
Seasonality of Aerosol Sources Calls for Distinct Air Quality Mitigation Strategies


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sampling Site
2.2. Instrument
2.3. Complementary Data
2.4. Source Apportionment of Organic Aerosol
3. Results
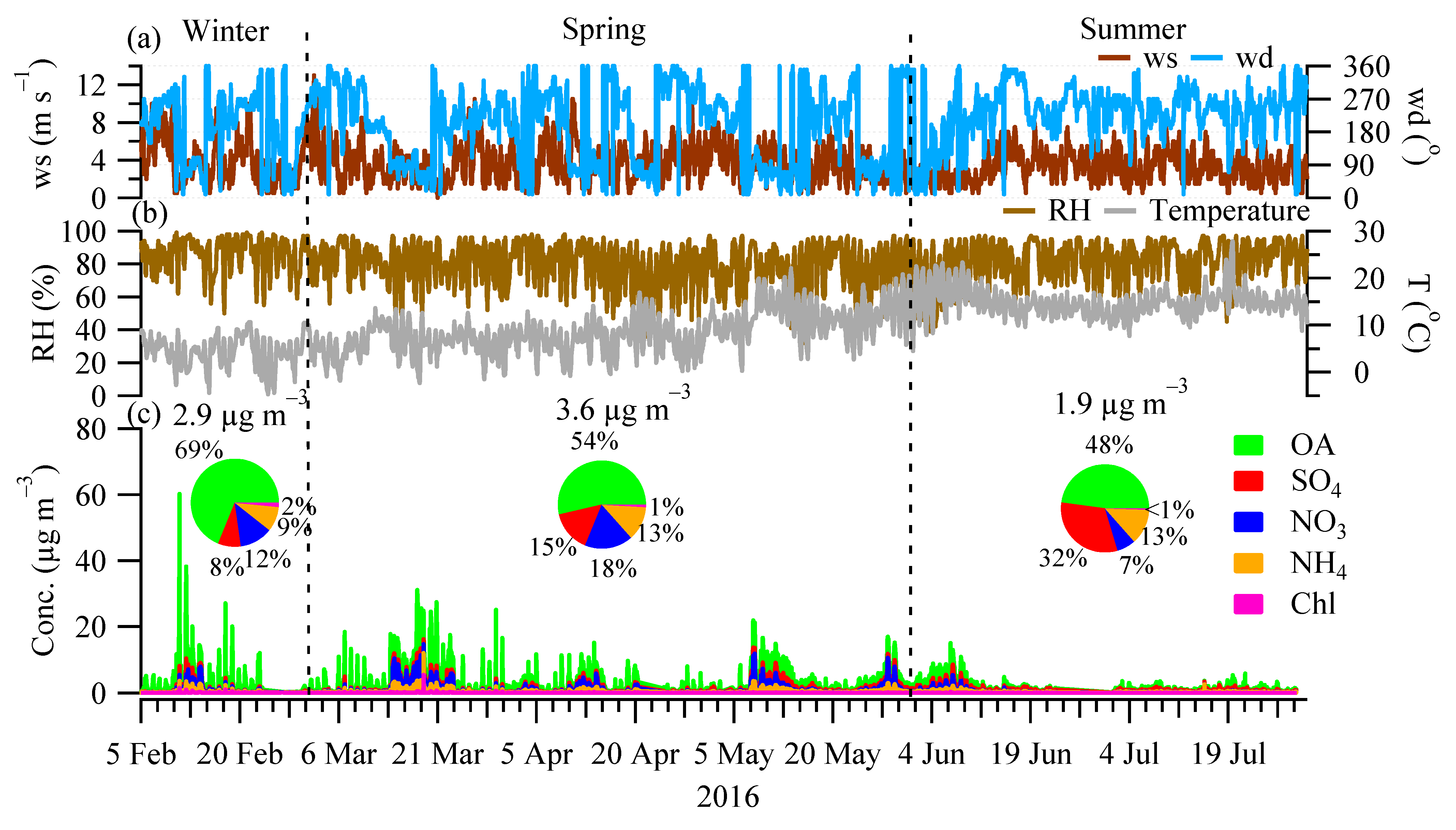
3.1. Chemical Composition and Seasonal Variations
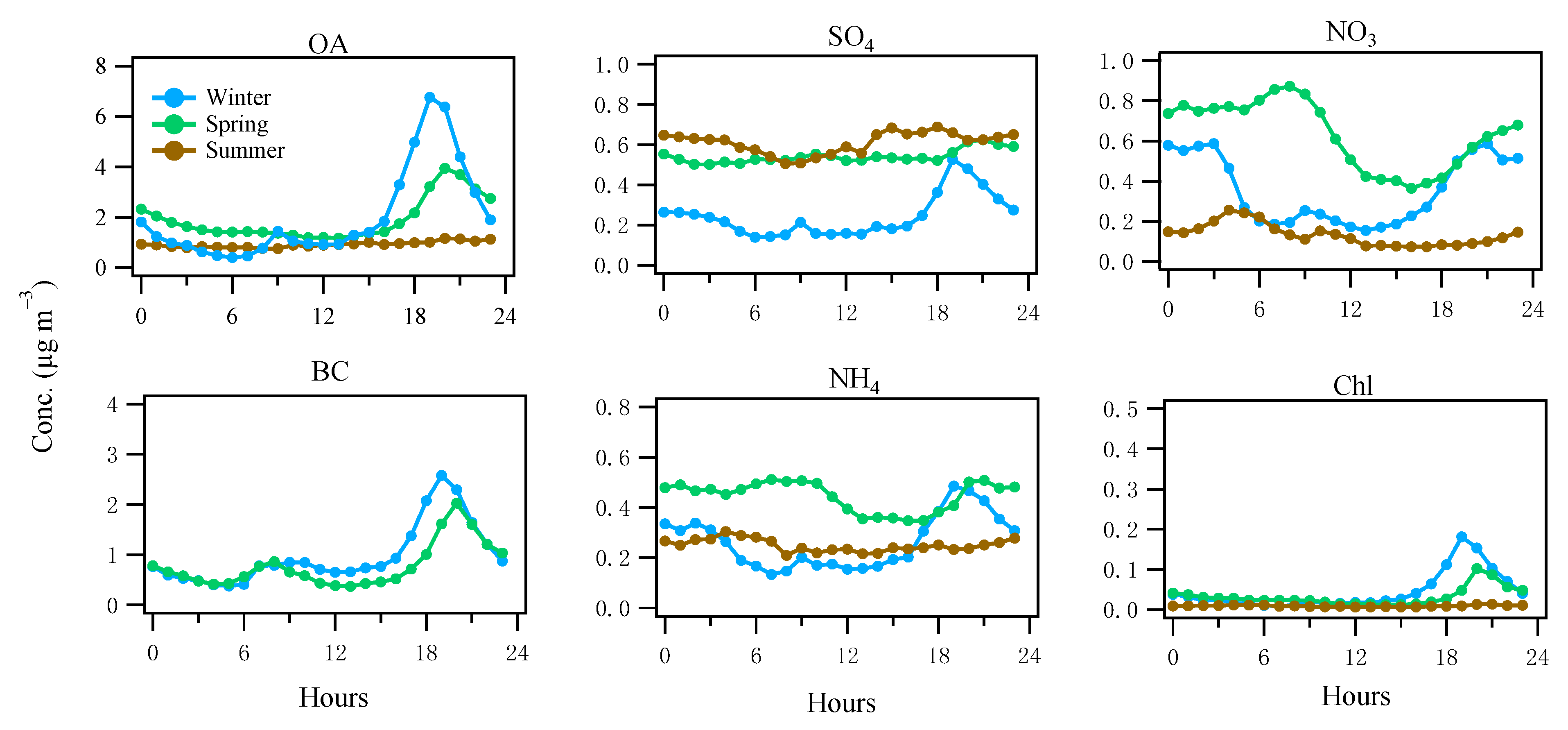
3.2. Diurnal Patterns of the Major Components of NR-PM1
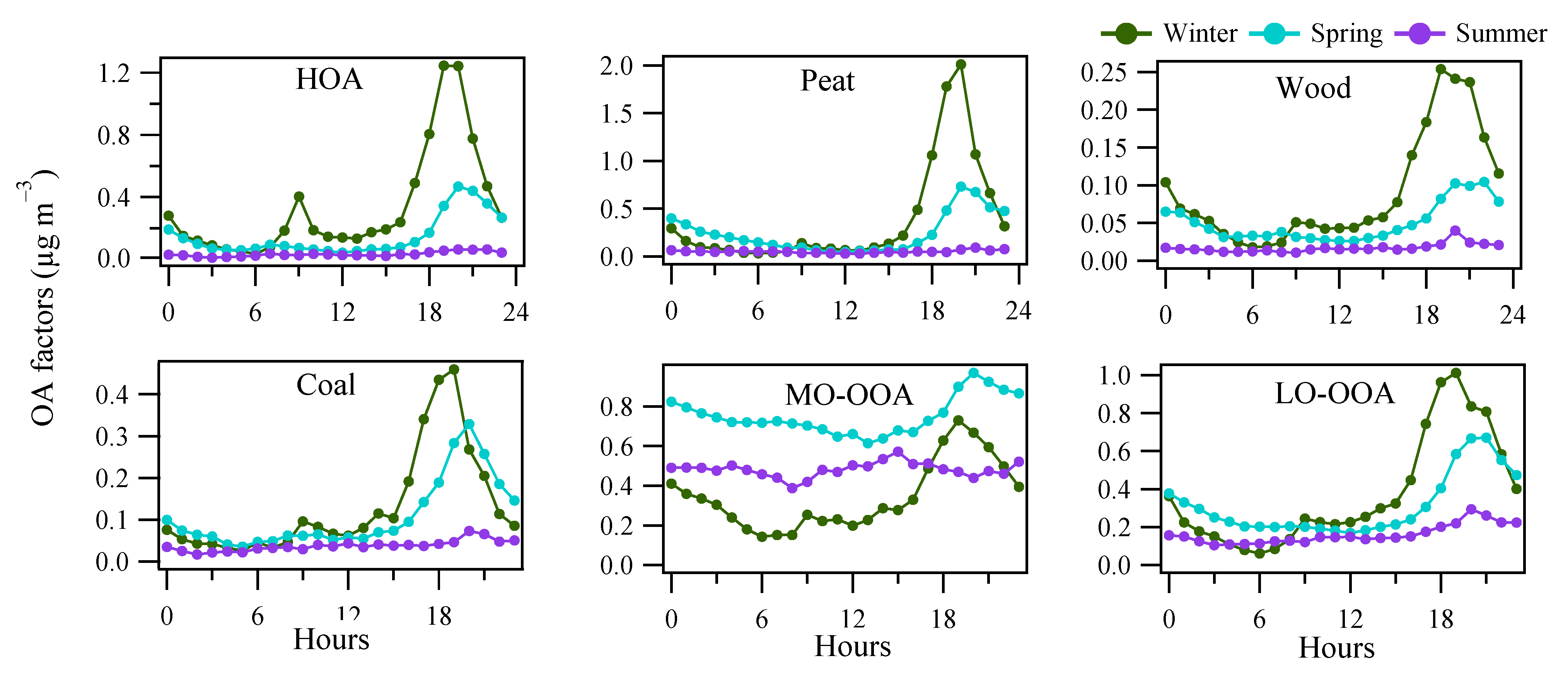
3.3. OA Source Apportionment
3.3.1. POA Factors
3.3.2. OOA Factors
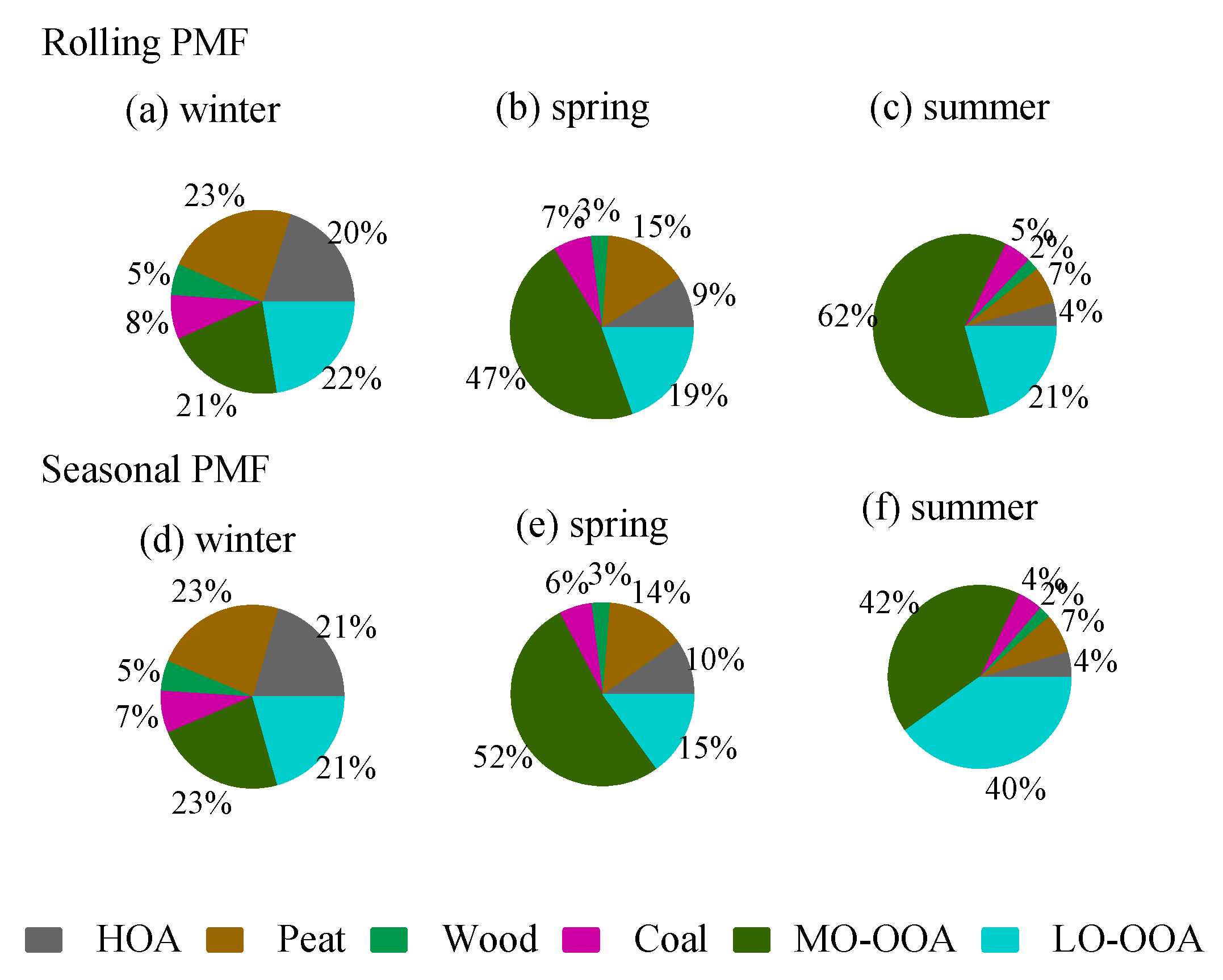
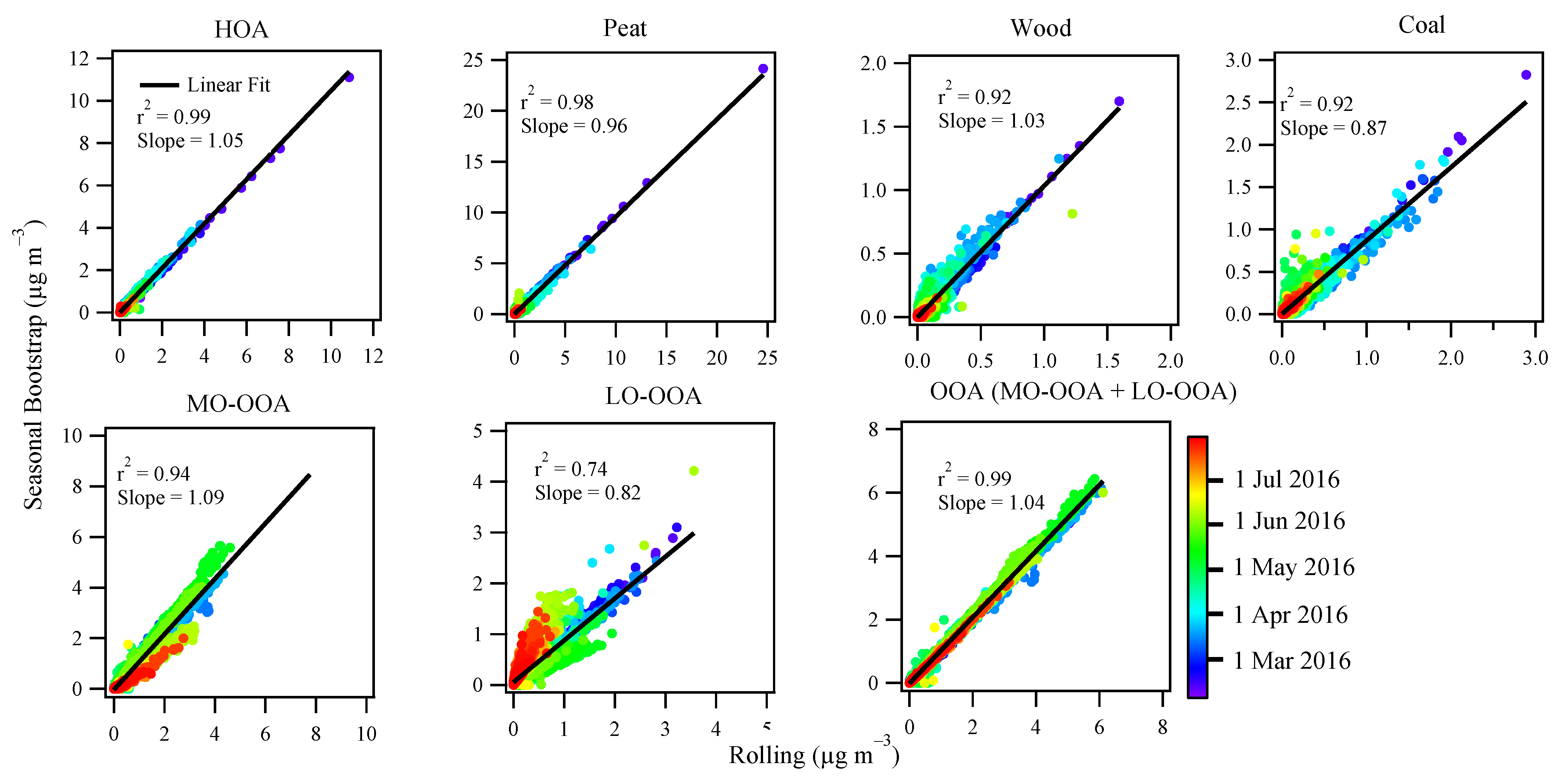
3.3.3. Comparison between Rolling and Seasonal PMF
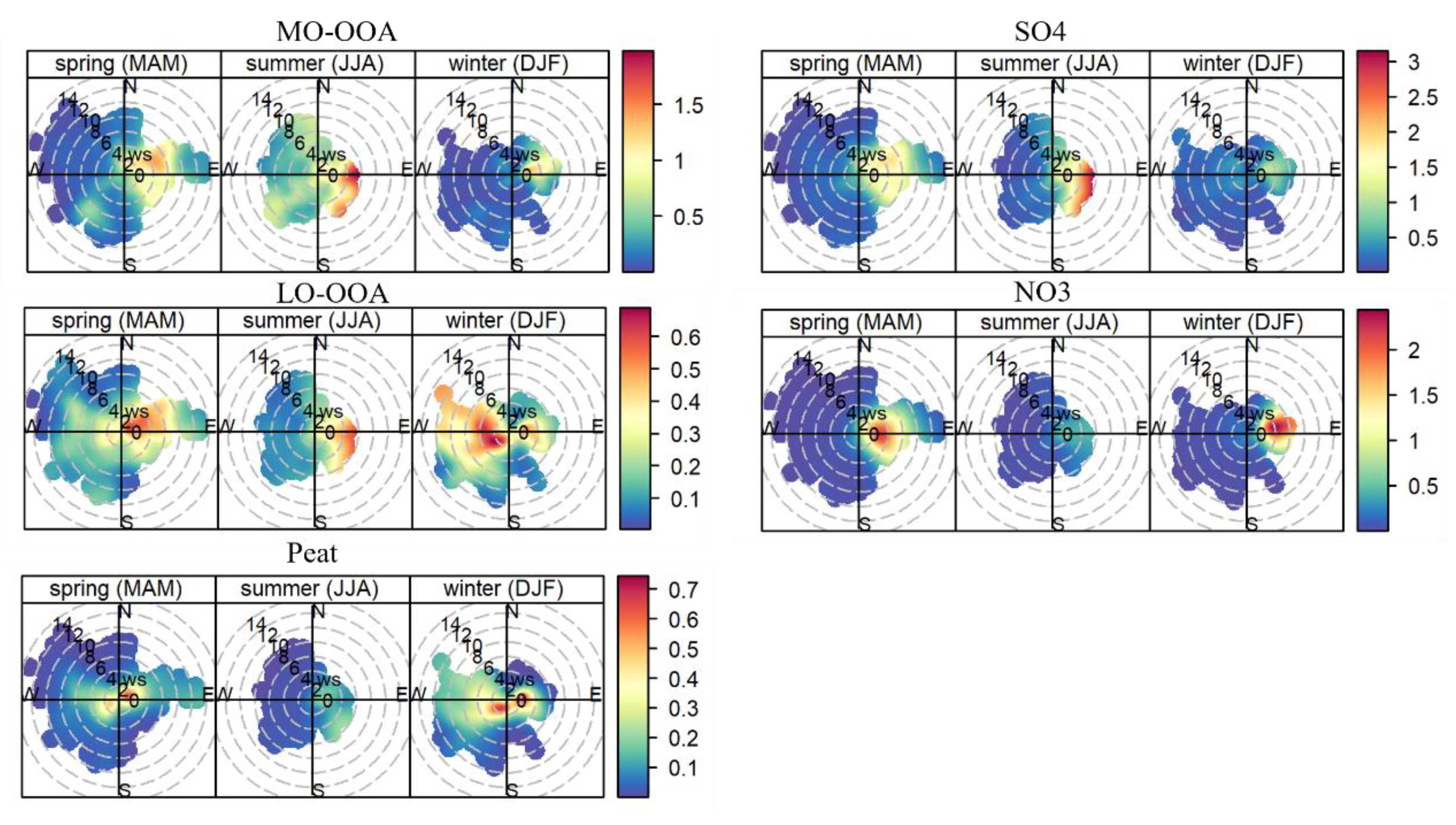
3.4. Spatial Origins of Primary and Secondary Aerosols: Local vs. Regional

4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tegen, I.; Lacis, A.A.; Fung, I. The influence on climate forcing of mineral aerosols from disturbed soils. Nature 1996, 380, 419–422. [Google Scholar] [CrossRef]
- Huang, R.-J.; Zhang, Y.; Bozzetti, C.; Ho, K.-F.; Cao, J.-J.; Han, Y.; Daellenbach, K.R.; Slowik, J.G.; Platt, S.M.; Canonaco, F. High secondary aerosol contribution to particulate pollution during haze events in China. Nature 2014, 514, 218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, X.; Tie, X.; Wu, D.; Zhou, X.; Bi, X.; Tan, H.; Li, F.; Jiang, C. Long-term trend of visibility and its characterizations in the Pearl River Delta (PRD) region, China. Atmos. Environ. 2008, 42, 1424–1435. [Google Scholar] [CrossRef]
- Pope, C.A., III; Burnett, R.T.; Thun, M.J.; Calle, E.E.; Krewski, D.; Ito, K.; Thurston, G.D. Lung cancer, cardiopulmonary mortality, and long-term exposure to fine particulate air pollution. JAMA 2002, 287, 1132–1141. [Google Scholar] [CrossRef] [Green Version]
- Cohen, A.J.; Brauer, M.; Burnett, R.; Anderson, H.R.; Frostad, J.; Estep, K.; Balakrishnan, K.; Brunekreef, B.; Dandona, L.; Dandona, R.; et al. Estimates and 25-year trends of the global burden of disease attributable to ambient air pollution: An analysis of data from the Global Burden of Diseases Study 2015. Lancet 2017, 389, 1907–1918. [Google Scholar] [CrossRef] [Green Version]
- Burnett, R.; Chen, H.; Szyszkowicz, M.; Fann, N.; Hubbell, B.; Pope, C.A.; Apte, J.S.; Brauer, M.; Cohen, A.; Weichenthal, S.; et al. Global estimates of mortality associated with long-term exposure to outdoor fine particulate matter. Proc. Natl. Acad. Sci. USA 2018, 115, 9592–9597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravina, M.; Panepinto, D.; Zanetti, M. Air Quality Planning and the Minimization of Negative Externalities. Resources 2019, 8, 15. [Google Scholar] [CrossRef] [Green Version]
- Decesari, S.; Sowlat, M.H.; Hasheminassab, S.; Sandrini, S.; Gilardoni, S.; Facchini, M.C.; Fuzzi, S.; Sioutas, C. Enhanced toxicity of aerosol in fog conditions in the Po Valley, Italy. Atmos. Chem. Phys. 2017, 17, 7721–7731. [Google Scholar] [CrossRef] [Green Version]
- Daellenbach, K.R.; Uzu, G.; Jiang, J.; Cassagnes, L.-E.; Leni, Z.; Vlachou, A.; Stefenelli, G.; Canonaco, F.; Weber, S.; Segers, A.; et al. Sources of particulate-matter air pollution and its oxidative potential in Europe. Nature 2020, 587, 414–419. [Google Scholar] [CrossRef]
- WHO. Air Quality Guidelines: Global Update 2005; World Health Organization: Geneva, Switzerland, 2006. [Google Scholar]
- Canagaratna, M.R.; Jayne, J.T.; Jimenez, J.L.; Allan, J.D.; Alfarra, M.R.; Zhang, Q.; Onasch, T.B.; Drewnick, F.; Coe, H.; Middlebrook, A.; et al. Chemical and microphysical characterization of ambient aerosols with the Aerodyne aerosol mass spectrometer. Mass Spectrom. Rev. 2007, 26, 185–222. [Google Scholar] [CrossRef]
- Drewnick, F.; Hings, S.S.; DeCarlo, P.; Jayne, J.T.; Gonin, M.; Fuhrer, K.; Weimer, S.; Jimenez, J.L.; Demerjian, K.L.; Borrmann, S.; et al. A new time-of-flight aerosol mass spectrometer (TOF-AMS)—Instrument description and first field deployment. Aerosol Sci. Technol. 2005, 39, 637–658. [Google Scholar] [CrossRef]
- DeCarlo, P.F.; Kimmel, J.R.; Trimborn, A.; Northway, M.J.; Jayne, J.T.; Aiken, A.C.; Gonin, M.; Fuhrer, K.; Horvath, T.; Docherty, K.S.; et al. Field-Deployable, High-Resolution, Time-of-Flight Aerosol Mass Spectrometer. Anal. Chem. 2006, 78, 8281–8289. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, J.L.; Canagaratna, M.R.; Donahue, N.M.; Prevot, A.S.H.; Zhang, Q.; Kroll, J.H.; DeCarlo, P.F.; Allan, J.D.; Coe, H.; Ng, N.L.; et al. Evolution of Organic Aerosols in the Atmosphere. Science 2009, 326, 1525–1529. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Zheng, H.; Li, Q.; Jin, L.; Lyu, R.; Ding, X.; Huo, Y.; Zhao, B.; Jiang, J.; Chen, J.; et al. Toxic potency-adjusted control of air pollution for solid fuel combustion. Nat. Energy 2022, 7, 194–202. [Google Scholar] [CrossRef]
- Thorpe, A.; Harrison, R.M. Sources and properties of non-exhaust particulate matter from road traffic: A review. Sci. Total Environ. 2008, 400, 270–282. [Google Scholar] [CrossRef]
- Lin, C.; Ceburnis, D.; Hellebust, S.; Buckley, P.; Wenger, J.; Canonaco, F.; Prévôt, A.S.H.; Huang, R.-J.; O’Dowd, C.; Ovadnevaite, J. Characterization of primary organic aerosol from domestic wood, peat, and coal burning in Ireland. Environ. Sci. Technol. 2017, 51, 10624–10632. [Google Scholar] [CrossRef]
- Parworth, C.; Fast, J.; Mei, F.; Shippert, T.; Sivaraman, C.; Tilp, A.; Watson, T.; Zhang, Q. Long-term measurements of submicrometer aerosol chemistry at the Southern Great Plains (SGP) using an Aerosol Chemical Speciation Monitor (ACSM). Atmos. Environ. 2015, 106, 43–55. [Google Scholar] [CrossRef] [Green Version]
- Petit, J.E.; Favez, O.; Sciare, J.; Crenn, V.; Sarda-Estève, R.; Bonnaire, N.; Močnik, G.; Dupont, J.C.; Haeffelin, M.; Leoz-Garziandia, E. Two years of near real-time chemical composition of submicron aerosols in the region of Paris using an Aerosol Chemical Speciation Monitor (ACSM) and a multi-wavelength Aethalometer. Atmos. Chem. Phys. 2015, 15, 2985–3005. [Google Scholar] [CrossRef] [Green Version]
- Ripoll, A.; Minguillón, M.C.; Pey, J.; Jimenez, J.L.; Day, D.A.; Sosedova, Y.; Canonaco, F.; Prévôt, A.S.H.; Querol, X.; Alastuey, A. Long-term real-time chemical characterization of submicron aerosols at Montsec (southern Pyrenees, 1570 m a.s.l.). Atmos. Chem. Phys. 2015, 15, 2935–2951. [Google Scholar] [CrossRef] [Green Version]
- Tiitta, P.; Vakkari, V.; Croteau, P.; Beukes, J.P.; Van Zyl, P.G.; Josipovic, M.; Venter, A.D.; Jaars, K.; Pienaar, J.J.; Ng, N.L.; et al. Chemical composition, main sources and temporal variability of PM1 aerosols in southern African grassland. Atmos. Chem. Phys. 2014, 14, 1909–1927. [Google Scholar] [CrossRef] [Green Version]
- Ng, N.L.; Herndon, S.C.; Trimborn, A.; Canagaratna, M.R.; Croteau, P.L.; Onasch, T.B.; Sueper, D.; Worsnop, D.R.; Zhang, Q.; Sun, Y.L.; et al. An Aerosol Chemical Speciation Monitor (ACSM) for routine monitoring of the composition and mass concentrations of ambient aerosol. Aerosol Sci. Technol. 2011, 45, 780–794. [Google Scholar] [CrossRef]
- Paatero, P.; Tapper, U. Positive matrix factorization: A non-negative factor model with optimal utilization of error estimates of data values. Environmetrics 1994, 5, 111–126. [Google Scholar] [CrossRef]
- Canonaco, F.; Crippa, M.; Slowik, J.G.; Baltensperger, U.; Prévôt, A.S.H. SoFi, an IGOR-based interface for the efficient use of the generalized multilinear engine (ME-2) for the source apportionment: ME-2 application to aerosol mass spectrometer data. Atmos. Meas. Technol. 2013, 6, 3649–3661. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Wang, Z.; Dong, H.; Yang, T.; Li, J.; Pan, X.; Chen, P.; Jayne, J.T. Characterization of summer organic and inorganic aerosols in Beijing, China with an Aerosol Chemical Speciation Monitor. Atmos. Environ. 2012, 51, 250–259. [Google Scholar] [CrossRef]
- Via, M.; Minguillón, M.C.; Reche, C.; Querol, X.; Alastuey, A. Increase in secondary organic aerosol in an urban environment. Atmos. Chem. Phys. 2021, 21, 8323–8339. [Google Scholar] [CrossRef]
- Chen, G.; Sosedova, Y.; Canonaco, F.; Fröhlich, R.; Tobler, A.; Vlachou, A.; Daellenbach, K.R.; Bozzetti, C.; Hueglin, C.; Graf, P.; et al. Time-dependent source apportionment of submicron organic aerosol for a rural site in an alpine valley using a rolling positive matrix factorisation (PMF) window. Atmos. Chem. Phys. 2021, 21, 15081–15101. [Google Scholar] [CrossRef]
- Lin, C.; Ceburnis, D.; Huang, R.-J.; Canonaco, F.; Prévôt, A.S.H.; O’Dowd, C.; Ovadnevaite, J. Summertime Aerosol over the West of Ireland Dominated by Secondary Aerosol during Long-Range Transport. Atmosphere 2019, 10, 59. [Google Scholar] [CrossRef] [Green Version]
- Canonaco, F.; Tobler, A.; Chen, G.; Sosedova, Y.; Slowik, J.G.; Bozzetti, C.; Daellenbach, K.R.; El Haddad, I.; Crippa, M.; Huang, R.J.; et al. A new method for long-term source apportionment with time-dependent factor profiles and uncertainty assessment using SoFi Pro: Application to 1 year of organic aerosol data. Atmos. Meas. Technol. 2021, 14, 923–943. [Google Scholar] [CrossRef]
- Allan, J.D.; Delia, A.E.; Coe, H.; Bower, K.N.; Alfarra, M.R.; Jimenez, J.L.; Middlebrook, A.M.; Drewnick, F.; Onasch, T.B.; Canagaratna, M.R.; et al. A generalised method for the extraction of chemically resolved mass spectra from Aerodyne aerosol mass spectrometer data. J. Aerosol Sci. 2004, 35, 909–922. [Google Scholar] [CrossRef]
- Drinovec, L.; Močnik, G.; Zotter, P.; Prévôt, A.S.H.; Ruckstuhl, C.; Coz, E.; Rupakheti, M.; Sciare, J.; Müller, T.; Wiedensohler, A.; et al. The “dual-spot” Aethalometer: An improved measurement of aerosol black carbon with real-time loading compensation. Atmos. Meas. Technol. 2015, 8, 1965–1979. [Google Scholar] [CrossRef] [Green Version]
- Paatero, P. The multilinear engine-a table-driven, least squares program for solving multilinear problems, including the n-way parallel factor analysis model. J. Comput. Graph. Stat. 1999, 8, 854–888. [Google Scholar]
- Kourtchev, I.; Hellebust, S.; Bell, J.M.; O’Connor, I.P.; Healy, R.M.; Allanic, A.; Healy, D.; Wenger, J.C.; Sodeau, J.R. The use of polar organic compounds to estimate the contribution of domestic solid fuel combustion and biogenic sources to ambient levels of organic carbon and PM2.5 in Cork Harbour, Ireland. Sci. Total Environ. 2011, 409, 2143–2155. [Google Scholar] [CrossRef]
- Dall’Osto, M.; Ovadnevaite, J.; Ceburnis, D.; Martin, D.; Healy, R.M.; O’Connor, I.P.; Kourtchev, I.; Sodeau, J.R.; Wenger, J.C.; O’Dowd, C. Characterization of urban aerosol in Cork city (Ireland) using aerosol mass spectrometry. Atmos. Chem. Phys. 2013, 13, 4997–5015. [Google Scholar] [CrossRef] [Green Version]
- CSO (Central Statistics Office). Private Households in Permanent Housing Units. 2016. Available online: https://www.cso.ie/px/pxeirestat/Statire/SelectVarVal/Define.asp?maintable=E4015&PLanguage=0 (accessed on 1 September 2021).
- Crippa, M.; Decarlo, P.F.; Slowik, J.G.; Mohr, C.; Heringa, M.F.; Chirico, R.; Poulain, L.; Freutel, F.; Sciare, J.; Cozic, J.; et al. Wintertime aerosol chemical composition and source apportionment of the organic fraction in the metropolitan area of Paris. Atmos. Chem. Phys. 2013, 13, 961–981. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.; Ceburnis, D.; Trubetskaya, A.; Xu, W.; Smith, W.; Hellebust, S.; Wenger, J.; O’Dowd, C.; Ovadnevaite, J. On the use of reference mass spectra for reducing uncertainty in source apportionment of solid-fuel burning in ambient organic aerosol. Atmos. Meas. Technol. 2021, 14, 6905–6916. [Google Scholar] [CrossRef]
- Cubison, M.J.; Ortega, A.M.; Hayes, P.L.; Farmer, D.K.; Day, D.; Lechner, M.J.; Brune, W.H.; Apel, E.; Diskin, G.S.; Fisher, J.A.; et al. Effects of aging on organic aerosol from open biomass burning smoke in aircraft and laboratory studies. Atmos. Chem. Phys. 2011, 11, 12049–12064. [Google Scholar] [CrossRef] [Green Version]
- Sandradewi, J.; Prévôt, A.S.H.; Szidat, S.; Perron, N.; Alfarra, M.R.; Lanz, V.A.; Weingartner, E.; Baltensperger, U. Using aerosol light absorption measurements for the quantitative determination of wood burning and traffic emission contributions to particulate matter. Environ. Sci. Technol. 2008, 42, 3316–3323. [Google Scholar] [CrossRef]
- Lin, C.; Ceburnis, D.; Xu, W.; Heffernan, E.; Hellebust, S.; Gallagher, J.; Huang, R.J.; O’Dowd, C.; Ovadnevaite, J. The impact of traffic on air quality in Ireland: Insights from the simultaneous kerbside and suburban monitoring of submicron aerosols. Atmos. Chem. Phys. 2020, 20, 10513–10529. [Google Scholar] [CrossRef]
- Alfarra, M.R.; Prevot, A.S.H.; Szidat, S.; Sandradewi, J.; Weimer, S.; Lanz, V.A.; Schreiber, D.; Mohr, M.; Baltensperger, U. Identification of the mass spectral signature of organic aerosols from wood burning emissions. Environ. Sci. Technol. 2007, 41, 5770–5777. [Google Scholar] [CrossRef]
- Ng, N.L.; Canagaratna, M.R.; Jimenez, J.L.; Chhabra, P.S.; Seinfeld, J.H.; Worsnop, D.R. Changes in organic aerosol composition with aging inferred from aerosol mass spectra. Atmos. Chem. Phys. 2011, 11, 6465–6474. [Google Scholar] [CrossRef] [Green Version]
- Tiitta, P.; Leskinen, A.; Hao, L.; Yli-Pirilä, P.; Kortelainen, M.; Grigonyte, J.; Tissari, J.; Lamberg, H.; Hartikainen, A.; Kuuspalo, K.; et al. Transformation of logwood combustion emissions in a smog chamber: Formation of secondary organic aerosol and changes in the primary organic aerosol upon daytime and nighttime aging. Atmos. Chem. Phys. 2016, 16, 13251–13269. [Google Scholar] [CrossRef] [Green Version]
- Ovadnevaite, J.; Ceburnis, D.; Leinert, S.; Dall’Osto, M.; Canagaratna, M.; O’Doherty, S.; Berresheim, H.; O’Dowd, C. Submicron NE Atlantic marine aerosol chemical composition and abundance: Seasonal trends and air mass categorization. J. Geophys. Res. Atmos. 2014, 119, 11850–11863. [Google Scholar] [CrossRef] [Green Version]
- Carslaw, D.C.; Ropkins, K. openair - An R package for air quality data analysis. Environ. Model. Soft. 2012, 27–28, 52–61. [Google Scholar] [CrossRef]
- Trubetskaya, A.; Lin, C.; Ovadnevaite, J.; Ceburnis, D.; O’Dowd, C.; Leahy, J.J.; Monaghan, R.F.D.; Johnson, R.; Layden, P.; Smith, W. Study of Emissions from Domestic Solid-Fuel Stove Combustion in Ireland. Energy Fuels 2021, 35, 4966–4978. [Google Scholar] [CrossRef]
- Lin, C.; Huang, R.-J.; Ceburnis, D.; Buckley, P.; Preissler, J.; Wenger, J.; Rinaldi, M.; Facchini, M.C.; O’Dowd, C.; Ovadnevaite, J. Extreme air pollution from residential solid fuel burning. Nat. Sustain. 2018, 1, 512–517. [Google Scholar] [CrossRef]
- Ng, N.L.; Brown, S.S.; Archibald, A.T.; Atlas, E.; Cohen, R.C.; Crowley, J.N.; Day, D.A.; Donahue, N.M.; Fry, J.L.; Fuchs, H.; et al. Nitrate radicals and biogenic volatile organic compounds: Oxidation, mechanisms, and organic aerosol. Atmos. Chem. Phys. 2017, 17, 2103–2162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, S.; Guenther, A.; Faiola, C. Effects of Anthropogenic and Biogenic Volatile Organic Compounds on Los Angeles Air Quality. Environ. Sci. Technol. 2021, 55, 12191–12201. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, C.; Ceburnis, D.; O’Dowd, C.; Ovadnevaite, J. Seasonality of Aerosol Sources Calls for Distinct Air Quality Mitigation Strategies. Toxics 2022, 10, 121. https://doi.org/10.3390/toxics10030121
Lin C, Ceburnis D, O’Dowd C, Ovadnevaite J. Seasonality of Aerosol Sources Calls for Distinct Air Quality Mitigation Strategies. Toxics. 2022; 10(3):121. https://doi.org/10.3390/toxics10030121
Chicago/Turabian StyleLin, Chunshui, Darius Ceburnis, Colin O’Dowd, and Jurgita Ovadnevaite. 2022. "Seasonality of Aerosol Sources Calls for Distinct Air Quality Mitigation Strategies" Toxics 10, no. 3: 121. https://doi.org/10.3390/toxics10030121
APA StyleLin, C., Ceburnis, D., O’Dowd, C., & Ovadnevaite, J. (2022). Seasonality of Aerosol Sources Calls for Distinct Air Quality Mitigation Strategies. Toxics, 10(3), 121. https://doi.org/10.3390/toxics10030121