Scholar] [CrossRef]
- Guidi, L.R.; Santos, F.A.; Ribeiro, A.; Fernandes, C.; Silva, L.H.M.; Gloria, M.B.A. Quinolones and tetracyclines in aquaculture fish by a simple and rapid LC-MS/MS method. Food Chem. 2018, 245, 1232–1238. [Google Scholar] [CrossRef]
- Bajkacz, S.; Felis, E.; Kycia-Slocka, E.; Harnisz, M.; Korzeniewska, E. Development of a new SLE-SPE-HPLC-MS/MS method for the determination of selected antibiotics and their transformation products in anthropogenically altered solid environmental matrices. Sci. Total Environ. 2020, 726, 138071. [Google Scholar] [CrossRef] [PubMed]
- de Groot, J.C.; Fiers, M.W.; van Ham, R.C.; America, A.H. Post alignment clustering procedure for comparative quantitative proteomics LC-MS data. Proteomics 2008, 8, 32–36. [Google Scholar] [CrossRef] [PubMed]
- Gu, H.D.Z.Y.; DeMichele, M.; Zheng, N.; Zhang, Y.; Pillutla, R.; Zeng, J. Eliminating preparation of multisample external calibration curves and dilution of study samples using the multiple isotop-ologue reaction monitoring (MIRM) technique in quantitative LC-MS/MS bioanalysis. Anal. Chem. 2008, 80, 4200–4207. [Google Scholar] [CrossRef]
- Heudi, O.B.S.; Zimmer, D.; Schmidt, J.; Bill, K.; Lehmann, N.; Bauer, C.; Kretz, O. Towards absolute quantification of therapeutic monoclonal antibody in serum by LC-MS/MS using isotope-labeled anti-body standard and protein cleavage isotope dilution mass spectrometry. Anal. Chem. 2019, 91, 8652–8659. [Google Scholar] [CrossRef]
- Long, T.E.; Keding, L.C.; Lewis, D.D.; Anstead, M.I.; Withers, T.R.; Yu, H.D. Anionic fluoroquinolones as antibacterials against biofilm-producing Pseudomonas aeruginosa. Bioorg. Med. Chem. Lett. 2016, 26, 1305–1309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Svahn, O.B.E. Thermal stability assessment of antibiotics in moderate temperature and subcritical water using a pressurized dynamic flow-through system. Int. J. Innov. Appl. Stud. 2015, 11, 872–880. [Google Scholar]
- Hermo, M.P.N.E.; Kır, S.; BarrÓn, D.; Barbosa, J. Improved determination of quinolones in milk at their MRL levels using LC–UV, LC–FD, LC–MS and LC–MS/MS and validation in line with regulation 2002/657/EC. Anal. Chim. Acta 2008, 613, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.H.; Cho, K.; Kim, S.; Jeon, S.H.; Shin, J.H.; Lee, J.; Ahn, Y.G. Inter-laboratory validation of method to determine residual enrofloxacin in chicken meat. Int. J. Anal. Chem. 2018, 2018, 6019549. [Google Scholar] [CrossRef]
- No 1077-1-2008; Announcement of the Ministry of Agriculture of the People’s Republic of China. Simultaneous determination of 17 sulfonamides and 15 quinolones residues in aquatic by LC-MS/MS method. Ministry of Agriculture of the People’s Republic of China: Beijing, China, 2008.
- Vogeser, M.; Seger, C. Pitfalls associated with the use of liquid chromatography-tandem mass spectrometry in the clinical laboratory. Clin. Chem. 2010, 56, 1234–1244. [Google Scholar] [CrossRef] [Green Version]
- Samanidou, V.; Evaggelopoulou, E.; Trotzmuller, M.; Guo, X.; Lankmayr, E. Multi-residue determination of seven quinolones antibiotics in gilthead seabream using liquid chromatography-tandem mass spectrometry. J. Chromatogr. A 2008, 1203, 115–123. [Google Scholar] [CrossRef]
- Zhang, X.; Fang, C.; Huang, D.; Yang, G.; Tang, Y.; Shi, Y.; Kong, C.; Cao, P.; Cai, Y. Determination of 8 biogenic amines in aquatic products and their derived products by high-performance liquid chromatog-raphy-tandem mass spectrometry without derivatization. Food Chem. 2021, 361, 130044. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Precursor Ion (m/z) and Adduct | Product Ions (m/z) and Theoretical Fragments | CE (eV) | Calibration Curves | Linear Range (ng/mL) | r2 |
|---|---|---|---|---|---|---|
| ENR | 360 ([M + H]+) | 316 * (C18H23FN3O+) | 19 | y = 0.295501 + 0.231345 *× (ENR-d5) | 1–243 | 0.9997 |
| y = 0.0231436 + 0.011281 *× (ENR-d3) | 27−6561 | 0.9996 | ||||
| 245 (C14H14FN2O+) | 26 | − | − | − | ||
| ENR-d5 | 365 ([M + H]+) | 321 (C18H17D5FN3O+) | 19 | − | − | − |
| ENR-d3 | 363 ([M + H]+) | 319 (C18H20D3FN3O+) | 19 | − | − | − |
| Spike Amount (µg/kg) | ENR-d5 as Internal Surrogate | ENR-d3 as Internal Surrogate | ||||||
|---|---|---|---|---|---|---|---|---|
| Detected in Blank (µg/kg) | Detected After Spiking (µg/kg) | Recovery (%) | RSD (%) | Detected in Blank (µg/kg) | Detected After Spiking (µg/kg) | Recovery (%) | RSD (%) | |
| 10,000 | 58.7 | 7632 | 75.7 | 12.3 | 64.1 | 10,207 | 101 | 3.81 |
| Calibration Method | Instrument | Linear Range (μg/L) | LOQ (μg/kg) | Stability |
|---|---|---|---|---|
| Internal calibration | LC–MS/MS | 0.2–100 | 1.0 | 6.7% (spiked at 2 μg/kg) [9] |
| External calibration | LC–MS/MS | 12.5–75.0 | 12.5 | 9.2% (spiked at 25 μg/kg) [29] |
| External calibration | LC–MS/MS | 0.5–600 | 0.5 | 3% (spike amount not mentioned) [8] |
| Internal calibration | LC-Q-Orbitrap MS | 5–500 | 1.0 | 12.8% (spiked at 1 μg/kg) [1] |
| Internal calibration | LC–MS/MS | 1–6561 | 2.0 | 2.14% (spiked at 2 μg/kg) (this study) |
| Sample Type | ENR-d5 as Isotope Surrogate | ENR-d3 as Isotope Surrogate | ||
|---|---|---|---|---|
| Concentration ± SD (μg/kg) | RSD (%) | Concentration ± SD (μg/kg) | RSD (%) | |
| Bluntnose black bream | 108 ± 7.25 | 6.72 | 99.1 ± 0.173 | 0.175 |
| Common carp | 681 ± 35.7 | 5.24 | 624 ± 4.95 | 0.794 |
| Bullfrog | 3903 ± 433 | 11.1 | 4340 ± 21.2 | 0.489 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tang, Y.; Yang, G.; Fodjo, E.K.; Wang, S.; Zhai, W.; Si, W.; Xia, L.; Kong, C. Improved LC/MS/MS Quantification Using Dual Deuterated Isomers as the Surrogates: A Case Analysis of Enrofloxacin Residue in Aquatic Products. Foods 2023, 12, 224. https://doi.org/10.3390/foods12010224
Tang Y, Yang G, Fodjo EK, Wang S, Zhai W, Si W, Xia L, Kong C. Improved LC/MS/MS Quantification Using Dual Deuterated Isomers as the Surrogates: A Case Analysis of Enrofloxacin Residue in Aquatic Products. Foods. 2023; 12(1):224. https://doi.org/10.3390/foods12010224
Chicago/Turabian StyleTang, Yunyu, Guangxin Yang, Essy Kouadio Fodjo, Shouying Wang, Wenlei Zhai, Wenshuai Si, Lian Xia, and Cong Kong. 2023. "Improved LC/MS/MS Quantification Using Dual Deuterated Isomers as the Surrogates: A Case Analysis of Enrofloxacin Residue in Aquatic Products" Foods 12, no. 1: 224. https://doi.org/10.3390/foods12010224
APA StyleTang, Y., Yang, G., Fodjo, E. K., Wang, S., Zhai, W., Si, W., Xia, L., & Kong, C. (2023). Improved LC/MS/MS Quantification Using Dual Deuterated Isomers as the Surrogates: A Case Analysis of Enrofloxacin Residue in Aquatic Products. Foods, 12(1), 224. https://doi.org/10.3390/foods12010224