

PM2.5 Extracts Induce INFγ-Independent Activation of CIITA, MHCII, and Increases Inflammation in Human Bronchial Epithelium

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
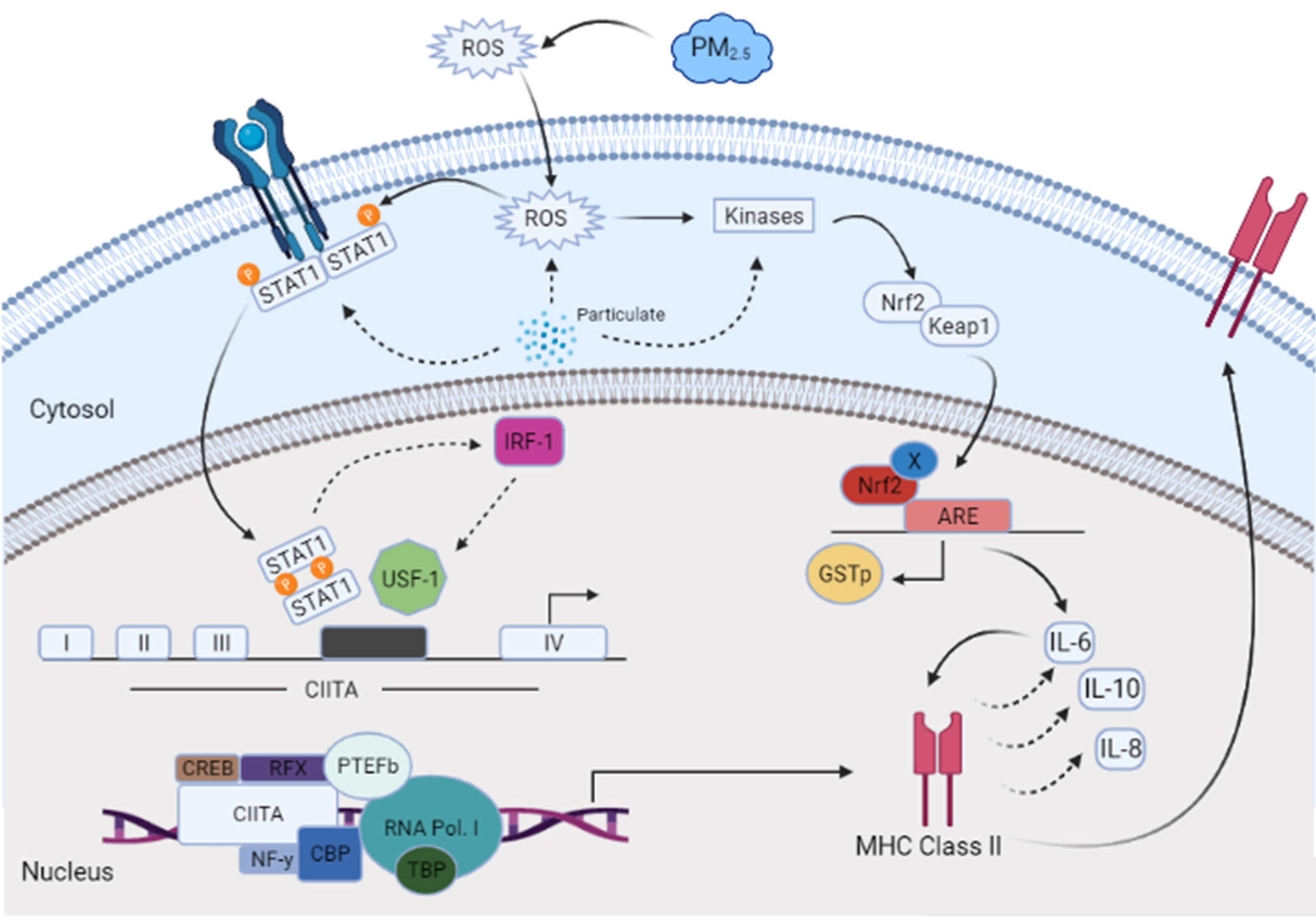
:1. Introduction
2. Materials and Methods
2.1. Particulate Matter Collection
2.2. Particulate Matter Extraction
2.3. Cell Culture and Exposure to PM2.5 Extract and Deferoxamine
2.4. Cell viability (Cytotoxicity)
2.5. Gene Expression
2.6. Cytokine Assay
2.7. STAT1 Phosphorylation
2.8. Statistical Analysis
3. Results
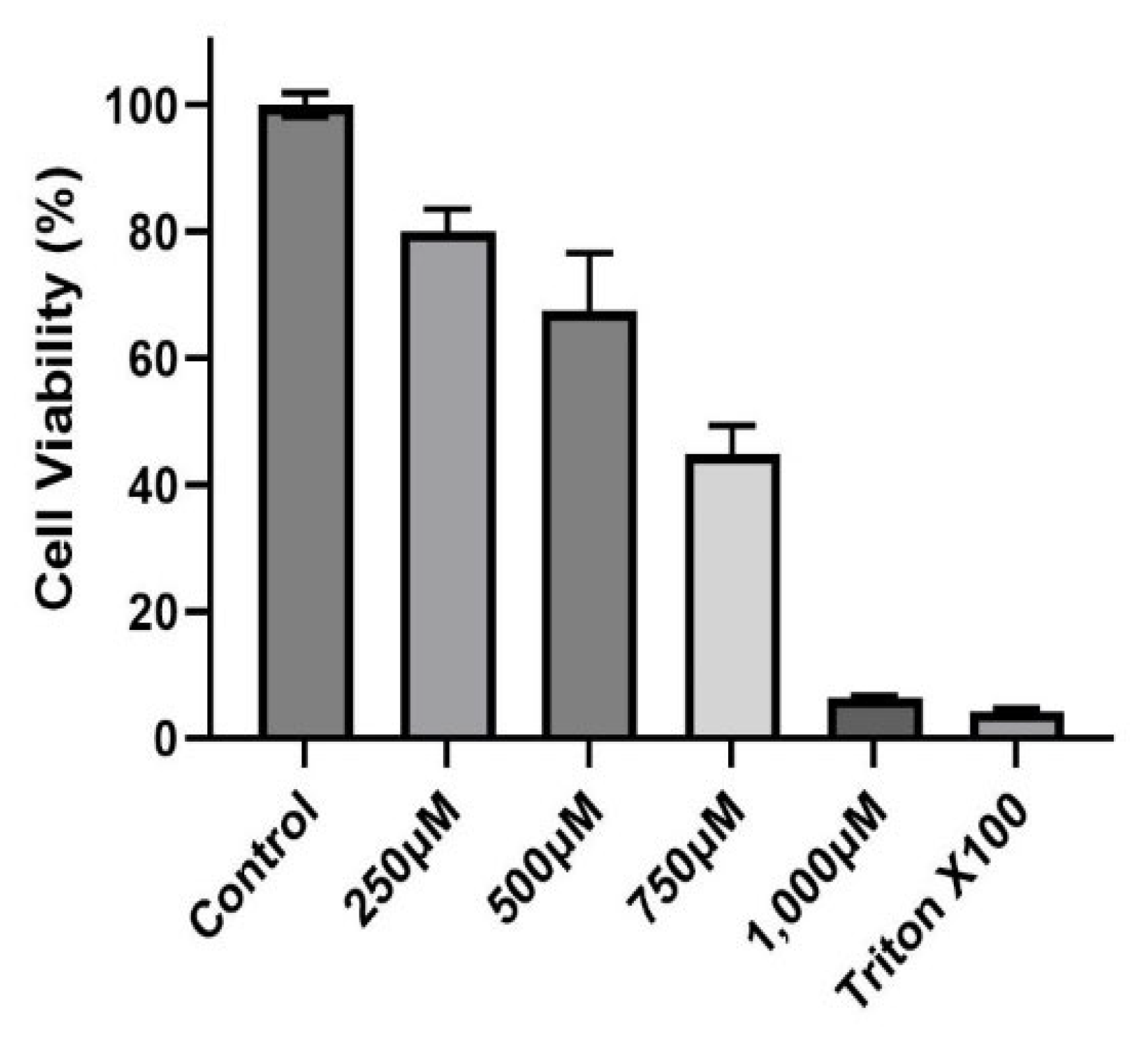
3.1. Evaluating CuSO4 and PM2.5 Composite Extract on Human Bronchial Epithelial Cells
3.2. Effect of CuSO4 Exposure on Immune Function
3.2.1. CIITA and HLA-DRα Immune Responses
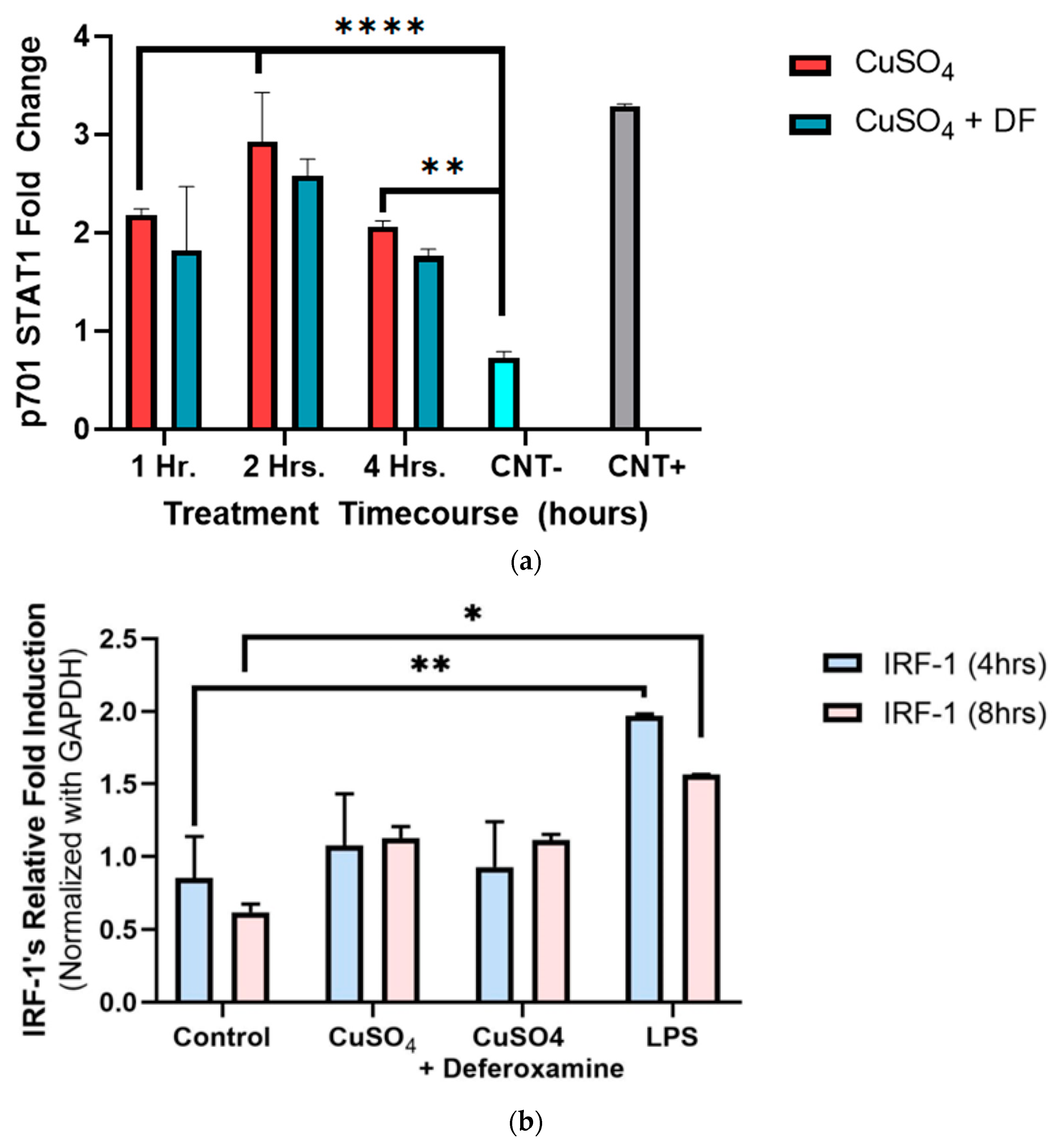
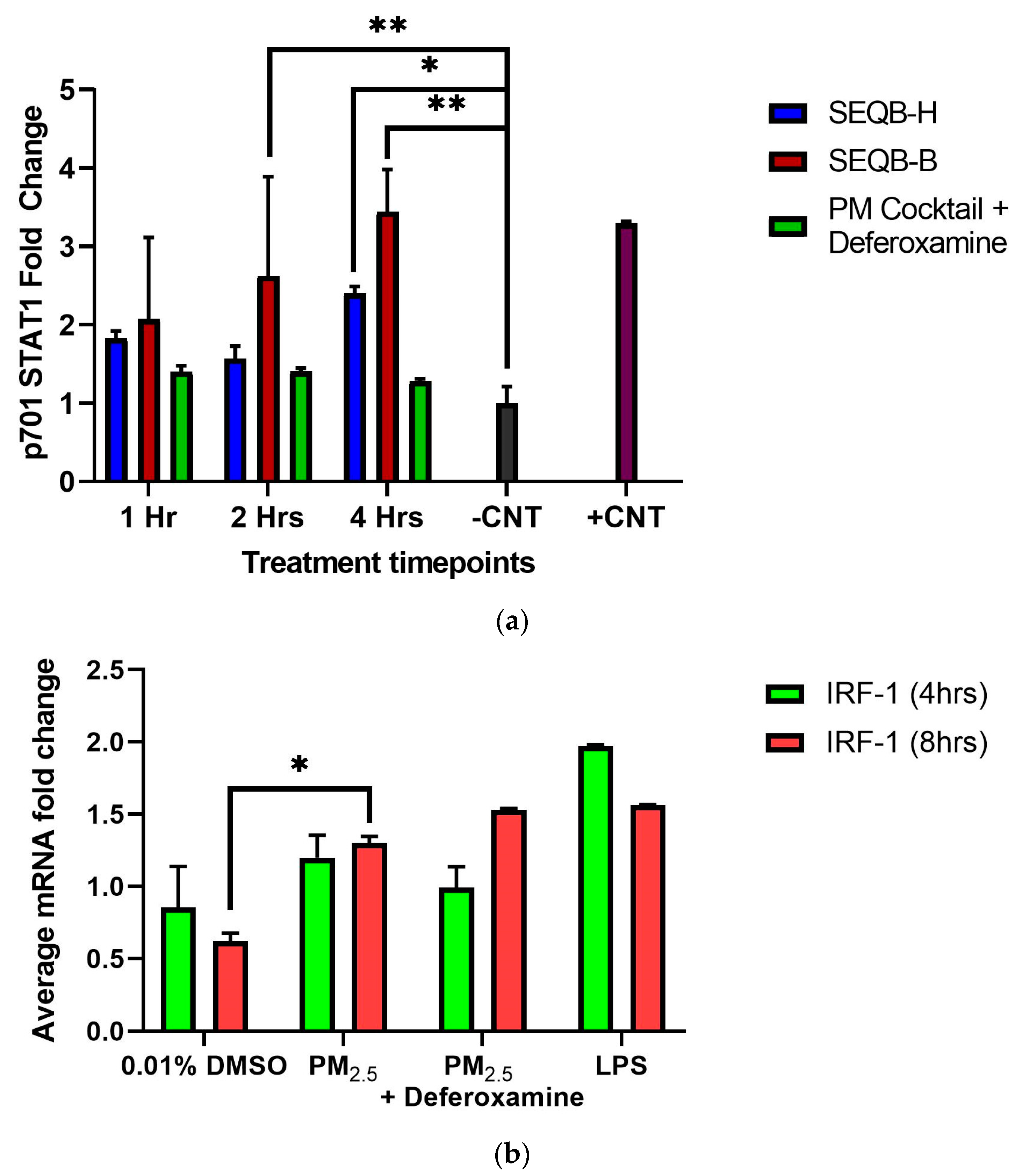
3.2.2. STAT1 p701 Phosphorylation, and IRF-1 mRNA Expression in BEAS-2B Cells
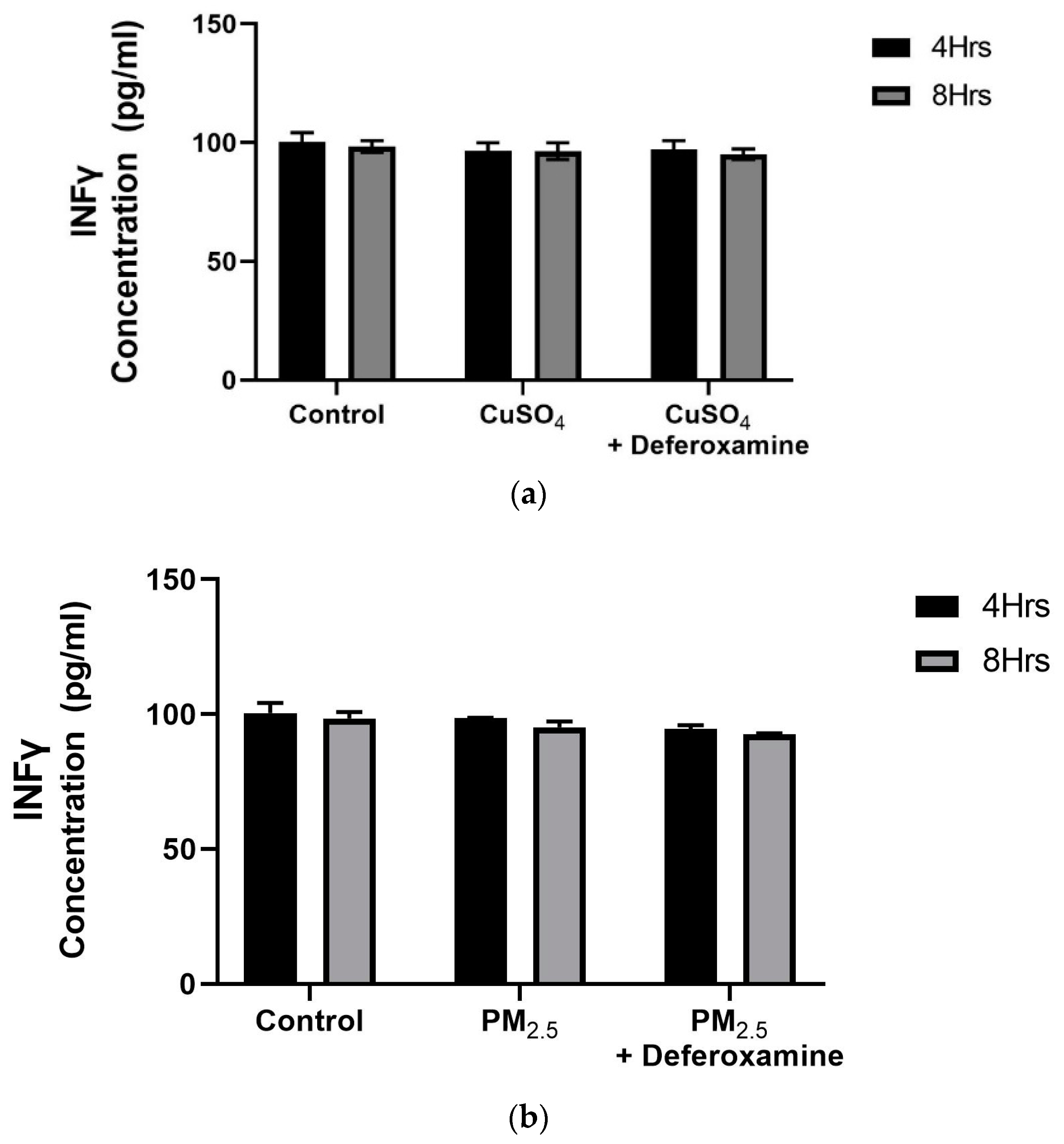
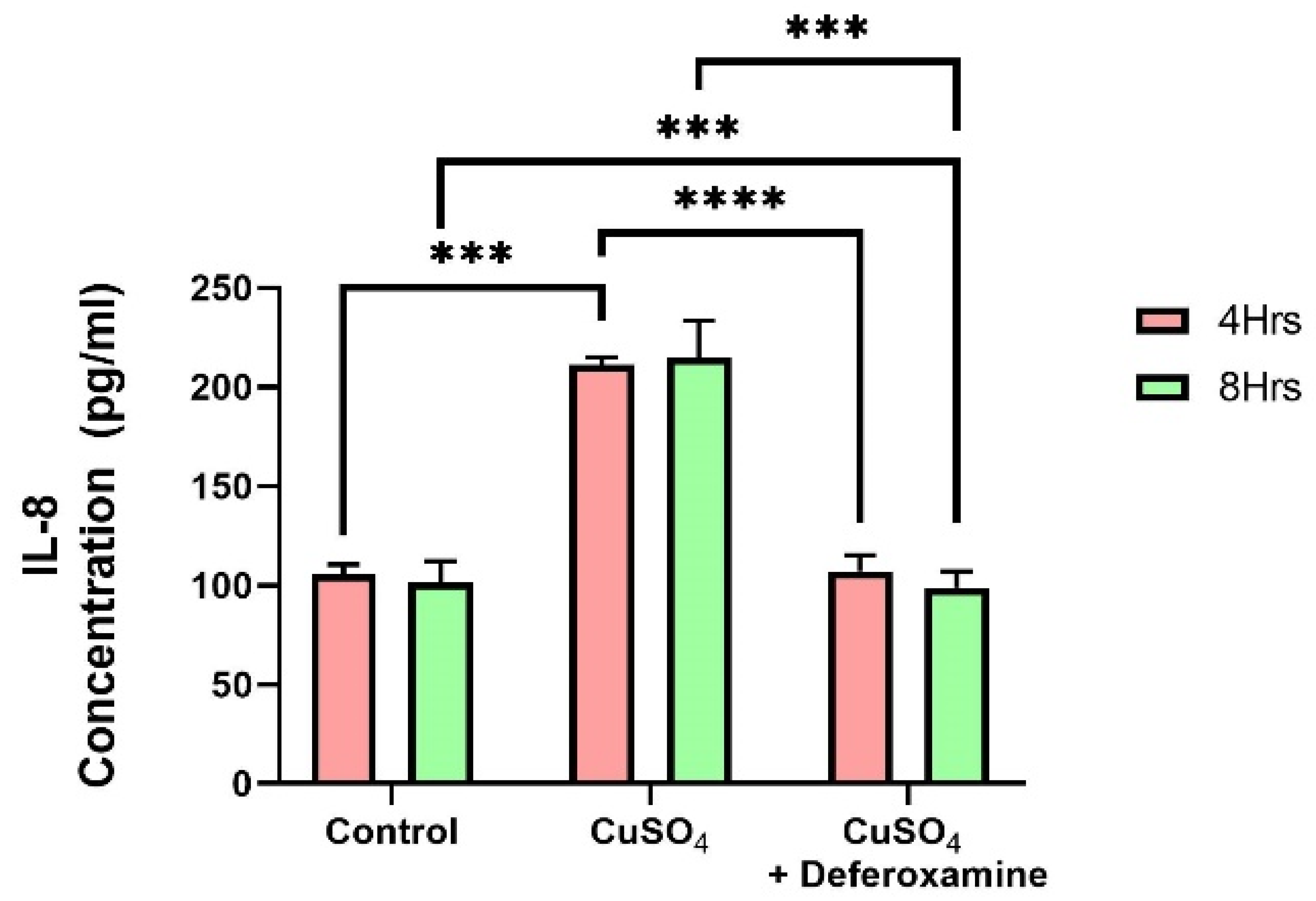
3.2.3. Quantification of Pro-Inflammatory Cytokines after CuSO4 Treatment in BEAS-2B
3.3. Effect of PM2.5 Exposure on Immune Function
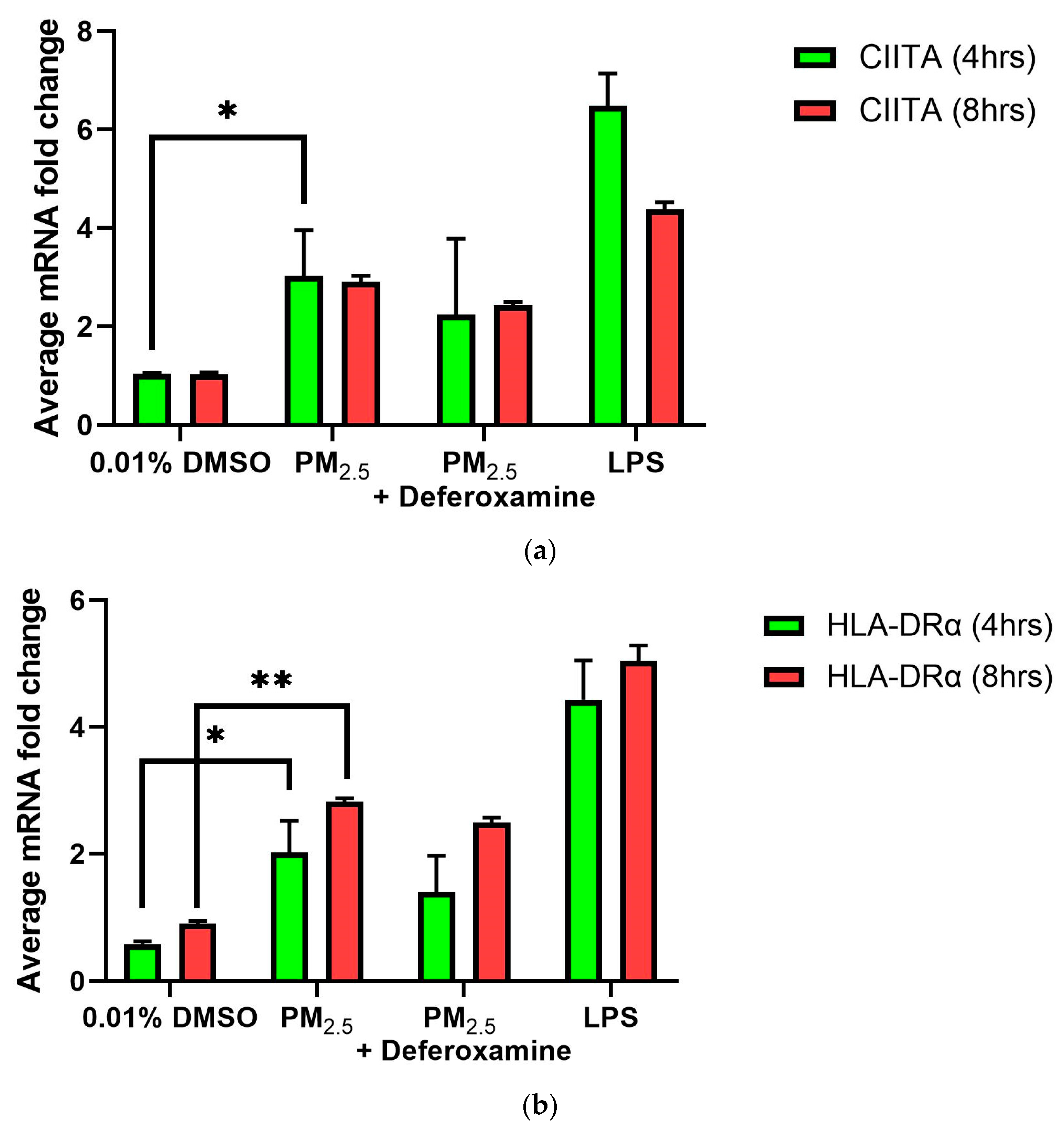
3.3.1. CIITA and HLA-DRα Immune Responses
3.3.2. Effects on pY701, STAT1, and Deferoxamine Mesylate by Urban SEQB-B and Rural SEQB-H Airborne PM2.5 Extract
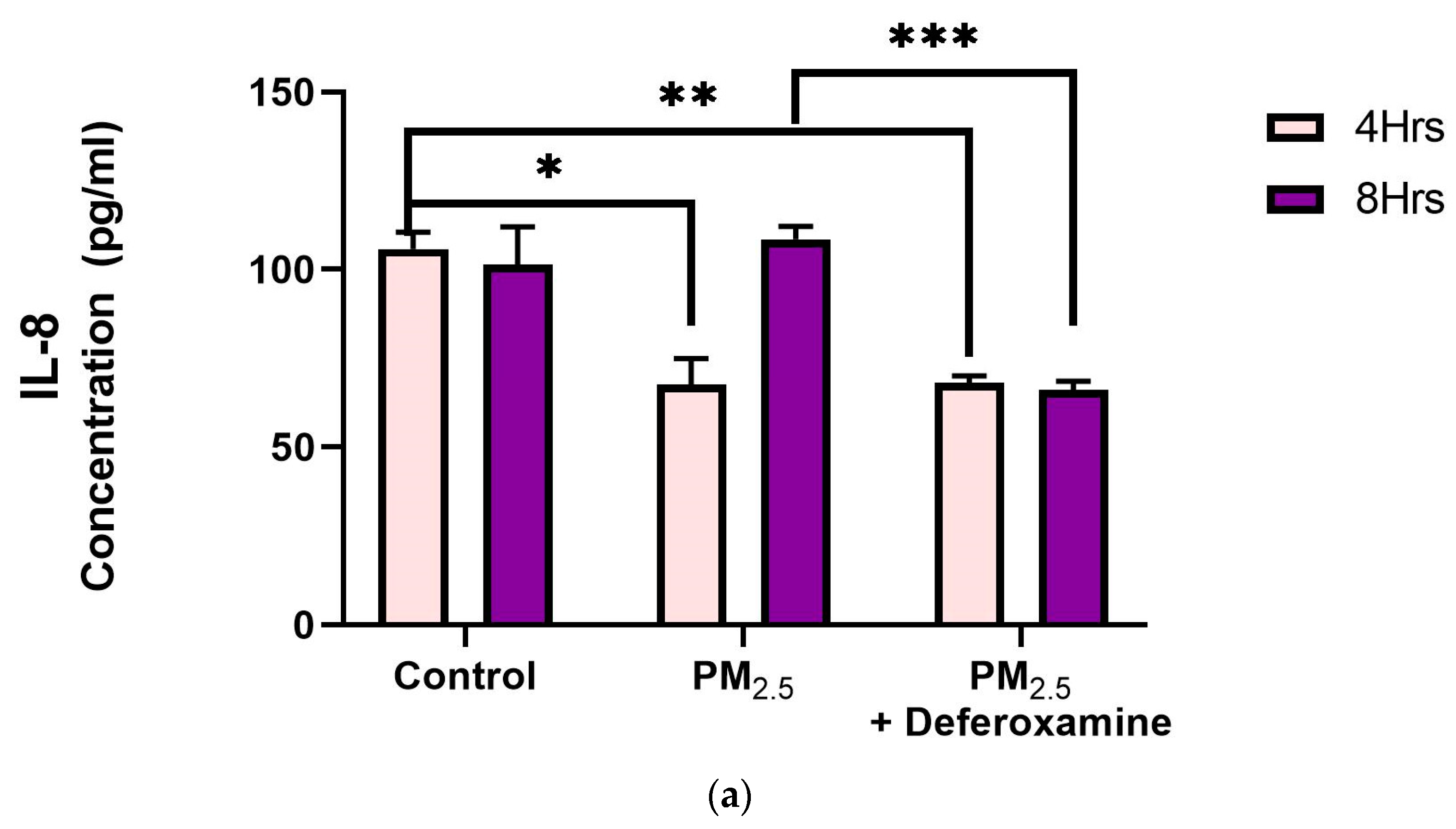
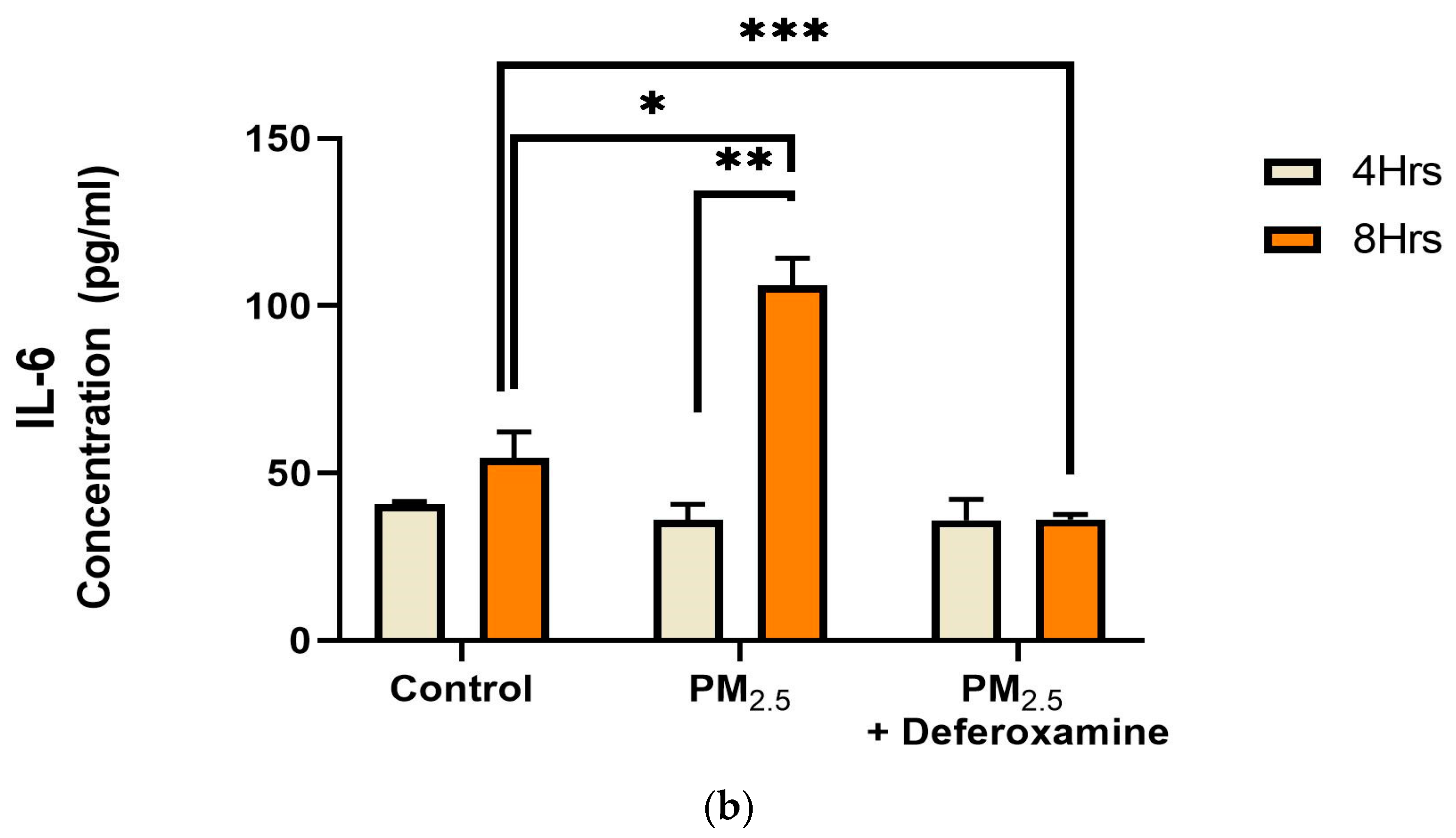
3.3.3. Pro-Inflammatory Cytokines IL-6 and IL-8 after PM2.5 Exposure and Deferoxamine Mesylate Treatment
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Air Pollution. World Health Organization. Available online: https://www.who.int/health-topics/air-pollution#tab=tab_1 (accessed on 10 March 2024).
- World Health Organization. Ambient Air Pollution. World Health Organization. Available online: https://www.who.int/airpollution/data/cities/en/ (accessed on 10 March 2024).
- Chowdhury, Z.; Hughes, L.S.; Salmon, L.G.; Cass, G.R. Atmospheric particle size and composition measurements to support light extinction calculations over the Indian Ocean. J. Geophys. Res. Atmos. 2001, 106, 28597–28605. [Google Scholar] [CrossRef]
- Wolf, K.; Popp, A.; Schneider, A.; Breitner, S.; Hampel, R.; Rathmann, W.; Herder, C.; Roden, M.; Koenig, W.; Meisinger, C.; et al. Association between longterm exposure to air pollution and biomarkers related to insulin resistance, Subclinical inflammation, and Adipokines. Diabetes 2016, 65, 3314–3326. [Google Scholar] [CrossRef] [PubMed]
- Mandal, S.; Jaganathan, S.; Kondal, D.; Schwartz, J.D.; Tandon, N.; Mohan, V.; Prabhakaran, D.; Narayan, K.M.V. PM2.5exposure, glycemic markers and incidence of type 2 diabetes in two large Indian cities. BMJ Open Diabetes Res. Care 2023, 11, e003333. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.Y.; Ma, J.K. The dual effect of the particulate and organic components of diesel exhaust particles on the alteration of pulmonary immune/inflammatory responses and metabolic enzymes. J. Environ. Sci. Health C Environ. Carcinog. Ecotoxicol. Rev. 2002, 20, 117–147. [Google Scholar] [CrossRef]
- Perez, L.; Tobías, A.; Querol, X.; Pey, J.; Alastuey, A. Saharan dust, particulate matter, and cause-specific mortality: A case-crossover study in Barcelona (Spain). Environ. Int. 2012, 48, 150–155. [Google Scholar] [CrossRef] [PubMed]
- Guaita, R.; Pichiule, M.; Mate, T.; Linares, C.; Diaz, J. Short-term impact of particulate matter (PM2.5) on respiratory mortality in Madrid. Int. J. Environ. Health Res. 2011, 21, 260–274. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, U.S.; Mcwhinney, R.D.; Rastogi, N.; Abbatt, J.P.; Evans, G.J.; Scott, J.A. Cytotoxic and pro-inflammatory effects of am-bient and source-related particulate matter (PM) in relation to the production of reactive oxygen species (ROS) and cytokine adsorption by particles. Inhal. Toxicol. 2010, 22, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Brauer, M.; Amann, M.; Burnett, R.T.; Cohen, A.; Dentener, F.; Ezzati, M.; Henderson, S.B.; Krzyzanowski, M.; Martin, R.V.; Van Dingenen, R.; et al. Exposure assessment for estimation of the global burden of disease attributable to outdoor air pollution. Environ. Sci. Technol. 2012, 46, 652–660. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.C.; Thurston, G.D. Air pollution, oxidative stress, and diabetes: A life course epidemiologic perspective. Curr. Diabetes Rep. 2019, 19, 58. [Google Scholar] [CrossRef]
- Li, Y.; Xu, L.; Shan, Z.; Teng, W.; Han, C. Association between air pollution and type 2 diabetes: An updated review of the literature. Ther. Adv. Endocrinol. Metab. 2019, 10, 2042018819897046. [Google Scholar] [CrossRef]
- Lonkar, P.C. Dedon. Reactive species and DNA damage in chronic inflammation: Reconciling chemical mechanisms and biological fates. Int. J. Cancer 2011, 128, 1999–2009. [Google Scholar] [CrossRef] [PubMed]
- Gualtieri, M.; Ovrevik, J.; Mollerup, S.; Asare, N.; Longhin, E.; Dahlman, H.J.; Camatini, M.; Holme, J.A. Airborne urban particles (Milan winter-PM2.5) cause mitotic arrest and cell death: Effects on DNA mitochondria AhR binding and spindle organization. Mutat. Res. 2011, 713, 18–31. [Google Scholar] [CrossRef] [PubMed]
- Perrone, M.; Gualtieri, M.; Consonni, V.; Ferrero, L.; Sangiorgi, G.; Longhin, E.; Ballabio, D.; Bolzacchini, E.; Camatini, M. Particle size, chemical composition, seasons of the year and urban, rural or remote site origins as determinants of biological effects of particulate matter on pulmonary cells. Environ. Pollut. 2013, 176, 215–227. [Google Scholar] [CrossRef] [PubMed]
- Ghio, A.J.; Kim, C.; Devlin, R.B. Concentrated Ambient Air Particles Induce Mild Pulmonary Inflammation in Healthy Human Volunteers. Am. J. Respir. Crit. Care Med. 2001, 162, 981–988. [Google Scholar] [CrossRef] [PubMed]
- Wold, L.E.; Simkhovich, B.Z.; Kleinman, M.T.; Nordlie, M.A.; Dow, J.S.; Sioutas, C.; Kloner, R.A. In vivo and in vitro models to test the hypothesis of particle-induced effects on cardiac function and arrhythmias. Cardiovasc. Toxicol. 2006, 6, 69–78. [Google Scholar] [CrossRef]
- Hirano, S.; Furuyama, A.; Koike, E.; Kobayashi, T. Oxidative-stress potency of organic extracts of diesel exhaust and urban fine particles in rat heart microvessel endothelial cells. Toxicology 2003, 187, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, K.; Stone, V. Current hypotheses on the mechanisms of toxicity of ultrafine particles. Ann. Dell’istituto Super. Di Sanita 2003, 39, 405–410. [Google Scholar]
- Toro-Heredia, J.; Jirau-Colón, H.; Jiménez-Vélez, B.D. Linking PM2.5 organic constituents, relative toxicity and health effects in Puerto Rico. Environ. Chall. 2021, 5, 100350. [Google Scholar] [CrossRef]
- Delfino, R.J.; Staimer, N.; Tjoa, T.; Arhami, M.; Polidori, A.; Gillen, D.L.; Kleinman, M.T.; Schauer, J.J.; Sioutas, C. Association of biomarkers of systemic inflammation with organic components and source tracers in quasi-ultrafine particles. Environ. Health Perspect. 2010, 118, 756–762. [Google Scholar] [CrossRef]
- Dergham, M.; Lepers, C.; Verdin, A.; Billet, S.; Cazier, F.; Courcot, D.; Garçon, G. Prooxidant and pro-inflammatory potency of air pollution particulate matter (PM2.5–0.3) produced in rural, urban, or industrial surroundings in human bronchial epithelial cells (BEAS-2B). Chem. Res. Toxicol. 2012, 5, 904–919. [Google Scholar] [CrossRef]
- Dergham, M.; Lepers, C.; Verdin, A.; Cazier, F.; Billet, S.; Courcot, D.; Garçon, G. Temporal–spatial variations of the physico-chemical characteristics of air pollution particulate matter (PM2.5–0.3) and toxicological effects in human bronchial epithelial cells (BEAS-2B). Environ. Res. 2015, 137, 256–267. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Qiao, B.; Zhang, L.; Yang, F.; Jiang, X. Characteristics and sources of trace elements in PM2.5 in two megacities in Sichuan Basin of southwest China. Environ. Pollut. 2018, 242, 1577–1586. [Google Scholar] [CrossRef] [PubMed]
- Thacker, E.L. Lung inflammatory responses. Vet. Res. 2006, 37, 469–486. [Google Scholar] [CrossRef] [PubMed]
- van Eeden, S.F.; Tan, W.C.; Suwa, T.; Mukae, H.; Terashima, T.; Fujii, T.; Qui, D.; Vincent, R.; Hogg, J.C. Cytokines involved in the systemic inflammatory response induced by exposure to particulate matter air pollutants (PM10). Am. J. Respir. Crit. Care Med. 2001, 164, 826–830. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Vélez, B.D.; Gioda, A.; Fuentes-Mattei, E. Organic and aqueous extracts from particulate matter (PM2.5) and their effect on the immunological response of human bronchial epithelial cells BEAS-2B. In Proceedings of the International Symposium on Metal Ions in Biology and Medicine, Lisboa, Portugal, 21–24 May 2006; pp. 267–272. [Google Scholar]
- Fuentes-Mattei, E.; Rivera, E.; Gioda, A.; Sanchez-Rivera, D.; Roman-Velazquez, F.R.; Jimenez-Velez, B.D. Use of human bronchial epithelial cells (BEAS-2B) to study immunological markers resulting from exposure to PM2.5 organic extract from Puerto Rico. Toxicol. Appl. Pharmacol. 2010, 243, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Gioda, A.; Fuentes-Mattei, E.; Jiménez-Vélez, B. Evaluation of cytokine expression in BEAS cells exposed to fine particulate matter (PM2.5) from specialized indoor environments. Int. J. Environ. Health Res. 2011, 21, 106–119. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Cotto, R.I.; Ortiz-Martínez, M.G.; Rivera-Ramírez, E.; Méndez, L.B.; Dávila, J.C.; Jiménez-Vélez, B.D. African Dust Storms Reaching Puerto Rican Coast Stimulate the Secretion of IL-6 and IL-8 and Cause Cytotoxicity to Human Bronchial Epithelial Cells (BEAS-2B). Health 2013, 5, 14–28. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Cotto, R.I.; Ortiz-Martínez, M.G.; Rivera-Ramírez, E.; Mateus, V.L.; Amaral, B.S.; Jiménez-Vélez, B.D.; Gioda, A. Particle pollution in Rio de Janeiro, Brazil: Increase and decrease of pro-inflammatory cytokines IL-6 and IL-8 in human lung cells. Environ. Pollut. 2014, 194, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Cotto, R.I.; Ortiz-Martínez, M.G.; Jiménez-Vélez, B.D. Organic extracts from African dust storms stimulate oxidative stress and induce inflammatory responses in human lung cells through Nrf2 but not NF-κB. Environ. Toxicol. Pharmacol. 2015, 39, 845–856. [Google Scholar] [CrossRef]
- Ortiz-Martínez, M.G.; Frías-Belén, O.; Nazario-Jiménez, S.; López-Quintero, M.; Rodríguez-Cotto, R.I.; Jiménez-Vélez, B.D. A case-control study of innate immunity pathway gene polymorphisms in Puerto Ricans reveals association of toll-like receptor 2 +596 variant with asthma. BMC Pulm. Med. 2016, 16, 112. [Google Scholar] [CrossRef]
- Ortiz-Martínez, M.G.; Rodríguez-Cotto, R.I.; Ortiz-Rivera, M.A.; Pluguez-Turull, C.W.; Jiménez-Vélez, B.D. Linking endotoxins, African dust PM10 and asthma in an urban and rural environment of Puerto Rico. Mediat. Inflamm. 2015, 2015, 784212. [Google Scholar] [CrossRef] [PubMed]
- Molinelli, A.R.; Santacana, G.E.; Madden, M.C.; Jiménez, B.D. Toxicity and metal content of Organic Solvent Extracts from Airborne Particulate Matter in Puerto Rico. Environ. Res. 2006, 102, 314–325. [Google Scholar] [CrossRef] [PubMed]
- Eskilsson, C.S.; Björklund, E. Analytical-scale microwave-assisted extraction. J. Chromatogr. A 2000, 902, 227–250. [Google Scholar] [CrossRef] [PubMed]
- Steimle, V.; Siegrist, C.A.; Mottet, A.; Lisowska-Grospierre, B.; Mach, B. Regulation of MHC class II expression by interferon-gamma mediated by the transactivator gene CIITA. Science 1994, 265, 106–109. [Google Scholar] [CrossRef] [PubMed]
- Choi, N.M.; Majumder, P.; Boss, J.M. Regulation of major histocompatibility complex class II genes. Curr. Opin. Immunol. 2010, 23, 81–87. [Google Scholar] [CrossRef]
- Boss, J.M.; Jensen, P.E. Transcriptional regulation of the MHC class II antigen presentation pathway. Curr. Opin. Immunol. 2003, 15, 105–111. [Google Scholar] [CrossRef]
- Arancibia-Cárcamo, C.V.; Osawa, H.; Arnett, H.A.; Háskova, Z.; George, A.J.T.; Ono, S.J.; Ting, J.P.-Y.; Streilein, J.W. A CIITA-independent pathway that promotes expression of endogenous rather than exogenous peptides in immune-privileged sites. Eur. J. Immunol. 2004, 34, 471–480. [Google Scholar] [CrossRef]
- Zheng, K.; Zeng, Z.; Tian, Q.; Huang, J.; Zhong, Q.; Huo, X. Epidemiological evidence for the effect of environmental heavy metal exposure on the immune system in children. Sci. Total Environ. 2023, 868, 161691. [Google Scholar] [CrossRef]
- Yoshida, Y.; Furuta, S.; Niki, E. Effects of metal chelating agents on the oxidation of lipids induced by copper and iron. Biochim. Biophys. Acta (BBA) 1993, 1210, 81–88. [Google Scholar] [CrossRef]
- Arredondo, M.; Núñez, M.T. Iron and copper metabolism. Mol. Asp. Med. 2005, 26, 313–327. [Google Scholar] [CrossRef]
- Matsukura, S.; Kokubu, F.; Noda, H.; Tokunaga, H.; Adachi, M. Expression of IL-6, IL-8, and RANTES on human bronchial epithelial cells, NCI-H292, induced by influenza virus A. J. Allergy Clin. Immunol. 1996, 98, 1080–1087. [Google Scholar] [CrossRef] [PubMed]
- LeibundGut-Landmann, S.; Waldburger, J.M.; Krawczyk, M.; Otten, L.A.; Suter, T.; Fontana, A.; Acha-Orbea, H.; Reith, W. Mini-review: Specificity and expression of CIITA, the master regulator of MHC class II genes. Eur. J. Immunol. 2004, 34, 1513–1525. [Google Scholar] [CrossRef]
- Sadzak, I.; Schiff, M.; Gattermeier, I.; Glinitzer, R.; Sauer, I.; Saalmüller, A.; Yang, E.; Schaljo, B.; Kovarik, P. Recruitment of Stat1 to chromatin is required for interferon-induced serine phosphorylation of Stat1 transactivation domain. Proc. Natl. Acad. Sci. USA 2008, 105, 8944–8949. [Google Scholar] [CrossRef]
- Tur, J.; Farrera, C.; Sánchez-Tilló, E.; Vico, T.; Guerrero-Gonzalez, P.; Fernandez-Elorduy, A.; Lloberas, J.; Celada, A. Induction of CIITA by IFN-γ in macrophages involves STAT1 activation by JAK and JNK. Immunobiology 2021, 226, 152114. [Google Scholar] [CrossRef] [PubMed]
- Caramori, G.; Nucera, F.; Mumby, S.; Bello, F.L.; Adcock, I.M. Corticosteroid resistance in asthma: Cellular and molecular mechanisms. Mol. Asp. Med. 2022, 85, 100969. [Google Scholar] [CrossRef] [PubMed]
- Courcot, E.; Leclerc, J.; Lafitte, J.J.; Mensier, E.; Jaillard, S.; Gosset, P.; Shirali, P.; Pottier, N.; Broly, F.; Lo-Guidice, J.M. Xenobiotic metabolism and disposition in human lung cell models: Comparison with in vivo expression profiles. Drug Metab. Dispos. 2012, 40, 1953–1965. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Qin, H.; Benveniste, E.N. The IFNy-induced transcriptional program of the CIITA gene is inhibited by statins. Eur. J. Immunol. 2008, 38, 2325–2336. [Google Scholar] [CrossRef] [PubMed]
- Forman, H.J.; Torres, M. Reactive oxygen species and cell signaling: Respiratory burst in macrophage signaling. Am. J. Respir. Crit. Care Med. 2002, 166 (Suppl. 1), S4–S8. [Google Scholar] [CrossRef]
- Hancock, J.T.; Desikan, R.; Neill, S.J. Role of reactive oxygen species in cell signalling pathways. Biochem. Soc. Trans. 2001, 29, 345–349. [Google Scholar] [CrossRef]
- Jirau-Colón, H.; Toro-Heredia, J.; Layuno, J.; Calderon, E.D.; Gioda, A.; Jiménez-Vélez, B.D. Distribution of toxic metals and relative toxicity of airborne PM2.5 in Puerto Rico. Environ. Sci. Pollut. Res. 2021, 28, 16504–16516. [Google Scholar] [CrossRef]
- Figueroa, D.A.; Rodríguez-Sierra, C.J.; Jiménez-Velez, B.D. Concentrations of Ni and V, other heavy metals, arsenic, elemental and organic carbon in atmospheric fine particles (PM2.5) from Puerto Rico. Toxicol. Ind. Health 2006, 22, 87–99. [Google Scholar] [CrossRef] [PubMed]
- Gioda, A.; Pérez, U.; Rosa, Z.; Jiménez-Vélez, B.D. Concentration of heavy metals in airborne PM10 from Jobos Bay National Estuary, Puerto Rico. Water Air Soil Pollut. 2006, 174, 141–159. [Google Scholar] [CrossRef]
- Gioda, A.; Peréz, U.; Rosa, Z.; Jiménez-Velez, B.D. Particulate matter (PM10 and PM2.5) from different areas of Puerto Rico. Fresenius Environ. Bull. 2007, 16, 861. [Google Scholar]
- Jiménez, B.D.; Maldonado, L.; Dahl, R.H.; Quattrochi, L.C.; Guzelian, P.S. Ectopic expression of MHC class II genes (RT1. B (I) β/α) in rat hepatocytes in vivo and in culture can be elicited by treatment with the pregnane X receptor agonists pregnenolone 16α-carbonitrile and dexamethasone. Life Sci. 2002, 71, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Taché, Y.; Taché, J.; Mécs, I.; Ruisseau, P.D.; Selye, H. Regulation of resistance to various toxicants by PCN (pregne-nolone-16alpha-carbonitrile) and thyroxine. J. Med. 1976, 7, 471–479. [Google Scholar] [PubMed]
- Lindeborg, T.; Glaumann, H. Effect of a catatoxic steroid pregnenolone-16α-carbonitrile, on rat liver microsomal subfractions. Exp. Mol. Pathol. 1974, 21, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Burk, O.; Koch, I.; Raucy, J.; Hustert, E.; Eichelbaum, M.; Brockmöller, J.; Zanger, U.M.; Wojnowski, L. The induction of cytochrome P450 3A5 (CYP3A5) in the human liver and intestine is mediated by the xenobiotic sensors pregnane X receptor (PXR) and constitutively activated receptor (CAR). J. Biol. Chem. 2004, 279, 38379–38385. [Google Scholar] [CrossRef] [PubMed]
- Cooper, B.W.; Cho, T.M.; Thompson, P.M.; Wallace, A.D. Phthalate induction of CYP3A4 is dependent on gluco-corticoid regulation of PXR expression. Toxicol. Sci. 2008, 103, 268–277. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.; Cunningham, M.; Kim, S.; Burn, T.; Lin, J.; Sinz, M.; Hamilton, G.; Rizzo, C.; Jolley, S.; Gilbert, D.; et al. CYP3A4 induction by drugs: Correlation between a pregnane X receptor reporter gene assay and CYP3A4 expression in human hepatocytes. Drug Metab. Dispos. 2002, 30, 795–804. [Google Scholar] [CrossRef]
- Ma, Y.; Liu, D. Activation of pregnane x receptor by pregnenolone 16 α-carbonitrile prevents high-fat diet-induced obesity in AKR/J mice. PLoS ONE 2012, 7, e38734. [Google Scholar] [CrossRef]
- Guzelian, J.; Barwick, J.L.; Hunter, L.; Phang, T.L.; Quattrochi, L.C.; Guzelian, P.S. Identification of Genes Controlled by the Pregnane X Receptor by Microarray Analysis of mRNAs from Pregnenolone 16α-Carbonitrile–Treated Rats. Toxicol. Sci. 2006, 94, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Amersfoort, J.; Eelen, G.; Carmeliet, P. Immunomodulation by endothelial cells—partnering up with the immune system? Nat. Rev. Immunol. 2022, 22, 576–588. [Google Scholar] [CrossRef] [PubMed]
- Wosen, J.E.; Mukhopadhyay, D.; Macaubas, C.; Mellins, E.D. Epithelial MHC class II expression and its role in antigen presentation in the gastrointestinal and respiratory tracts. Front. Immunol. 2018, 9, 408694. [Google Scholar] [CrossRef] [PubMed]
- Papazian, D.; Würtzen, P.A.; Hansen, S.W. Polarized airway epithelial models for immunological co-culture studies. Int. Arch. Allergy Immunol. 2016, 170, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Glanville, A.R.; Tazelaar, H.D.; Theodore, J.; Imoto, E.; Rouse, R.V.; Baldwin, J.C.; Robin, E.D. The distribution of MHC class I and II antigens on bronchial epithelium. Am. Rev. Respir. Dis. 1989, 139, 330–334. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, A.C.; Milne, D.S.; Wilkes, J.; Dark, J.H.; Tetley, T.D.; Kirby, J.A. Constitutive expression of mhc and adhesion molecules by alveolar epithelial cells (type ii pneumocytes) isolated from human lung and comparison with immunocytochemical findings. J. Cell Sci. 1994, 107, 443–449. [Google Scholar] [CrossRef] [PubMed]
- Longhin, E.; Holme, J.A.; Gutzkow, K.B.; Arlt, V.M.; Kucab, J.E.; Camatini, M.; Gualtieri, M. Cell cycle alterations induced by urban PM2.5 in bronchial epithelial cells: Characterization of the process and possible mechanisms involved. Part Fibre Toxicol. 2013, 10, 63. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Jiang, H.; Wang, J.; Zhou, J. A hybrid model for PM2. 5 forecasting based on ensemble empirical mode decomposition and a general regression neural network. Sci. Total Environ. 2014, 496, 264–274. [Google Scholar] [CrossRef] [PubMed]
- Bell, M.L.; Ebisu, K.; Peng, R.D.; Samet, J.M.; Dominici, F. Hospital admissions and chemical composition of fine particle air pollution. Am. J. Respir. Crit. Care Med. 2009, 179, 1115–1120. [Google Scholar] [CrossRef]
- Ruvolo, V.; Navarro, L.; Sample, C.E.; David, M.; Sung, S.; Swaminathan, S. The Epstein-Barr virus SM protein induces STAT1 and interferon-stimulated gene expression. J. Virol. 2003, 77, 3690–3701. [Google Scholar] [CrossRef]
- Collins-McMillen, D.; Stevenson, E.V.; Kim, J.H.; Lee, B.-J.; Cieply, S.J.; Nogalski, M.T.; Chan, G.C.; Frost, R.W.; Spohn, C.R.; Yurochko, A.D. Human cytomegalovirus utilizes a nontraditional signal transducer and activator of transcription 1 activation cascade via signaling through epidermal growth factor receptor and integrins to efficiently promote the motility, differentiation, and polarization of infected monocytes. J. Virol. 2017, 91, e00622-17. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.E.; O’Keefe, R.A.; Grandis, J.R. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat. Rev. Clin. Oncol. 2018, 15, 234–248. [Google Scholar] [CrossRef] [PubMed]
- Geist, L.J.; Dai, L.Y. Cytomegalovirus modulates interleukin-6 gene expression1. Transplantation 1996, 62, 653–658. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Wen, Z.; Darnell, J.E., Jr. Stat3: A STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science 1994, 264, 95–98. [Google Scholar] [CrossRef] [PubMed]
- Reitsma, J.M.; Sato, H.; Nevels, M.; Terhune, S.S.; Paulus, C. Human cytomegalovirus IE1 protein disrupts interleukin-6 signaling by sequestering STAT3 in the nucleus. J. Virol. 2013, 87, 10763–10776. [Google Scholar] [CrossRef] [PubMed]
- Harwardt, T.; Lukas, S.; Zenger, M.; Reitberger, T.; Danzer, D.; Übner, T.; Munday, D.C.; Nevels, M.; Paulus, C. Human cytomegalovirus immediate-early 1 protein rewires upstream STAT3 to downstream STAT1 signaling switching an IL6-type to an IFNγ-like response. PLoS Pathog. 2016, 12, e1005748. [Google Scholar] [CrossRef]
- Nemec, A.A.; Barchowsky, A. Signal transducer and activator of transcription 1 (STAT1) is essential for chromium silencing of gene induction in human airway epithelial cells. Toxicol. Sci. 2009, 110, 212–223. [Google Scholar] [CrossRef]
- Risom, L.; Møller, P.; Loft, S. Oxidative stress-induced DNA damage by particulate air pollution. Mutat. Res. Mol. Mech. Mutagen. 2005, 592, 119–137. [Google Scholar] [CrossRef] [PubMed]
- Andersson, H.; Piras, E.; Demma, J.; Hellman, B.; Brittebo, E. Low levels of the air pollutant 1-nitropyrene induce DNA damage, increased levels of reactive oxygen species and endoplasmic reticulum stress in human endothelial cells. Toxicology 2009, 262, 57–64. [Google Scholar] [CrossRef]
- Kernagis, D.N.; Hall, A.H.; Datto, M.B. Genes with bimodal expression are robust diagnostic targets that define distinct subtypes of epithelial ovarian cancer with different overall survival. J. Mol. Diagn. 2012, 14, 214–222. [Google Scholar] [CrossRef]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jirau-Colón, H.; Jiménez-Vélez, B.D. PM2.5 Extracts Induce INFγ-Independent Activation of CIITA, MHCII, and Increases Inflammation in Human Bronchial Epithelium. Toxics 2024, 12, 292. https://doi.org/10.3390/toxics12040292
Jirau-Colón H, Jiménez-Vélez BD. PM2.5 Extracts Induce INFγ-Independent Activation of CIITA, MHCII, and Increases Inflammation in Human Bronchial Epithelium. Toxics. 2024; 12(4):292. https://doi.org/10.3390/toxics12040292
Chicago/Turabian StyleJirau-Colón, Héctor, and Braulio D. Jiménez-Vélez. 2024. "PM2.5 Extracts Induce INFγ-Independent Activation of CIITA, MHCII, and Increases Inflammation in Human Bronchial Epithelium" Toxics 12, no. 4: 292. https://doi.org/10.3390/toxics12040292
APA StyleJirau-Colón, H., & Jiménez-Vélez, B. D. (2024). PM2.5 Extracts Induce INFγ-Independent Activation of CIITA, MHCII, and Increases Inflammation in Human Bronchial Epithelium. Toxics, 12(4), 292. https://doi.org/10.3390/toxics12040292