Tris(2-chloroethyl) Phosphate (TCEP) Elicits Hepatotoxicity by Activating Human Cancer Pathway Genes in HepG2 Cells


 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Selection of TCEP Exposure Concentrations
2.2. MTT and NRU Cytotoxicity Assays
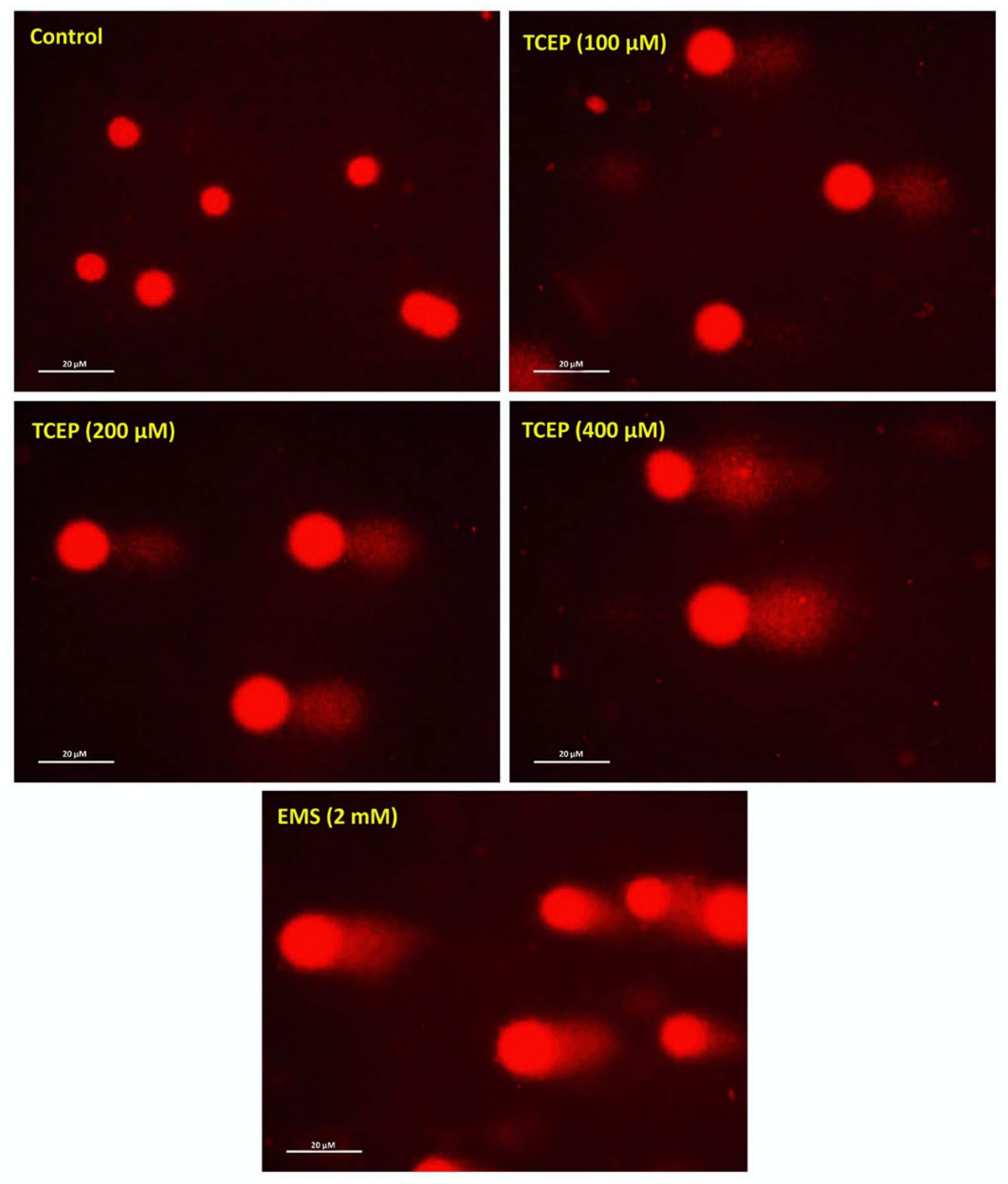
2.3. Alkaline Comet Assay
2.4. Flow Cytometric Studies
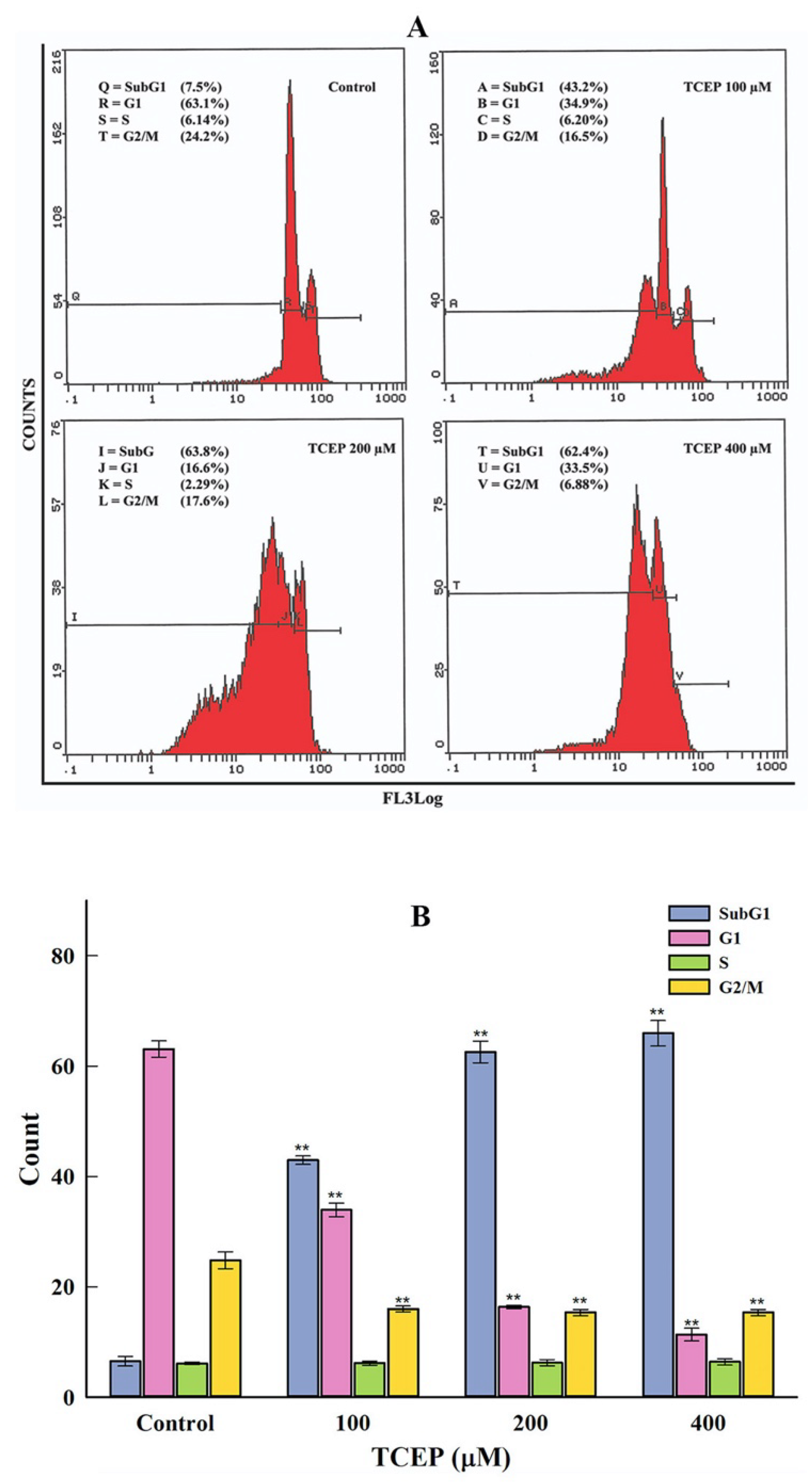
2.4.1. Cell Cycle Analysis
2.4.2. Ca2+ Influx and Mitochondrial Membrane Potential (ΔΨm)
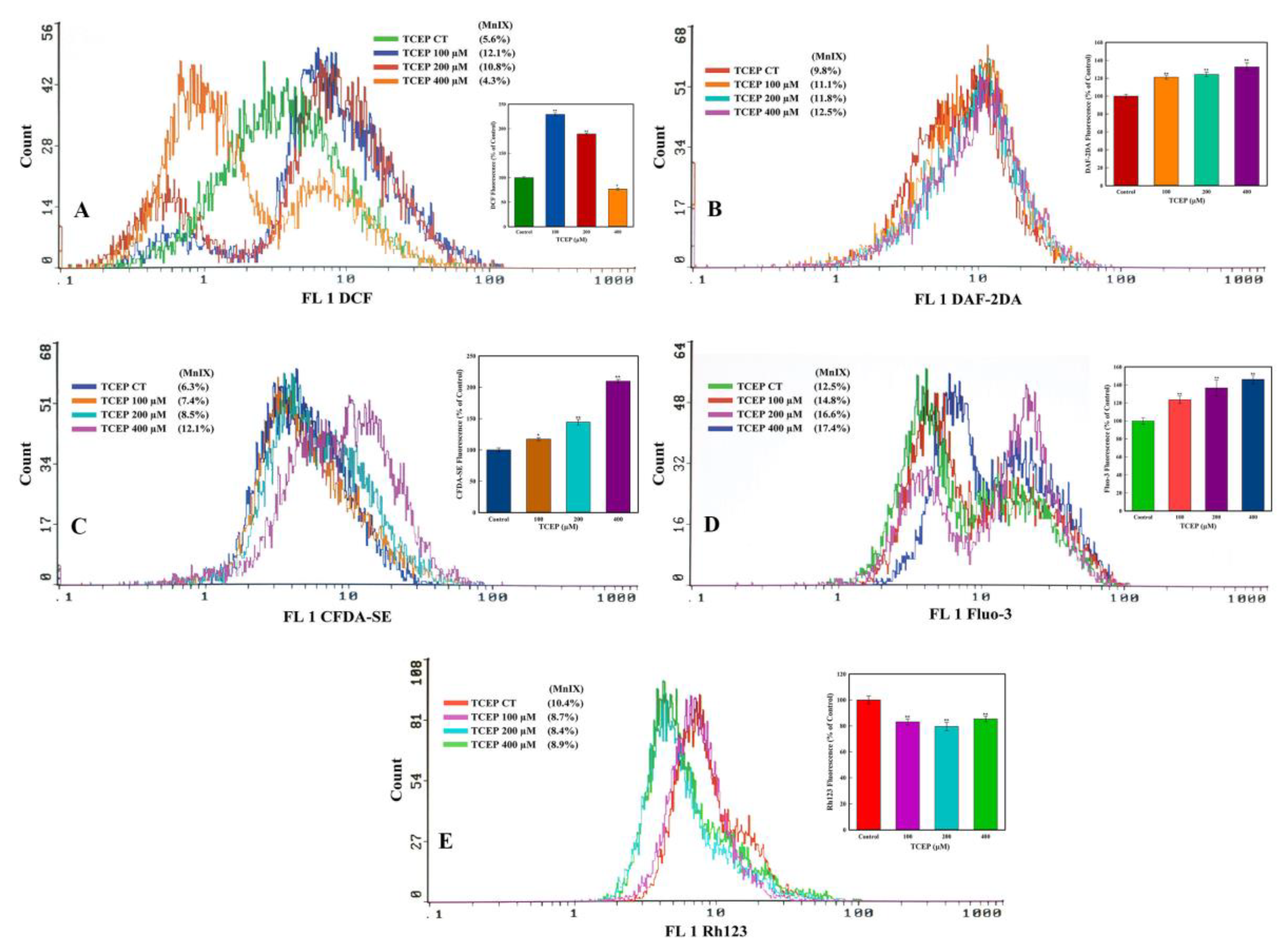
2.4.3. Reactive Oxygen Species (ROS), Nitric Oxide (NO), and Esterase Activities
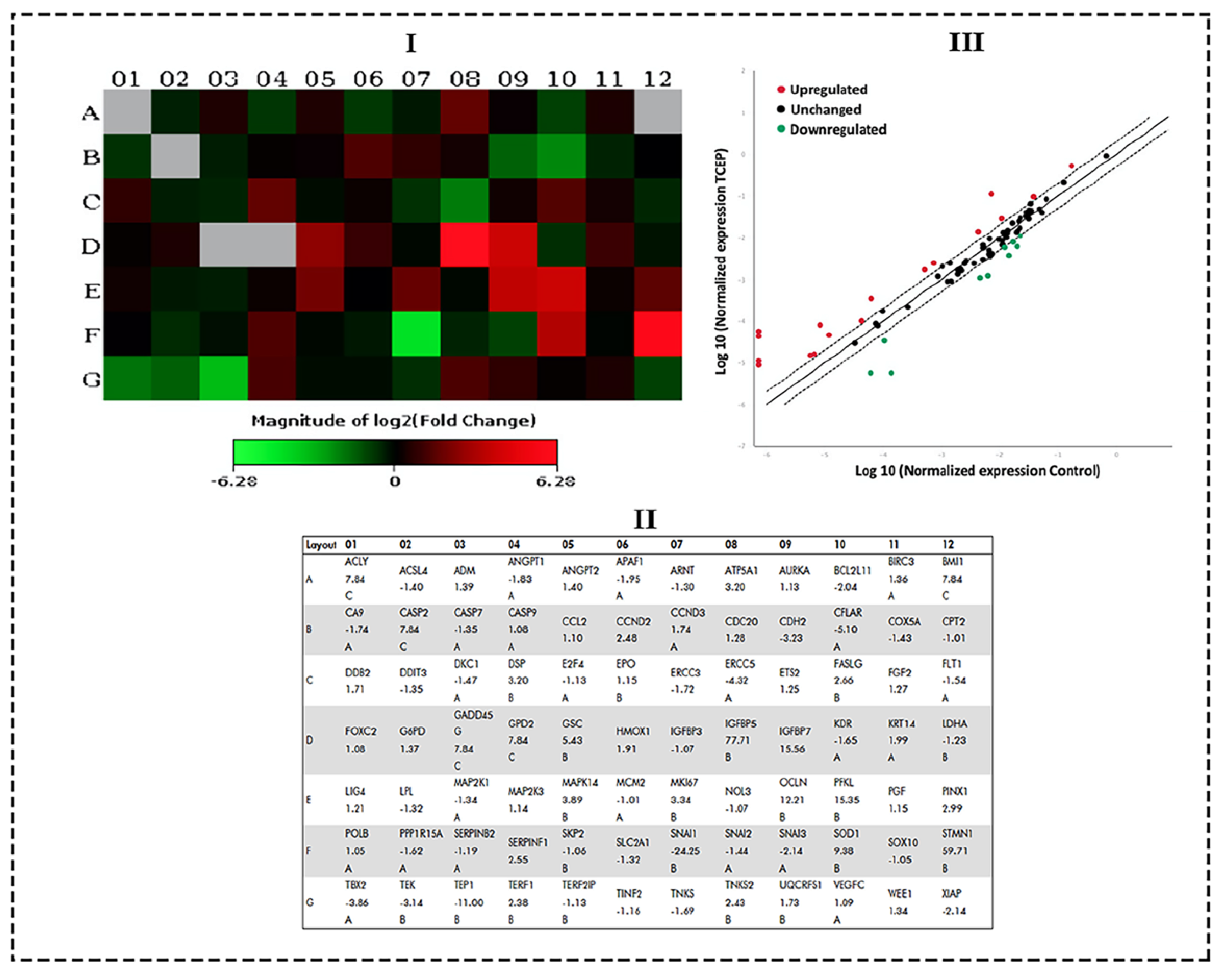
2.5. Transcriptomic Analysis by qPCR Array
2.6. Statistical Analysis
3. Results
3.1. TCEP Induced Cytotoxicity in HepG2 Cells
3.2. Quantitation of DNA Damage
3.3. Flow Cytometric Data
3.3.1. HepG2 Cell Cycle Dysfunction by TCEP
3.3.2. TCEP Induced Oxidative Stress and Esterase Activity
3.3.3. Ca2+ Influx and ΔΨm Alteration by TCEP
3.4. Transcriptomic Changes in HepG2 by TCEP Exposure
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Marklund, A.; Andersson, B.; Haglund, P. Screening of organophosphorus compounds and their distribution in various indoor environments. Chemosphere 2003, 53, 1137–1146. [Google Scholar] [CrossRef]
- Zhou, L.; Hiltscher, M.; Gruber, D.; Püttmann, W. Organophosphate flame retardants (OPFRs) in indoor and outdoor air in the Rhine/Main area, Germany: Comparison of concentrations and distribution profiles in different microenvironments. Environ. Sci. Pollut. Res. 2017, 24, 10992–11005. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Liu, L.-Y.; Sverko, E.; Li, Y.-F.; Li, H.-L.; Huo, C.-Y.; Ma, W.-L.; Song, W.-W.; Zhang, Z.-F. Organophosphate flame retardants in college dormitory dust of northern Chinese cities: Occurrence, human exposure and risk assessment. Sci. Total Environ. 2019, 665, 731–738. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Su, H.; Ya, M.; Li, J.; Ho, S.-H.; Zhao, L.; Jian, K.; Letcher, R.J.; Su, G. Distribution of flame retardants in smartphones and identification of current-use organic chemicals including three novel aryl organophosphate esters. Sci. Total Environ. 2019, 693, 133654. [Google Scholar] [CrossRef]
- Tokumura, M.; Hatayama, R.; Tatsu, K.; Naito, T.; Takeda, T.; Raknuzzaman, M.; Habibullah-Al-Mamun, M.; Masunaga, S. Organophosphate flame retardants in the indoor air and dust in cars in Japan. Environ. Monit. Assess. 2017, 189, 48. [Google Scholar] [CrossRef]
- Hoffman, K.; Lorenzo, A.; Butt, C.M.; Hammel, S.C.; Henderson, B.B.; Roman, S.A.; Scheri, R.P.; Stapleton, H.M.; Sosa, J.A. Exposure to flame retardant chemicals and occurrence and severity of papillary thyroid cancer: A case-control study. Environ. Int. 2017, 107, 235–242. [Google Scholar] [CrossRef]
- Ma, Y.; Jin, J.; Li, P.; Xu, M.; Sun, Y.; Wang, Y.; Yuan, H. Organophosphate ester flame retardant concentrations and distributions in serum from inhabitants of Shandong, China, and changes between 2011 and 2015. Environ. Toxicol. Chem. 2017, 36, 414–421. [Google Scholar] [CrossRef]
- He, C.; Toms, L.-M.L.; Thai, P.; Van den Eede, N.; Wang, X.; Li, Y.; Baduel, C.; Harden, F.A.; Heffernan, A.L.; Hobson, P. Urinary metabolites of organophosphate esters: Concentrations and age trends in Australian children. Environ. Int. 2018, 111, 124–130. [Google Scholar] [CrossRef]
- Romano, M.E.; Hawley, N.L.; Eliot, M.; Calafat, A.M.; Jayatilaka, N.K.; Kelsey, K.; McGarvey, S.; Phipps, M.G.; Savitz, D.A.; Werner, E.F. Variability and predictors of urinary concentrations of organophosphate flame retardant metabolites among pregnant women in Rhode Island. Environ. Health 2017, 16, 40. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Bai, X.-Y.; Lu, S.-Y.; Zhang, B.; Xie, L.; Zheng, H.-C.; Jiang, Y.-C.; Zhou, M.-Z.; Zhou, Z.-Q.; Song, S.-M. Urinary metabolites of organophosphate flame retardants in China: Health risk from tris (2-chloroethyl) phosphate (TCEP) exposure. Environ. Int. 2018, 121, 1363–1371. [Google Scholar] [CrossRef]
- Bastiaensen, M.; Malarvannan, G.; Been, F.; Yin, S.; Yao, Y.; Huygh, J.; Clotman, K.; Schepens, T.; Jorens, P.G.; Covaci, A. Metabolites of phosphate flame retardants and alternative plasticizers in urine from intensive care patients. Chemosphere 2019, 233, 590–596. [Google Scholar] [CrossRef] [PubMed]
- Lai, N.L.; Kwok, K.Y.; Wang, X.-H.; Yamashita, N.; Liu, G.; Leung, K.M.; Lam, P.K.; Lam, J.C. Assessment of organophosphorus flame retardants and plasticizers in aquatic environments of China (Pearl River Delta, South China Sea, Yellow River Estuary) and Japan (Tokyo Bay). J. Hazard. Mater. 2019, 371, 288–294. [Google Scholar] [CrossRef] [PubMed]
- Government of Canada. Ethanol, 2-Chloro-, Phosphate (3:1) (Tris(2-Chloroethyl)Phosphate) (TCEP); Government of Canada: Ottawa, ON, Canada, 2009.
- U.S. Department of Health and Human Services. Toxicological Profile for Phosphate Ester Falme Retardants; U.S. Department of Health and Human Services: Washington, DC, USA, 2012. Available online: https://www.atsdr.cdc.gov/toxprofiles/tp202.pdf (accessed on 17 November 2020).
- Yang, W.; Zhao, F.; Fang, Y.; Li, L.; Li, C.; Ta, N. 1H-nuclear magnetic resonance metabolomics revealing the intrinsic relationships between neurochemical alterations and neurobehavioral and neuropathological abnormalities in rats exposed to tris (2-chloroethyl) phosphate. Chemosphere 2018, 200, 649–659. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zhang, Y.; Hou, J.; Xu, T.; Yin, W.; Xiong, W.; Lu, W.; Zheng, H.; Chen, J.; Yuan, J. Tris (2-chloroethyl) phosphate induces senescence-like phenotype of hepatocytes via the p21Waf1/Cip1-Rb pathway in a p53-independent manner. Environ. Toxicol. Pharmacol. 2017, 56, 68–75. [Google Scholar] [CrossRef]
- Mennillo, E.; Cappelli, F.; Arukwe, A. Biotransformation and oxidative stress responses in rat hepatic cell-line (H4IIE) exposed to organophosphate esters (OPEs). Toxicol. Appl. Pharmacol. 2019, 371, 84–94. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, W.; Hou, J.; Wang, X.; Zheng, H.; Xiong, W.; Yuan, J. Combined effect of tris(2-chloroethyl)phosphate and benzo (a) pyrene on the release of IL-6 and IL-8 from HepG2 cells via the EGFR-ERK1/2 signaling pathway. RSC Adv. 2017, 7, 54281–54290. [Google Scholar] [CrossRef] [Green Version]
- Krivoshiev, B.V.; Beemster, G.T.S.; Sprangers, K.; Blust, R.; Husson, S.J. A toxicogenomics approach to screen chlorinated flame retardants tris(2-chloroethyl) phosphate and tris(2-chloroisopropyl) phosphate for potential health effects. J. Appl. Toxicol. 2018, 38, 459–470. [Google Scholar] [CrossRef]
- Zhang, W.; Zhang, Y.; Wang, Z.; Xu, T.; Huang, C.; Yin, W.; Wang, J.; Xiong, W.; Lu, W.; Zheng, H.; et al. Tris(2-chloroethyl)phosphate-induced cell growth arrest via attenuation of SIRT1-independent PI3K/Akt/mTOR pathway. J. Appl. Toxicol. 2016, 36, 914–924. [Google Scholar] [CrossRef]
- Mokra, K.; Bukowski, K.; Woźniak, K. Effects of tris (1-chloro-2-propyl) phosphate and tris (2-chloroethyl) phosphate on cell viability and morphological changes in peripheral blood mononuclear cells (in vitro study). Hum. Exp. Toxicol. 2018, 37, 1336–1345. [Google Scholar] [CrossRef]
- Yu, X.; Yin, H.; Peng, H.; Lu, G.; Liu, Z.; Dang, Z. OPFRs and BFRs induced A549 cell apoptosis by caspase-dependent mitochondrial pathway. Chemosphere 2019, 221, 693–702. [Google Scholar] [CrossRef]
- Gu, Y.; Yang, Y.; Wan, B.; Li, M.; Guo, L.-H. Inhibition of O-linked N-acetylglucosamine transferase activity in PC12 cells–A molecular mechanism of organophosphate flame retardants developmental neurotoxicity. Biochem. Pharmacol. 2018, 152, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Gerets, H.; Tilmant, K.; Gerin, B.; Chanteux, H.; Depelchin, B.; Dhalluin, S.; Atienzar, F. Characterization of primary human hepatocytes, HepG2 cells, and HepaRG cells at the mRNA level and CYP activity in response to inducers and their predictivity for the detection of human hepatotoxins. Cell Biol. Toxicol. 2012, 28, 69–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saquib, Q.; Musarrat, J.; Siddiqui, M.A.; Dutta, S.; Dasgupta, S.; Giesy, J.P.; Al-Khedhairy, A.A. Cytotoxic and necrotic responses in human amniotic epithelial (WISH) cells exposed to organophosphate insecticide phorate. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2012, 744, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Saquib, Q.; Al-Khedhairy, A.A.; Al-Arifi, S.; Dhawan, A.; Musarrat, J. Assessment of methyl thiophanate–Cu (II) induced DNA damage in human lymphocytes. Toxicol. In Vitro 2009, 23, 848–854. [Google Scholar] [CrossRef]
- Saquib, Q.; Al-Khedhairy, A.A.; Ahmad, J.; Siddiqui, M.A.; Dwivedi, S.; Khan, S.T.; Musarrat, J. Zinc ferrite nanoparticles activate IL-1b, NFKB1, CCL21 and NOS2 signaling to induce mitochondrial dependent intrinsic apoptotic pathway in WISH cells. Toxicol. Appl. Pharmacol. 2013, 273, 289–297. [Google Scholar] [CrossRef]
- Al-Salem, A.M.; Saquib, Q.; Siddiqui, M.A.; Ahmad, J.; Wahab, R.; Al-Khedhairy, A.A. Organophosphorus flame retardant (tricresyl phosphate) trigger apoptosis in HepG2 cells: Transcriptomic evidence on activation of human cancer pathways. Chemosphere 2019, 237, 124519. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, W.; Hou, J.; Wang, X.; Lu, W.; Zheng, H.; Xiong, W.; Liu, J.; Yuan, J. Seasonal variations of tris (2-chloroethyl) phosphate and cytotoxicity of organic extracts in water samples from Wuhan, China. J. Environ. Sci. 2019, 76, 299–309. [Google Scholar] [CrossRef]
- Aslantürk, Ö.S. In Vitro Cytotoxicity and Cell Viability Assays: Principles, Advantages, and Disadvantages; InTech Open: London, UK, 2018; Volume 2. [Google Scholar]
- Saquib, Q.; Siddiqui, M.A.; Ahmad, J.; Ansari, S.M.; Al-Wathnani, H.A.; Rensing, C. 6-OHBDE-47 induces transcriptomic alterations of CYP1A1, XRCC2, HSPA1A, EGR1 genes and trigger apoptosis in HepG2 cells. Toxicology 2018, 400, 40–47. [Google Scholar] [CrossRef]
- Zhang, W.; Zhang, Y.; Xu, T.; Wang, Z.; Wang, J.; Xiong, W.; Lu, W.; Zheng, H.; Yuan, J. Involvement of ROS-mediated mitochondrial dysfunction and SIRT3 down-regulation in tris (2-chloroethyl) phosphate-induced cell cycle arrest. Toxicol. Res. 2016, 5, 461–470. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Xiao, Y.; Chang, Y.; Cui, Y.; Klobučar, G.; Li, M. Intestinal damage, neurotoxicity and biochemical responses caused by tris (2-chloroethyl) phosphate and tricresyl phosphate on earthworm. Ecotoxicol. Environ. Saf. 2018, 158, 78–86. [Google Scholar] [CrossRef]
- Lubos, E.; Handy, D.E.; Loscalzo, J. Role of oxidative stress and nitric oxide in atherothrombosis. Front. Biosci. A J. Virtual Libr. 2008, 13, 5323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saquib, Q.; Siddiqui, M.A.; Ahmed, J.; Al-Salim, A.; Ansari, S.M.; Faisal, M.; Al-Khedhairy, A.A.; Musarrat, J.; AlWathnani, H.A.; Alatar, A.A. Hazards of low dose flame-retardants (BDE-47 and BDE-32): Influence on transcriptome regulation and cell death in human liver cells. J. Hazard. Mater. 2016, 308, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Rasola, A.; Bernardi, P. Mitochondrial permeability transition in Ca2+-dependent apoptosis and necrosis. Cell Calcium 2011, 50, 222–233. [Google Scholar] [CrossRef]
- Wei, T.; Chen, C.; Hou, J.; Xin, W.; Mori, A. Nitric oxide induces oxidative stress and apoptosis in neuronal cells. Biochim. Biophys. Acta BBA Mol. Cell Res. 2000, 1498, 72–79. [Google Scholar] [CrossRef] [Green Version]
- Almeida, A.; Bolaños, J.P. A transient inhibition of mitochondrial ATP synthesis by nitric oxide synthase activation triggered apoptosis in primary cortical neurons. J. Neurochem. 2001, 77, 676–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marshman, E.; Green, K.A.; Flint, D.J.; White, A.; Streuli, C.H.; Westwood, M. Insulin-like growth factor binding protein 5 and apoptosis in mammary epithelial cells. J. Cell Sci. 2003, 116, 675–682. [Google Scholar] [CrossRef] [Green Version]
- Lochrie, J.D.; Phillips, K.; Tonner, E.; Flint, D.J.; Allan, G.J.; Price, N.C.; Beattie, J. Insulin-like growth factor binding protein (IGFBP)-5 is upregulated during both differentiation and apoptosis in primary cultures of mouse mammary epithelial cells. J. Cell. Physiol. 2006, 207, 471–479. [Google Scholar] [CrossRef]
- Burger, A.M.; Leyland-Jones, B.; Banerjee, K.; Spyropoulos, D.D.; Seth, A.K. Essential roles of IGFBP-3 and IGFBP-rP1 in breast cancer. Eur. J. Cancer 2005, 41, 1515–1527. [Google Scholar] [CrossRef]
- Chen, Z.; Zhang, W.; Zhang, N.; Zhou, Y.; Hu, G.; Xue, M.; Liu, J.; Li, Y. Down-regulation of insulin-like growth factor binding protein 5 is involved in intervertebral disc degeneration via the ERK signalling pathway. J. Cell. Mol. Med. 2019, 23, 6368–6377. [Google Scholar] [CrossRef]
- Gleason, C.E.; Ning, Y.; Cominski, T.P.; Gupta, R.; Kaestner, K.H.; Pintar, J.E.; Birnbaum, M.J. Role of Insulin-Like Growth Factor-Binding Protein 5 (IGFBP5) in Organismal and Pancreatic β-Cell Growth. Mol. Endocrinol. 2010, 24, 178–192. [Google Scholar] [CrossRef] [Green Version]
- Han, W.; Fang, W.; Zhang, G.; Jia, L.; Lei, W.; Jia, R.; Li, T. Studies on cataclastic lithofacies and copper metallogenic regularity for the glutenite-type sareke copper deposit, Xinjiang. Acta Geol. Sin. Engl. Ed. 2017, 91, 210–211. [Google Scholar] [CrossRef] [Green Version]
- Akaogi, K.; Sato, J.; Okabe, Y.; Sakamoto, Y.; Yasumitsu, H.; Miyazaki, K. Synergistic growth stimulation of mouse fibroblasts by tumor-derived adhesion factor with insulin-like growth factors and insulin. Cell Growth Differ. Publ. Am. Assoc. Cancer Res. 1996, 7, 1671–1678. [Google Scholar]
- Wilson, H.-M.P.; Birnbaum, R.S.; Poot, M.; Quinn, L.S.; Swisshelm, K. Insulin-like growth factor binding protein-related protein 1 inhibits proliferation of MCF-7 breast cancer cells via a senescence-like mechanism. Cell Growth Differ. Publ. Am. Assoc. Cancer Res. 2002, 13, 205–214. [Google Scholar]
- Walker, G.; Antoniono, R.; Ross, H.J.; Paisley, T.; Oh, Y. Neuroendocrine-like differentiation of non-small cell lung carcinoma cells: Regulation by cAMP and the interaction of mac25/IGFBP-rP1 and 25.1. Oncogene 2006, 25, 1943–1954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, Y.; Chen, Z.; Miyazaki, K. Strong suppression of tumor growth by insulin-like growth factor-binding protein-related protein 1/tumor-derived cell adhesion factor/mac25. Cancer Sci. 2007, 98, 1055–1063. [Google Scholar] [CrossRef]
- Severino, V.; Alessio, N.; Farina, A.; Sandomenico, A.; Cipollaro, M.; Peluso, G.; Galderisi, U.; Chambery, A. Insulin-like growth factor binding proteins 4 and 7 released by senescent cells promote premature senescence in mesenchymal stem cells. Cell Death Dis. 2013, 4, e911. [Google Scholar] [CrossRef]
- Serachi, F.d.O.; Marie, S.K.N.; Oba-Shinjo, S.M. Relevant coexpression of STMN1, MELK and FOXM1 in glioblastoma and review of the impact of STMN1 in cancer biology. MedicalExpress 2017, 4, M170506. [Google Scholar] [CrossRef]
- Feng, Y.; Zhang, Y.; Cai, Y.; Liu, R.; Lu, M.; Li, T.; Fu, Y.; Guo, M.; Huang, H.; Ou, Y.; et al. A20 targets PFKL and glycolysis to inhibit the progression of hepatocellular carcinoma. Cell Death Dis. 2020, 11, 89. [Google Scholar] [CrossRef]
- Jonckheere, A.I.; Renkema, G.H.; Bras, M.; van den Heuvel, L.P.; Hoischen, A.; Gilissen, C.; Nabuurs, S.B.; Huynen, M.A.; de Vries, M.C.; Smeitink, J.A. A complex V ATP5A1 defect causes fatal neonatal mitochondrial encephalopathy. Brain 2013, 136, 1544–1554. [Google Scholar] [CrossRef] [Green Version]
- Pustišek, N.; Šitum, M. UV-radiation, apoptosis and skin. Coll. Antropol. 2011, 35, 339–341. [Google Scholar]
- Sayers, T.J. Targeting the extrinsic apoptosis signaling pathway for cancer therapy. Cancer Immunol. Immunother. 2011, 60, 1173–1180. [Google Scholar] [CrossRef] [PubMed]
- Rachow, S.; Zorn-Kruppa, M.; Ohnemus, U.; Kirschner, N.; Vidal-y-Sy, S.; von den Driesch, P.; Börnchen, C.; Eberle, J.; Mildner, M.; Vettorazzi, E. Occludin is involved in adhesion, apoptosis, differentiation and Ca2+-homeostasis of human keratinocytes: Implications for tumorigenesis. PLoS ONE 2013, 8, e55116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrallo-Gimeno, A.; Nieto, M.A. Evolutionary history of the Snail/Scratch superfamily. Trends Genet. 2009, 25, 248–252. [Google Scholar] [CrossRef] [PubMed]
- Chiang, C.; Ayyanathan, K. Snail/Gfi-1 (SNAG) family zinc finger proteins in transcription regulation, chromatin dynamics, cell signaling, development, and disease. Cytokine Growth Factor Rev. 2013, 24, 123–131. [Google Scholar] [CrossRef] [Green Version]
- Barrallo-Gimeno, A.; Nieto, M.A. The Snail genes as inducers of cell movement and survival: Implications in development and cancer. Development 2005, 132, 3151–3161. [Google Scholar] [CrossRef] [Green Version]
- Alimperti, S.; Andreadis, S.T. CDH2 and CDH11 act as regulators of stem cell fate decisions. Stem Cell Res. 2015, 14, 270–282. [Google Scholar] [CrossRef] [Green Version]
- Thompson, S.A.; Blazeski, A.; Copeland, C.R.; Cohen, D.M.; Chen, C.S.; Reich, D.M.; Tung, L. Acute slowing of cardiac conduction in response to myofibroblast coupling to cardiomyocytes through N-cadherin. J. Mol. Cell. Cardiol. 2014, 68, 29–37. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Groups | Olive Tail Moment (OTM) (Arbitrary Unit) | Tail Length (TL)(µm) | Tail Intensity (TI) (%) |
|---|---|---|---|
| Control | 0.43 ± 0.03 | 43.84 ± 1.83 | 2.35 ± 0.13 |
| EMS (2 mM) | 11.49 ± 0.72 ** | 107.43 ± 2.61 ** | 33.81 ± 1.65 ** |
| TCEP (µM) | |||
| 100 | 3.09 ± 0.24 ** | 84.39 ± 3.70 ** | 7.91 ± 0.32 ** |
| 200 | 5.05 ± 0.59 ** | 105.19 ± 3.86 ** | 11.28 ± 0.81 ** |
| 400 | 8.64 ± 0.77 ** | 123.71 ± 2.51 ** | 19.04 ± 0.40 ** |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
M. Al-Salem, A.; Saquib, Q.; Siddiqui, M.A.; Ahmad, J.; Al-Khedhairy, A.A. Tris(2-chloroethyl) Phosphate (TCEP) Elicits Hepatotoxicity by Activating Human Cancer Pathway Genes in HepG2 Cells. Toxics 2020, 8, 109. https://doi.org/10.3390/toxics8040109
M. Al-Salem A, Saquib Q, Siddiqui MA, Ahmad J, Al-Khedhairy AA. Tris(2-chloroethyl) Phosphate (TCEP) Elicits Hepatotoxicity by Activating Human Cancer Pathway Genes in HepG2 Cells. Toxics. 2020; 8(4):109. https://doi.org/10.3390/toxics8040109
Chicago/Turabian StyleM. Al-Salem, Abdullah, Quaiser Saquib, Maqsood A. Siddiqui, Javed Ahmad, and Abdulaziz A. Al-Khedhairy. 2020. "Tris(2-chloroethyl) Phosphate (TCEP) Elicits Hepatotoxicity by Activating Human Cancer Pathway Genes in HepG2 Cells" Toxics 8, no. 4: 109. https://doi.org/10.3390/toxics8040109
APA StyleM. Al-Salem, A., Saquib, Q., Siddiqui, M. A., Ahmad, J., & Al-Khedhairy, A. A. (2020). Tris(2-chloroethyl) Phosphate (TCEP) Elicits Hepatotoxicity by Activating Human Cancer Pathway Genes in HepG2 Cells. Toxics, 8(4), 109. https://doi.org/10.3390/toxics8040109