
Cognitive Impairment Induced by Lead Exposure during Lifespan: Mechanisms of Lead Neurotoxicity

 , , , , , , and
, , , , , , and 
Abstract
:1. Introduction
2. Lead Exposure during Developing Brain, Early Life, and Adulthood: Clinical Evidence
2.1. Cognitive Effects Associated with Prenatal and Early Postnatal Pb Exposure in Humans
2.2. Cognitive Impairments in Children Pb-Exposed
2.3. Adulthood Pb Exposure
3. Mechanisms Related to Cognitive Impairment Induced by Pb Exposure in Experimental Models
3.1. Neurotransmission and Long-Term Potentiation (LTP) Impairment by Pb Toxicity
3.2. Behavioral and Structural Alterations Induced by Pb Exposure in Animal Models
3.3. Redox and Energetic Imbalance Induced by Pb Toxicity
3.4. Strategies Used against Pb Neurotoxicity in Experimental Models
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Glass, T.A.; Bandeen-Roche, K.; McAtee, M.; Bolla, K.; Todd, A.C.; Schwartz, B.S. Neighborhood psychosocial hazards and the association of cumulative lead dose with cognitive function in older adults. Am. J. Epidemiol. 2009, 169, 683–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welton, M.; Rodriguez-Lainz, A.; Loza, O.; Brodine, S.; Fraga, M. Use of lead-glazed ceramic ware and lead-based folk remedies in a rural community of Baja California, Mexico. Glob. Health Promot. 2018, 25, 6–14. [Google Scholar] [CrossRef] [PubMed]
- Kordas, K.; Ravenscroft, J.; Cao, Y.; McLean, E.V. Lead Exposure in Low and Middle-Income Countries: Perspectives and Lessons on Patterns, Injustices, Economics, and Politics. Int. J. Environ. Res. Public Health 2018, 15, 2351. [Google Scholar] [CrossRef] [Green Version]
- Obeng-Gyasi, E. Sources of lead exposure in various countries. Rev. Environ. Health 2019, 34, 25–34. [Google Scholar] [CrossRef] [PubMed]
- O’Flaherty, E.J. Physiologically based models for bone-seeking elements. III. Human skeletal and bone growth. Toxicol. Appl. Pharmacol. 1991, 111, 332–341. [Google Scholar] [CrossRef]
- Papanikolaou, N.C.; Hatzidaki, E.G.; Belivanis, S.; Tzanakakis, G.N.; Tsatsakis, A.M. Lead toxicity update. A brief review. Med. Sci. Monit. 2005, 11, RA329–RA336. [Google Scholar]
- Goyer, R.A.; Mahaffey, K.R. Susceptibility to lead toxicity. Environ. Health Perspect. 1972, 2, 73–80. [Google Scholar] [CrossRef]
- White, R.F.; Diamond, R.; Proctor, S.; Morey, C.; Hu, H. Residual cognitive deficits 50 years after lead poisoning during childhood. Br. J. Ind. Med. 1993, 50, 613–622. [Google Scholar] [CrossRef] [Green Version]
- Nagpal, A.G.; Brodie, S.E. Supranormal electroretinogram in a 10-year-old girl with lead toxicity. Doc. Ophthalmol. 2009, 118, 163–166. [Google Scholar] [CrossRef]
- Lidsky, T.I.; Schneider, J.S. Lead neurotoxicity in children: Basic mechanisms and clinical correlates. Brain 2003, 126, 5–19. [Google Scholar] [CrossRef]
- Hou, S.; Yuan, L.; Jin, P.; Ding, B.; Qin, N.; Li, L.; Liu, X.; Wu, Z.; Zhao, G.; Deng, Y. A clinical study of the effects of lead poisoning on the intelligence and neurobehavioral abilities of children. Theor. Biol. Med. Model. 2013, 10, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sciarillo, W.G.; Alexander, G.; Farrell, K.P. Lead exposure and child behavior. Am. J. Public Health 1992, 82, 1356–1360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winneke, G.; Kraemer, U. Neuropsychological effects of lead in children: Interactions with social background variables. Neuropsychobiology 1984, 11, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Mostafa, G.A.; El-Shahawi, H.H.; Mokhtar, A. Blood lead levels in Egyptian children from high and low lead-polluted areas: Impact on cognitive function. Acta Neurol. Scand. 2009, 120, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Herman, D.S.; Geraldine, M.; Venkatesh, T. Evaluation, diagnosis, and treatment of lead poisoning in a patient with occupational lead exposure: A case presentation. J. Occup. Med. Toxicol. 2007, 2, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barth, A.; Schaffer, A.W.; Osterode, W.; Winker, R.; Konnaris, C.; Valic, E.; Wolf, C.; Rudiger, H.W. Reduced cognitive abilities in lead-exposed men. Int. Arch. Occup. Environ. Health 2002, 75, 394–398. [Google Scholar] [CrossRef] [PubMed]
- Maizlish, N.A.; Parra, G.; Feo, O. Neurobehavioural evaluation of Venezuelan workers exposed to inorganic lead. Occup. Environ. Med. 1995, 52, 408–414. [Google Scholar] [CrossRef] [Green Version]
- Weisskopf, M.G.; Hu, H.; Mulkern, R.V.; White, R.; Aro, A.; Oliveira, S.; Wright, R.O. Cognitive deficits and magnetic resonance spectroscopy in adult monozygotic twins with lead poisoning. Environ. Health Perspect. 2004, 112, 620–625. [Google Scholar] [CrossRef] [Green Version]
- Seo, J.; Lee, B.K.; Jin, S.U.; Park, J.W.; Kim, Y.T.; Ryeom, H.K.; Lee, J.; Suh, K.J.; Kim, S.H.; Park, S.J.; et al. Lead-induced impairments in the neural processes related to working memory function. PLoS ONE 2014, 9, e105308. [Google Scholar] [CrossRef]
- Ahamed, M.; Fareed, M.; Kumar, A.; Siddiqui, W.A.; Siddiqui, M.K. Oxidative stress and neurological disorders in relation to blood lead levels in children. Redox Rep. 2008, 13, 117–122. [Google Scholar] [CrossRef]
- Ziegler, E.E.; Edwards, B.B.; Jensen, R.L.; Mahaffey, K.R.; Fomon, S.J. Absorption and retention of lead by infants. Pediatr. Res. 1978, 12, 29–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabinowitz, M.B.; Kopple, J.D.; Wetherill, G.W. Effect of food intake and fasting on gastrointestinal lead absorption in humans. Am. J. Clin. Nutr. 1980, 33, 1784–1788. [Google Scholar] [CrossRef] [PubMed]
- Gulson, B.L.; Mahaffey, K.R.; Jameson, C.W.; Mizon, K.J.; Korsch, M.J.; Cameron, M.A.; Eisman, J.A. Mobilization of lead from the skeleton during the postnatal period is larger than during pregnancy. J. Lab. Clin. Med. 1998, 131, 324–329. [Google Scholar] [CrossRef]
- Choi, J.; Tanaka, T.; Koren, G.; Ito, S. Lead exposure during breastfeeding. Can. Fam. Physician 2008, 54, 515–516. [Google Scholar]
- Counter, S.A.; Buchanan, L.H.; Ortega, F. Neurophysiologic and neurocognitive case profiles of Andean patients with chronic environmental lead poisoning. J. Toxicol. Environ. Health A 2009, 72, 1150–1159. [Google Scholar] [CrossRef] [PubMed]
- Pottier, G.; Viau, M.; Ricoul, M.; Shim, G.; Bellamy, M.; Cuceu, C.; Hempel, W.M.; Sabatier, L. Lead Exposure Induces Telomere Instability in Human Cells. PLoS ONE 2013, 8, e67501. [Google Scholar] [CrossRef] [Green Version]
- Kasperczyk, S.; Dobrakowski, M.; Kasperczyk, A.; Ostalowska, A.; Birkner, E. The administration of N-acetylcysteine reduces oxidative stress and regulates glutathione metabolism in the blood cells of workers exposed to lead. Clin. Toxicol. (Phila.) 2013, 51, 480–486. [Google Scholar] [CrossRef]
- Kim, P.; Leckman, J.F.; Mayes, L.C.; Feldman, R.; Wang, X.; Swain, J.E. The plasticity of human maternal brain: Longitudinal changes in brain anatomy during the early postpartum period. Behav. Neurosci. 2010, 124, 695–700. [Google Scholar] [CrossRef] [Green Version]
- Barry, P.S.I. A Comparison of Concentrations of Lead in Human Tissues. Br. J. Ind. Med. 1975, 32, 119–139. [Google Scholar] [CrossRef]
- Gross, S.B.; Pfitzer, E.A.; Yeager, D.W.; Kehoe, R.A. Lead in human tissues. Toxicol. Appl. Pharmacol. 1975, 32, 638–651. [Google Scholar] [CrossRef]
- Schroeder, H.A.; Tipton, I.H. The human body burden of lead. Arch. Environ. Health 1968, 17, 965–978. [Google Scholar] [CrossRef] [PubMed]
- Goyer, R.A. Transplacental transport of lead. Environ. Health Perspect. 1990, 89, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Korpela, H.; Loueniva, R.; Yrjanheikki, E.; Kauppila, A. Lead and cadmium concentrations in maternal and umbilical cord blood, amniotic fluid, placenta, and amniotic membranes. Am. J. Obstet. Gynecol. 1986, 155, 1086–1089. [Google Scholar] [CrossRef]
- Ernhart, C.B.; Wolf, A.W.; Kennard, M.J.; Erhard, P.; Filipovich, H.F.; Sokol, R.J. Intrauterine exposure to low levels of lead: The status of the neonate. Arch. Environ. Health 1986, 41, 287–291. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.M.; Dai, Y.H.; Xie, X.H.; Fan, Z.Y.; Tan, Z.W.; Zhang, Y.F. Surveillance of childhood blood lead levels in 14 cities of China in 2004–2006. Biomed. Environ. Sci. 2009, 22, 288–296. [Google Scholar] [CrossRef]
- Wang, S.; Zhang, J. Blood lead levels in children, China. Environ. Res. 2006, 101, 412–418. [Google Scholar] [CrossRef]
- Gulson, B.; Taylor, A.; Eisman, J. Bone remodeling during pregnancy and post-partum assessed by metal lead levels and isotopic concentrations. Bone 2016, 89, 40–51. [Google Scholar] [CrossRef]
- Gulson, B.L.; Mizon, K.J.; Korsch, M.J.; Palmer, J.M.; Donnelly, J.B. Mobilization of lead from human bone tissue during pregnancy and lactation—A summary of long-term research. Sci. Total Environ. 2003, 303, 79–104. [Google Scholar] [CrossRef]
- Ettinger, A.S.; Roy, A.; Amarasiriwardena, C.J.; Smith, D.; Lupoli, N.; Mercado-Garcia, A.; Lamadrid-Figueroa, H.; Tellez-Rojo, M.M.; Hu, H.; Hernandez-Avila, M. Maternal blood, plasma, and breast milk lead: Lactational transfer and contribution to infant exposure. Environ. Health Perspect. 2014, 122, 87–92. [Google Scholar] [CrossRef]
- Neal, A.P.; Guilarte, T.R. Mechanisms of lead and manganese neurotoxicity. Toxicol. Res. (Camb.) 2013, 2, 99–114. [Google Scholar] [CrossRef]
- Baranowska-Bosiacka, I.; Kosinska, I.; Jamiol, D.; Gutowska, I.; Prokopowicz, A.; Rebacz-Maron, E.; Goschorska, M.; Olszowski, T.; Chlubek, D. Environmental Lead (Pb) Exposure Versus Fatty Acid Content in Blood and Milk of the Mother and in the Blood of Newborn Children. Biol. Trace Elem. Res. 2016, 170, 279–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanning, R.M.; Sandhu, R.; MacMillan, A.; Moss, L.; Tsuji, L.J.; Nieboer, E. Impact on blood Pb levels of maternal and early infant feeding practices of First Nation Cree in the Mushkegowuk Territory of northern Ontario, Canada. J. Environ. Monit. 2003, 5, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Zhuang, T.; Shi, J.; Liang, Y.; Song, M. Heavy metals in maternal and cord blood in Beijing and their efficiency of placental transfer. J. Environ. Sci. (China) 2019, 80, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Ozel, S.; Ozyer, S.; Aykut, O.; Cinar, M.; Yilmaz, O.H.; Caglar, A.; Engin-Ustun, Y. Maternal second trimester blood levels of selected heavy metals in pregnancies complicated with neural tube defects. J. Matern. Fetal Neonatal. Med. 2019, 32, 2547–2553. [Google Scholar] [CrossRef]
- Castro-Bedrinana, J.; Chirinos-Peinado, D.; Rios-Rios, E. Lead levels in pregnant women and newborns in la Oroya City, Peru. Rev. Peru. Med. Exp. Salud Publica 2013, 30, 393–398. [Google Scholar]
- Wojdon-Machala, H. Pattern of arterial blood pressure changes in repeated blood pressure determinations in a young female population. Pol. Tyg. Lek. 1976, 31, 441–444. [Google Scholar]
- Parajuli, R.P.; Fujiwara, T.; Umezaki, M.; Furusawa, H.; Ser, P.H.; Watanabe, C. Cord blood levels of toxic and essential trace elements and their determinants in the Terai region of Nepal: A birth cohort study. Biol. Trace Elem. Res. 2012, 147, 75–83. [Google Scholar] [CrossRef]
- Aelion, C.M.; Davis, H.T. Blood lead levels in children in urban and rural areas: Using multilevel modeling to investigate impacts of gender, race, poverty, and the environment. Sci. Total Environ. 2019, 694, 133783. [Google Scholar] [CrossRef]
- Ji, X.; He, H.; Ren, L.; Liu, J.; Han, C. Evaluation of blood zinc, calcium and blood lead levels among children aged 1–36 months. Nutr. Hosp. 2014, 30, 548–551. [Google Scholar] [CrossRef]
- Olympio, K.P.K.; Silva, J.; Silva, A.S.D.; Souza, V.C.O.; Buzalaf, M.A.R.; Barbosa, F., Jr.; Cardoso, M.R.A. Blood lead and cadmium levels in preschool children and associated risk factors in Sao Paulo, Brazil. Environ. Pollut. 2018, 240, 831–838. [Google Scholar] [CrossRef]
- Allen Counter, S.; Buchanan, L.H.; Ortega, F. Blood Lead Levels in Andean Infants and Young Children in Ecuador: An International Comparison. J. Toxicol. Environ. Health A 2015, 78, 778–787. [Google Scholar] [CrossRef] [PubMed]
- Yabe, J.; Nakayama, S.M.; Nakata, H.; Toyomaki, H.; Yohannes, Y.B.; Muzandu, K.; Kataba, A.; Zyambo, G.; Hiwatari, M.; Narita, D.; et al. Current trends of blood lead levels, distribution patterns and exposure variations among household members in Kabwe, Zambia. Chemosphere 2020, 243, 125412. [Google Scholar] [CrossRef] [PubMed]
- Seifu, S.; Tanabe, K.; Hauck, F.R. The Prevalence of Elevated Blood Lead Levels in Foreign-Born Refugee Children Upon Arrival to the U.S. and the Adequacy of Follow-up Treatment. J. Immigr. Minority Health 2020, 22, 10–16. [Google Scholar] [CrossRef]
- Caini, S.; Bendinelli, B.; Masala, G.; Saieva, C.; Assedi, M.; Querci, A.; Lundh, T.; Kyrtopoulos, S.A.; Palli, D. Determinants of Erythrocyte Lead Levels in 454 Adults in Florence, Italy. Int. J. Environ. Res. Public Health 2019, 16, 425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Victory, K.R.; Braun, C.R.; de Perio, M.A.; Calvert, G.M.; Alarcon, W. Elevated blood lead levels in adults-Missouri, 2013. Am. J. Ind. Med. 2019, 62, 347–351. [Google Scholar] [CrossRef]
- Specht, A.J.; Lin, Y.; Xu, J.; Weisskopf, M.; Nie, L.H. Bone lead levels in an environmentally exposed elderly population in shanghai, China. Sci. Total Environ. 2018, 626, 96–98. [Google Scholar] [CrossRef]
- Batra, J.; Thakur, A.; Meena, S.K.; Singh, L.; Kumar, J.; Juyal, D. Blood lead levels among the occupationally exposed workers and its effect on calcium and vitamin D metabolism: A case-control study. J. Fam. Med. Prim. Care 2020, 9, 2388–2393. [Google Scholar] [CrossRef]
- Kasperczyk, S.; Dobrakowski, M.; Kasperczyk, A.; Machnik, G.; Birkner, E. Effect of N-acetylcysteine administration on the expression and activities of antioxidant enzymes and the malondialdehyde level in the blood of lead-exposed workers. Environ. Toxicol. Pharmacol. 2014, 37, 638–647. [Google Scholar] [CrossRef]
- Nouioui, M.A.; Araoud, M.; Milliand, M.L.; Bessueille-Barbier, F.; Amira, D.; Ayouni-Derouiche, L.; Hedhili, A. Biomonitoring chronic lead exposure among battery manufacturing workers in Tunisia. Environ. Sci. Pollut. Res. Int. 2019, 26, 7980–7993. [Google Scholar] [CrossRef]
- Abdulah, D.M.; Al-Dosky, A.H.A.; Mohammed, A.H. Lead and zinc exposure in the blood of workers in municipal waste management. Environ. Sci. Pollut. Res. Int. 2020, 27, 11147–11154. [Google Scholar] [CrossRef]
- Barton, J.C.; Conrad, M.E.; Harrison, L.; Nuby, S. Effects of calcium on the absorption and retention of lead. J. Lab. Clin. Med. 1978, 91, 366–376. [Google Scholar] [PubMed]
- Ragan, H.A. The bioavailability of iron, lead and cadmium via gastrointestinal absorption: A review. Sci. Total Environ. 1983, 28, 317–326. [Google Scholar] [CrossRef]
- Tiwari, A.K.; Mahdi, A.A.; Zahra, F.; Sharma, S.; Negi, M.P. Evaluation of Low Blood Lead Levels and Its Association with Oxidative Stress in Pregnant Anemic Women: A Comparative Prospective Study. Indian J. Clin. Biochem. 2012, 27, 246–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jamiol-Milc, D.; Stachowska, E.; Janus, T.; Barcz, A.; Chlubek, D. Elaidic acid and vaccenic acid in the plasma of pregnant women and umbilical blood plasma. Pomeranian J. Life Sci. 2015, 61, 51–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamayo, Y.O.M.; Tellez-Rojo, M.M.; Trejo-Valdivia, B.; Schnaas, L.; Osorio-Valencia, E.; Coull, B.; Bellinger, D.; Wright, R.J.; Wright, R.O. Maternal stress modifies the effect of exposure to lead during pregnancy and 24-month old children’s neurodevelopment. Environ. Int. 2017, 98, 191–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Saleh, I.; Shinwari, N.; Nester, M.; Mashhour, A.; Moncari, L.; El Din Mohamed, G.; Rabah, A. Longitudinal study of prenatal and postnatal lead exposure and early cognitive development in Al-Kharj, Saudi Arabia: A preliminary results of cord blood lead levels. J. Trop. Pediatr. 2008, 54, 300–307. [Google Scholar] [CrossRef] [Green Version]
- Mamtani, M.; Patel, A.; Kulkarni, H. Association of the pattern of transition between arousal states in neonates with the cord blood lead level. Early Hum. Dev. 2008, 84, 231–235. [Google Scholar] [CrossRef]
- Gomaa, A.; Hu, H.; Bellinger, D.; Schwartz, J.; Tsaih, S.W.; Gonzalez-Cossio, T.; Schnaas, L.; Peterson, K.; Aro, A.; Hernandez-Avila, M. Maternal bone lead as an independent risk factor for fetal neurotoxicity: A prospective study. Pediatrics 2002, 110, 110–118. [Google Scholar] [CrossRef]
- Bellinger, D.; Leviton, A.; Sloman, J. Antecedents and correlates of improved cognitive performance in children exposed in utero to low levels of lead. Environ. Health Perspect. 1990, 89, 5–11. [Google Scholar] [CrossRef]
- Bellinger, D.; Leviton, A.; Waternaux, C.; Needleman, H.; Rabinowitz, M. Longitudinal analyses of prenatal and postnatal lead exposure and early cognitive development. N. Engl. J. Med. 1987, 316, 1037–1043. [Google Scholar] [CrossRef]
- Wasserman, G.A.; Liu, X.; Popovac, D.; Factor-Litvak, P.; Kline, J.; Waternaux, C.; LoIacono, N.; Graziano, J.H. The Yugoslavia Prospective Lead Study: Contributions of prenatal and postnatal lead exposure to early intelligence. Neurotoxicol. Teratol. 2000, 22, 811–818. [Google Scholar] [CrossRef]
- Wasserman, G.A.; Musabegovic, A.; Liu, X.; Kline, J.; Factor-Litvak, P.; Graziano, J.H. Lead exposure and motor functioning in 4(1/2)-year-old children: The Yugoslavia prospective study. J. Pediatr. 2000, 137, 555–561. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Hu, H.; Sanchez, B.N.; Peterson, K.E.; Ettinger, A.S.; Lamadrid-Figueroa, H.; Schnaas, L.; Mercado-Garcia, A.; Wright, R.O.; Basu, N.; et al. Childhood Blood Lead Levels and Symptoms of Attention Deficit Hyperactivity Disorder (ADHD): A Cross-Sectional Study of Mexican Children. Environ. Health Perspect. 2016, 124, 868–874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, S.C.; Kim, B.N.; Hong, Y.C.; Shin, M.S.; Yoo, H.J.; Kim, J.W.; Bhang, S.Y.; Cho, I.H.; Kim, H.W. Effect of environmental exposure to lead and tobacco smoke on inattentive and hyperactive symptoms and neurocognitive performance in children. J. Child Psychol. Psychiatry 2010, 51, 1050–1057. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Hu, H.; Wright, R.; Sanchez, B.N.; Schnaas, L.; Bellinger, D.C.; Park, S.K.; Martinez, S.; Hernandez-Avila, M.; Tellez-Rojo, M.M.; et al. Prenatal Lead Exposure Modifies the Impact of Maternal Self-Esteem on Children’s Inattention Behavior. J. Pediatr. 2015, 167, 435–441. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Cho, S.C.; Kim, B.N.; Hong, Y.C.; Shin, M.S.; Yoo, H.J.; Kim, J.W.; Bhang, S.Y. Association between blood lead levels (<5 mug/dL) and inattention-hyperactivity and neurocognitive profiles in school-aged Korean children. Sci. Total Environ. 2010, 408, 5737–5743. [Google Scholar] [CrossRef]
- Nigg, J.T.; Knottnerus, G.M.; Martel, M.M.; Nikolas, M.; Cavanagh, K.; Karmaus, W.; Rappley, M.D. Low blood lead levels associated with clinically diagnosed attention-deficit/hyperactivity disorder and mediated by weak cognitive control. Biol. Psychiatry 2008, 63, 325–331. [Google Scholar] [CrossRef] [Green Version]
- Kponee-Shovein, K.Z.; Weisskopf, M.G.; Grashow, R.; Rotem, R.S.; Coull, B.A.; Schnaas, L.; Hernandez-Chavez, M.D.C.; Sanchez, B.; Peterson, K.; Hu, H.; et al. Estimating the causal effect of prenatal lead exposure on prepulse inhibition deficits in children and adolescents. Neurotoxicology 2020, 78, 116–126. [Google Scholar] [CrossRef]
- Jansen, E.C.; Dunietz, G.L.; Dababneh, A.; Peterson, K.E.; Chervin, R.D.; Baek, J.; O’Brien, L.; Song, P.X.K.; Cantoral, A.; Hu, H.; et al. Cumulative Childhood Lead Levels in Relation to Sleep During Adolescence. J. Clin. Sleep Med. 2019, 15, 1443–1449. [Google Scholar] [CrossRef]
- Markowitz, M. Lead poisoning. Pediatr. Rev. 2000, 21, 327–335. [Google Scholar] [CrossRef]
- Gorospe, E.C.; Gerstenberger, S.L. Atypical sources of childhood lead poisoning in the United States: A systematic review from 1966-2006. Clin. Toxicol. (Phila.) 2008, 46, 728–737. [Google Scholar] [CrossRef] [PubMed]
- Tamayo y Ortiz, M.; Tellez-Rojo, M.M.; Hu, H.; Hernandez-Avila, M.; Wright, R.; Amarasiriwardena, C.; Lupoli, N.; Mercado-Garcia, A.; Pantic, I.; Lamadrid-Figueroa, H. Lead in candy consumed and blood lead levels of children living in Mexico City. Environ. Res. 2016, 147, 497–502. [Google Scholar] [CrossRef]
- Maxwell, E.D.; Neumann, C.M. Lead-tainted candy: A possible source of lead exposure to children. Toxicol. Environ. Chem. 2008, 90, 301–313. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention (CDC). Childhood lead poisoning associated with tamarind candy and folk remedies—California, 1999–2000. MMWR Morb. Mortal. Wkly. Rep. 2002, 51, 684–686. [Google Scholar]
- Bose, A.; Vashistha, K.; O’Loughlin, B.J. Azarcon por empacho—Another cause of lead toxicity. Pediatrics 1983, 72, 106–108. [Google Scholar]
- Baghurst, P.A.; McMichael, A.J.; Wigg, N.R.; Vimpani, G.V.; Robertson, E.F.; Roberts, R.J.; Tong, S.L. Environmental exposure to lead and children’s intelligence at the age of seven years. The Port Pirie Cohort Study. N. Engl. J. Med. 1992, 327, 1279–1284. [Google Scholar] [CrossRef]
- Wigg, N.R.; Vimpani, G.V.; McMichael, A.J.; Baghurst, P.A.; Robertson, E.F.; Roberts, R.J. Port Pirie Cohort study: Childhood blood lead and neuropsychological development at age two years. J. Epidemiol. Community Health 1988, 42, 213–219. [Google Scholar] [CrossRef] [Green Version]
- Kordas, K.; Casavantes, K.M.; Mendoza, C.; Lopez, P.; Ronquillo, D.; Rosado, J.L.; Vargas, G.G.; Stoltzfus, R.J. The association between lead and micronutrient status, and children’s sleep, classroom behavior, and activity. Arch. Environ. Occup. Health 2007, 62, 105–112. [Google Scholar] [CrossRef]
- Bhattacharya, A.; Shukla, R.; Dietrich, K.N.; Bornschein, R.L. Effect of early lead exposure on the maturation of children’s postural balance: A longitudinal study. Neurotoxicol. Teratol. 2006, 28, 376–385. [Google Scholar] [CrossRef]
- Bhattacharya, A.; Shukla, R.; Bornschein, R.L.; Dietrich, K.N.; Keith, R. Lead effects on postural balance of children. Environ. Health Perspect. 1990, 89, 35–42. [Google Scholar] [CrossRef]
- Needleman, H.L.; Gunnoe, C.; Leviton, A.; Reed, R.; Peresie, H.; Maher, C.; Barrett, P. Deficits in psychologic and classroom performance of children with elevated dentine lead levels. N. Engl. J. Med. 1979, 300, 689–695. [Google Scholar] [CrossRef] [PubMed]
- Needleman, H.L.; Riess, J.A.; Tobin, M.J.; Biesecker, G.E.; Greenhouse, J.B. Bone lead levels and delinquent behavior. JAMA 1996, 275, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Stroustrup, A.; Hsu, H.H.; Svensson, K.; Schnaas, L.; Cantoral, A.; Solano Gonzalez, M.; Torres-Calapiz, M.; Amarasiriwardena, C.; Bellinger, D.C.; Coull, B.A.; et al. Toddler temperament and prenatal exposure to lead and maternal depression. Environ. Health 2016, 15, 71. [Google Scholar] [CrossRef] [Green Version]
- Surkan, P.J.; Schnaas, L.; Wright, R.J.; Tellez-Rojo, M.M.; Lamadrid-Figueroa, H.; Hu, H.; Hernandez-Avila, M.; Bellinger, D.C.; Schwartz, J.; Perroni, E.; et al. Maternal self-esteem, exposure to lead, and child neurodevelopment. Neurotoxicology 2008, 29, 278–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moodie, S.; Ialongo, N.; Lopez, P.; Rosado, J.; Garcia-Vargas, G.; Ronquillo, D.; Kordas, K. The conjoint influence of home enriched environment and lead exposure on children’s cognition and behaviour in a Mexican lead smelter community. Neurotoxicology 2013, 34, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Pérez, H.; Nóbrega, D.; Aular, Y.; Núñez, C.; Pereira, K.; Gómez, M.E. Niveles de plomo en sangre, malondialdehido y vitaminas antioxidantes en escolares. Salus 2015, 19, 12–19. [Google Scholar]
- Meng, X.M.; Zhu, D.M.; Ruan, D.Y.; She, J.Q.; Luo, L. Effects of chronic lead exposure on 1H MRS of hippocampus and frontal lobes in children. Neurology 2005, 64, 1644–1647. [Google Scholar] [CrossRef] [PubMed]
- Shih, R.A.; Glass, T.A.; Bandeen-Roche, K.; Carlson, M.C.; Bolla, K.I.; Todd, A.C.; Schwartz, B.S. Environmental lead exposure and cognitive function in community-dwelling older adults. Neurology 2006, 67, 1556–1562. [Google Scholar] [CrossRef]
- Martin, D.; Glass, T.A.; Bandeen-Roche, K.; Todd, A.C.; Shi, W.; Schwartz, B.S. Association of blood lead and tibia lead with blood pressure and hypertension in a community sample of older adults. Am. J. Epidemiol. 2006, 163, 467–478. [Google Scholar] [CrossRef]
- Grashow, R.; Sparrow, D.; Hu, H.; Weisskopf, M.G. Cumulative lead exposure is associated with reduced olfactory recognition performance in elderly men: The Normative Aging Study. Neurotoxicology 2015, 49, 158–164. [Google Scholar] [CrossRef] [Green Version]
- Caruso, A.; Lucchini, R.; Toffoletto, F.; Porro, S.; Moroni, P.; Mascagni, P. Study of the olfactory function of a group of workers with significant lead exposure. G. Ital. Med. Lav. Ergon. 2007, 29 (Suppl. 3), 460–463. [Google Scholar] [PubMed]
- Fenga, C.; Gangemi, S.; Alibrandi, A.; Costa, C.; Micali, E. Relationship between lead exposure and mild cognitive impairment. J. Prev. Med. Hyg. 2016, 57, E205–E210. [Google Scholar] [PubMed]
- Kaminska, M.S.; Cybulska, A.M.; Panczyk, M.; Baranowska-Bosiacka, I.; Chlubek, D.; Grochans, E.; Stanislawska, M.; Jurczak, A. The Effect of Whole Blood Lead (Pb-B) Levels on Changes in Peripheral Blood Morphology and Selected Biochemical Parameters, and the Severity of Depression in Peri-Menopausal Women at Risk of Metabolic Syndrome or with Metabolic Syndrome. Int. J. Environ. Res. Public Health 2020, 17, 5033. [Google Scholar] [CrossRef] [PubMed]
- Jurczak, A.; Brodowska, A.; Szkup, M.; Prokopowicz, A.; Karakiewicz, B.; Loj, B.; Kotwas, A.; Brodowska, A.; Grochans, E. Influence of Pb and Cd levels in whole blood of postmenopausal women on the incidence of anxiety and depressive symptoms. Ann. Agric. Environ. Med. 2018, 25, 219–223. [Google Scholar] [CrossRef] [PubMed]
- Guilarte, T.R.; Opler, M.; Pletnikov, M. Is lead exposure in early life an environmental risk factor for Schizophrenia? Neurobiological connections and testable hypotheses. Neurotoxicology 2012, 33, 560–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, J.; Yan, L.; Guo, T.; Yang, S.; Guo, C.; Liu, Y.; Xie, Q.; Wang, J. Association of Typical Toxic Heavy Metals with Schizophrenia. Int. J. Environ. Res. Public Health 2019, 16, 4200. [Google Scholar] [CrossRef] [Green Version]
- Orisakwe, O.E. The role of lead and cadmium in psychiatry. N. Am. J. Med. Sci. 2014, 6, 370–376. [Google Scholar] [CrossRef] [Green Version]
- Bakulski, K.M.; Seo, Y.A.; Hickman, R.C.; Brandt, D.; Vadari, H.S.; Hu, H.; Park, S.K. Heavy Metals Exposure and Alzheimer’s Disease and Related Dementias. J. Alzheimer’s Dis. 2020, 76, 1215–1242. [Google Scholar] [CrossRef]
- Sadiq, S.; Ghazala, Z.; Chowdhury, A.; Busselberg, D. Metal toxicity at the synapse: Presynaptic, postsynaptic, and long-term effects. J. Toxicol. 2012, 2012, 132671. [Google Scholar] [CrossRef]
- Guilarte, T.R. Pb2+ inhibits NMDA receptor function at high and low affinity sites: Developmental and regional brain expression. Neurotoxicology 1997, 18, 43–51. [Google Scholar]
- Toscano, C.D.; Guilarte, T.R. Lead neurotoxicity: From exposure to molecular effects. Brain Res. Brain Res. Rev. 2005, 49, 529–554. [Google Scholar] [CrossRef] [PubMed]
- White, L.D.; Cory-Slechta, D.A.; Gilbert, M.E.; Tiffany-Castiglioni, E.; Zawia, N.H.; Virgolini, M.; Rossi-George, A.; Lasley, S.M.; Qian, Y.C.; Basha, M.R. New and evolving concepts in the neurotoxicology of lead. Toxicol. Appl. Pharmacol. 2007, 225, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Neal, A.P.; Worley, P.F.; Guilarte, T.R. Lead exposure during synaptogenesis alters NMDA receptor targeting via NMDA receptor inhibition. Neurotoxicology 2011, 32, 281–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guilarte, T.R.; McGlothan, J.L. Hippocampal NMDA receptor mRNA undergoes subunit specific changes during developmental lead exposure. Brain Res. 1998, 790, 98–107. [Google Scholar] [CrossRef]
- Liu, L.; Wong, T.P.; Pozza, M.F.; Lingenhoehl, K.; Wang, Y.; Sheng, M.; Auberson, Y.P.; Wang, Y.T. Role of NMDA receptor subtypes in governing the direction of hippocampal synaptic plasticity. Science 2004, 304, 1021–1024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toscano, C.D.; Hashemzadeh-Gargari, H.; McGlothan, J.L.; Guilarte, T.R. Developmental Pb2+ exposure alters NMDAR subtypes and reduces CREB phosphorylation in the rat brain. Brain Res. Dev. Brain Res. 2002, 139, 217–226. [Google Scholar] [CrossRef]
- Guilarte, T.R.; McGlothan, J.L.; Nihei, M.K. Hippocampal expression of N-methyl-D-aspartate receptor (NMDAR1) subunit splice variant mRNA is altered by developmental exposure to Pb(2+). Brain Res. Mol. Brain Res. 2000, 76, 299–305. [Google Scholar] [CrossRef]
- Nihei, M.K.; Guilarte, T.R. Molecular changes in glutamatergic synapses induced by Pb2+: Association with deficits of LTP and spatial learning. Neurotoxicology 2001, 22, 635–643. [Google Scholar] [CrossRef]
- Xiao, C.; Gu, Y.; Zhou, C.Y.; Wang, L.; Zhang, M.M.; Ruan, D.Y. Pb2+ impairs GABAergic synaptic transmission in rat hippocampal slices: A possible involvement of presynaptic calcium channels. Brain Res. 2006, 1088, 93–100. [Google Scholar] [CrossRef]
- Braga, M.F.; Pereira, E.F.; Albuquerque, E.X. Nanomolar concentrations of lead inhibit glutamatergic and GABAergic transmission in hippocampal neurons. Brain Res. 1999, 826, 22–34. [Google Scholar] [CrossRef]
- Busselberg, D. Calcium channels as target sites of heavy metals. Toxicol. Lett. 1995, 82–83, 255–261. [Google Scholar] [CrossRef]
- Simons, T.J.; Pocock, G. Lead enters bovine adrenal medullary cells through calcium channels. J. Neurochem. 1987, 48, 383–389. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.R.; Suszkiw, J.B. Extracellular inhibition and intracellular enhancement of Ca2+ currents by Pb2+ in bovine adrenal chromaffin cells. J. Neurophysiol. 1995, 74, 574–581. [Google Scholar] [CrossRef] [PubMed]
- Minnema, D.J.; Michaelson, I.A.; Cooper, G.P. Calcium efflux and neurotransmitter release from rat hippocampal synaptosomes exposed to lead. Toxicol. Appl. Pharmacol. 1988, 92, 351–357. [Google Scholar] [CrossRef]
- Bouton, C.M.; Frelin, L.P.; Forde, C.E.; Arnold Godwin, H.; Pevsner, J. Synaptotagmin I is a molecular target for lead. J. Neurochem. 2001, 76, 1724–1735. [Google Scholar] [CrossRef] [Green Version]
- Gill, K.D.; Gupta, V.; Sandhir, R. Ca2+/calmodulin-mediated neurotransmitter release and neurobehavioural deficits following lead exposure. Cell Biochem. Funct. 2003, 21, 345–353. [Google Scholar] [CrossRef]
- Toscano, C.D.; O’Callaghan, J.P.; Guilarte, T.R. Calcium/calmodulin-dependent protein kinase II activity and expression are altered in the hippocampus of Pb2+-exposed rats. Brain Res. 2005, 1044, 51–58. [Google Scholar] [CrossRef]
- Neal, A.P.; Stansfield, K.H.; Worley, P.F.; Thompson, R.E.; Guilarte, T.R. Lead exposure during synaptogenesis alters vesicular proteins and impairs vesicular release: Potential role of NMDA receptor-dependent BDNF signaling. Toxicol. Sci. 2010, 116, 249–263. [Google Scholar] [CrossRef] [Green Version]
- Ishihara, K.; Alkondon, M.; Montes, J.G.; Albuquerque, E.X. Nicotinic responses in acutely dissociated rat hippocampal neurons and the selective blockade of fast-desensitizing nicotinic currents by lead. J. Pharmacol. Exp. Ther. 1995, 273, 1471–1482. [Google Scholar]
- Braga, M.F.; Pereira, E.F.; Mike, A.; Albuquerque, E.X. Pb2+ via protein kinase C inhibits nicotinic cholinergic modulation of synaptic transmission in the hippocampus. J. Pharmacol. Exp. Ther. 2004, 311, 700–710. [Google Scholar] [CrossRef] [Green Version]
- Mike, A.; Pereira, E.F.; Albuquerque, E.X. Ca(2+)-sensitive inhibition by Pb(2+) of alpha7-containing nicotinic acetylcholine receptors in hippocampal neurons. Brain Res. 2000, 873, 112–123. [Google Scholar] [CrossRef]
- Ramirez Ortega, D.; Ovalle Rodriguez, P.; Pineda, B.; Gonzalez Esquivel, D.F.; Ramos Chavez, L.A.; Vazquez Cervantes, G.I.; Roldan Roldan, G.; de la Perez Cruz, G.; Diaz Ruiz, A.; Mendez Armenta, M.; et al. Kynurenine Pathway as a New Target of Cognitive Impairment Induced by Lead Toxicity During the Lactation. Sci. Rep. 2020, 10, 3184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golter, M.; Michaelson, I.A. Growth, behavior, and brain catecholamines in lead-exposed neonatal rats: A reappraisal. Science 1975, 187, 359–361. [Google Scholar] [CrossRef] [PubMed]
- Sobotka, T.J.; Cook, M.P. Postnatal lead acetate exposure in rats: Possible relationship to minimal brain dysfunction. Am. J. Ment. Defic. 1974, 79, 5–9. [Google Scholar]
- Sauerhoff, M.W.; Michaelson, I.A. Hyperactivity and brain catecholamines in lead-exposed developing rats. Science 1973, 182, 1022–1024. [Google Scholar] [CrossRef]
- Overmann, S.R. Behavioral effects of asymptomatic lead exposure during neonatal development in rats. Toxicol. Appl. Pharmacol. 1977, 41, 459–471. [Google Scholar] [CrossRef]
- Brady, K.; Herrera, Y.; Zenick, H. Influence of parental lead exposure on subsequent learning ability of offspring. Pharmacol. Biochem. Behav. 1975, 3, 561–565. [Google Scholar] [CrossRef]
- Padich, R.; Zenick, H. The effects of developmental and/or direct lead exposure on FR behavior in the rat. Pharmacol. Biochem. Behav. 1977, 6, 371–375. [Google Scholar] [CrossRef]
- Fan, G.; Feng, C.; Li, Y.; Wang, C.; Yan, J.; Li, W.; Feng, J.; Shi, X.; Bi, Y. Selection of nutrients for prevention or amelioration of lead-induced learning and memory impairment in rats. Ann. Occup. Hyg. 2009, 53, 341–351. [Google Scholar] [CrossRef] [Green Version]
- Rao Barkur, R.; Bairy, L.K. Evaluation of passive avoidance learning and spatial memory in rats exposed to low levels of lead during specific periods of early brain development. Int. J. Occup. Med. Environ. Health 2015, 28, 533–544. [Google Scholar] [CrossRef]
- Rahman, A.; Khan, K.M.; Al-Khaledi, G.; Khan, I.; Al-Shemary, T. Over activation of hippocampal serine/threonine protein phosphatases PP1 and PP2A is involved in lead-induced deficits in learning and memory in young rats. Neurotoxicology 2012, 33, 370–383. [Google Scholar] [CrossRef] [PubMed]
- Rice, D.C. Chronic low-lead exposure from birth produces deficits in discrimination reversal in monkeys. Toxicol. Appl. Pharmacol. 1985, 77, 201–210. [Google Scholar] [CrossRef]
- Rice, D.C.; Hayward, S. Comparison of visual function at adulthood and during aging in monkeys exposed to lead or methylmercury. Neurotoxicology 1999, 20, 767–784. [Google Scholar] [PubMed]
- Rice, D.C. Behavioral effects of lead in monkeys tested during infancy and adulthood. Neurotoxicol. Teratol. 1992, 14, 235–245. [Google Scholar] [CrossRef]
- Muller, Y.M.; Rivero, L.B.; Carvalho, M.C.; Kobus, K.; Farina, M.; Nazari, E.M. Behavioral impairments related to lead-induced developmental neurotoxicity in chicks. Arch. Toxicol. 2008, 82, 445–451. [Google Scholar] [CrossRef] [PubMed]
- Petit, T.L.; Alfano, D.P. Differential experience following developmental lead exposure: Effects on brain and behavior. Pharmacol. Biochem. Behav. 1979, 11, 165–171. [Google Scholar] [CrossRef]
- Tiffany-Castiglioni, E. Cell culture models for lead toxicity in neuronal and glial cells. Neurotoxicology 1993, 14, 513–536. [Google Scholar]
- Lorton, D.; Anderson, W.J. The effects of postnatal lead toxicity on the development of cerebellum in rats. Neurobehav. Toxicol. Teratol. 1986, 8, 51–59. [Google Scholar]
- Michaelson, I.A.; Sauerhoff, M.W. Animal models of human disease: Severe and mild lead encephalopathy in the neonatal rat. Environ. Health Perspect. 1974, 7, 201–225. [Google Scholar] [CrossRef] [Green Version]
- Krigman, M.R.; Druse, M.J.; Traylor, T.D.; Wilson, M.H.; Newell, L.R.; Hogan, E.L. Lead encephalopathy in the developing rat: Effect on cortical ontogenesis. J. Neuropathol. Exp. Neurol. 1974, 33, 671–686. [Google Scholar] [CrossRef]
- Patrick, G.W.; Anderson, W.J. Dendritic alterations of cortical pyramidal neurons in postnatally lead-exposed kittens: A Golgi-Cox study. Dev. Neurosci. 1995, 17, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Petit, T.L.; LeBoutillier, J.C. Effects of lead exposure during development on neocortical dendritic and synaptic structure. Exp. Neurol. 1979, 64, 482–492. [Google Scholar] [CrossRef]
- Lampert, P.W.; Schochet, S.S., Jr. Demyelination and remyelination in lead neuropathy. Electron microscopic studies. J. Neuropathol. Exp. Neurol. 1968, 27, 527–545. [Google Scholar] [PubMed]
- Baranowska-Bosiacka, I.; Struzynska, L.; Gutowska, I.; Machalinska, A.; Kolasa, A.; Klos, P.; Czapski, G.A.; Kurzawski, M.; Prokopowicz, A.; Marchlewicz, M.; et al. Perinatal exposure to lead induces morphological, ultrastructural and molecular alterations in the hippocampus. Toxicology 2013, 303, 187–200. [Google Scholar] [CrossRef]
- Gassowska, M.; Baranowska-Bosiacka, I.; Moczydlowska, J.; Frontczak-Baniewicz, M.; Gewartowska, M.; Struzynska, L.; Gutowska, I.; Chlubek, D.; Adamczyk, A. Perinatal exposure to lead (Pb) induces ultrastructural and molecular alterations in synapses of rat offspring. Toxicology 2016, 373, 13–29. [Google Scholar] [CrossRef]
- Gilbert, M.E.; Kelly, M.E.; Samsam, T.E.; Goodman, J.H. Chronic developmental lead exposure reduces neurogenesis in adult rat hippocampus but does not impair spatial learning. Toxicol. Sci. 2005, 86, 365–374. [Google Scholar] [CrossRef] [Green Version]
- Jaako-Movits, K.; Zharkovsky, T.; Romantchik, O.; Jurgenson, M.; Merisalu, E.; Heidmets, L.T.; Zharkovsky, A. Developmental lead exposure impairs contextual fear conditioning and reduces adult hippocampal neurogenesis in the rat brain. Int. J. Dev. Neurosci. 2005, 23, 627–635. [Google Scholar] [CrossRef]
- Fan, Y.; Zhao, X.; Yu, J.; Xie, J.; Li, C.; Liu, D.; Tang, C.; Wang, C. Lead-induced oxidative damage in rats/mice: A meta-analysis. J. Trace Elem. Med. Biol. 2020, 58, 126443. [Google Scholar] [CrossRef]
- Gargouri, M.; Ghorbel-Koubaa, F.; Bonenfant-Magne, M.; Magne, C.; Dauvergne, X.; Ksouri, R.; Krichen, Y.; Abdelly, C.; El Feki, A. Spirulina or dandelion-enriched diet of mothers alleviates lead-induced damages in brain and cerebellum of newborn rats. Food Chem. Toxicol. 2012, 50, 2303–2310. [Google Scholar] [CrossRef]
- Zhang, Y.M.; Liu, X.Z.; Lu, H.; Mei, L.; Liu, Z.P. Lipid peroxidation and ultrastructural modifications in brain after perinatal exposure to lead and/or cadmium in rat pups. Biomed. Environ. Sci. 2009, 22, 423–429. [Google Scholar] [CrossRef]
- Babu, M.S.; Gopal, N.V.; Reddy, K.P. Post natal antioxidant enzyme activity of rat brain regions during developmental lead exposure. J. Environ. Biol. 2007, 28, 21–27. [Google Scholar] [PubMed]
- Nehru, B.; Kanwar, S.S. N-acetylcysteine exposure on lead-induced lipid peroxidative damage and oxidative defense system in brain regions of rats. Biol. Trace Elem. Res. 2004, 101, 257–264. [Google Scholar] [CrossRef]
- Prasanthi, R.P.; Devi, C.B.; Basha, D.C.; Reddy, N.S.; Reddy, G.R. Calcium and zinc supplementation protects lead (Pb)-induced perturbations in antioxidant enzymes and lipid peroxidation in developing mouse brain. Int. J. Dev. Neurosci. 2010, 28, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Kang, J.C. Effects of sub-chronic exposure to lead (Pb) and ascorbic acid in juvenile rockfish: Antioxidant responses, MT gene expression, and neurotransmitters. Chemosphere 2017, 171, 520–527. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.H.; Shang, J.; Yan, L.; Wei, T.; Xiang, L.; Wang, H.L.; Cheng, J.; Xiao, G. Oxidative stress caused by lead (Pb) induces iron deficiency in Drosophila melanogaster. Chemosphere 2020, 243, 125428. [Google Scholar] [CrossRef] [PubMed]
- Holtzman, D.; Olson, J.E.; DeVries, C.; Bensch, K. Lead toxicity in primary cultured cerebral astrocytes and cerebellar granular neurons. Toxicol. Appl. Pharmacol. 1987, 89, 211–225. [Google Scholar] [CrossRef]
- Lindahl, L.S.; Bird, L.; Legare, M.E.; Mikeska, G.; Bratton, G.R.; Tiffany-Castiglioni, E. Differential ability of astroglia and neuronal cells to accumulate lead: Dependence on cell type and on degree of differentiation. Toxicol. Sci. 1999, 50, 236–243. [Google Scholar] [CrossRef] [Green Version]
- Peng, J.; Zhou, F.; Wang, Y.; Xu, Y.; Zhang, H.; Zou, F.; Meng, X. Differential response to lead toxicity in rat primary microglia and astrocytes. Toxicol. Appl. Pharmacol. 2019, 363, 64–71. [Google Scholar] [CrossRef]
- Liu, X.; Wu, J.; Shi, W.; Shi, W.; Liu, H.; Wu, X. Lead Induces Genotoxicity via Oxidative Stress and Promoter Methylation of DNA Repair Genes in Human Lymphoblastoid TK6 Cells. Med. Sci. Monit. 2018, 24, 4295–4304. [Google Scholar] [CrossRef]
- Ye, F.; Li, X.; Li, L.; Lyu, L.; Yuan, J.; Chen, J. The role of Nrf2 in protection against Pb-induced oxidative stress and apoptosis in SH-SY5Y cells. Food Chem. Toxicol. 2015, 86, 191–201. [Google Scholar] [CrossRef]
- Verma, S.K.; Dua, R.; Gill, K.D. Impaired energy metabolism after co-exposure to lead and ethanol. Basic Clin. Pharmacol. Toxicol. 2005, 96, 475–479. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Liu, J.Y.; Dong, J.X.; Xiao, Q.; Zhao, J.; Jiang, F.L. Toxicity of Pb(2+) on rat liver mitochondria induced by oxidative stress and mitochondrial permeability transition. Toxicol. Res. (Camb.) 2017, 6, 822–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, F.; Li, X.; Liu, Y.; Jiang, A.; Li, X.; Yang, L.; Chang, W.; Yuan, J.; Chen, J. CypD deficiency confers neuroprotection against mitochondrial abnormality caused by lead in SH-SY5Y cell. Toxicol. Lett. 2020, 323, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Baranowska-Bosiacka, I.; Gutowska, I.; Marchetti, C.; Rutkowska, M.; Marchlewicz, M.; Kolasa, A.; Prokopowicz, A.; Wiernicki, I.; Piotrowska, K.; Baskiewicz, M.; et al. Altered energy status of primary cerebellar granule neuronal cultures from rats exposed to lead in the pre- and neonatal period. Toxicology 2011, 280, 24–32. [Google Scholar] [CrossRef]
- Dabrowska, A.; Venero, J.L.; Iwasawa, R.; Hankir, M.K.; Rahman, S.; Boobis, A.; Hajji, N. PGC-1alpha controls mitochondrial biogenesis and dynamics in lead-induced neurotoxicity. Aging (Albany NY) 2015, 7, 629–647. [Google Scholar] [CrossRef] [Green Version]
- Breckenridge, D.G.; Stojanovic, M.; Marcellus, R.C.; Shore, G.C. Caspase cleavage product of BAP31 induces mitochondrial fission through endoplasmic reticulum calcium signals, enhancing cytochrome c release to the cytosol. J. Cell Biol. 2003, 160, 1115–1127. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Jiao, X.; An, Y.; Li, S.; Teng, X. Selenium against lead-induced apoptosis in chicken nervous tissues via mitochondrial pathway. Oncotarget 2017, 8, 108130–108145. [Google Scholar] [CrossRef] [Green Version]
- Gu, X.; Han, M.; Du, Y.; Wu, Y.; Xu, Y.; Zhou, X.; Ye, D.; Wang, H.L. Pb disrupts autophagic flux through inhibiting the formation and activity of lysosomes in neural cells. Toxicol. In Vitro 2019, 55, 43–50. [Google Scholar] [CrossRef]
- Habermann, E.; Crowell, K.; Janicki, P. Lead and other metals can substitute for Ca2+ in calmodulin. Arch. Toxicol. 1983, 54, 61–70. [Google Scholar] [CrossRef]
- Markovac, J.; Goldstein, G.W. Lead activates protein kinase C in immature rat brain microvessels. Toxicol. Appl. Pharmacol. 1988, 96, 14–23. [Google Scholar] [CrossRef] [Green Version]
- Long, G.J.; Rosen, J.F.; Schanne, F.A. Lead activation of protein kinase C from rat brain. Determination of free calcium, lead, and zinc by 19F NMR. J. Biol. Chem. 1994, 269, 834–837. [Google Scholar] [CrossRef]
- Azam, S.; Miksovska, J. Pb(2+) Binds to Downstream Regulatory Element Antagonist Modulator (DREAM) and Modulates Its Interactions with Binding Partners: A Link between Neuronal Calcium Sensors and Pb(2+) Neurotoxicity. ACS Chem. Neurosci. 2019, 10, 1263–1272. [Google Scholar] [CrossRef] [PubMed]
- Palanirajan, S.K.; Gummadi, S.N. Heavy-Metals-Mediated Phospholipids Scrambling by Human Phospholipid Scramblase 3: A Probable Role in Mitochondrial Apoptosis. Chem. Res. Toxicol. 2020, 33, 553–564. [Google Scholar] [CrossRef] [PubMed]
- Baranowska-Bosiacka, I.; Falkowska, A.; Gutowska, I.; Gassowska, M.; Kolasa-Wolosiuk, A.; Tarnowski, M.; Chibowska, K.; Goschorska, M.; Lubkowska, A.; Chlubek, D. Glycogen metabolism in brain and neurons—Astrocytes metabolic cooperation can be altered by pre- and neonatal lead (Pb) exposure. Toxicology 2017, 390, 146–158. [Google Scholar] [CrossRef] [PubMed]
- Gassowska, M.; Baranowska-Bosiacka, I.; Moczydlowska, J.; Tarnowski, M.; Pilutin, A.; Gutowska, I.; Struzynska, L.; Chlubek, D.; Adamczyk, A. Perinatal exposure to lead (Pb) promotes Tau phosphorylation in the rat brain in a GSK-3beta and CDK5 dependent manner: Relevance to neurological disorders. Toxicology 2016, 347–349, 17–28. [Google Scholar] [CrossRef]
- El-Missiry, M.A. Prophylactic effect of melatonin on lead-induced inhibition of heme biosynthesis and deterioration of antioxidant systems in male rats. J. Biochem. Mol. Toxicol. 2000, 14, 57–62. [Google Scholar] [CrossRef]
- Thangarajan, S.; Vedagiri, A.; Somasundaram, S.; Sakthimanogaran, R.; Murugesan, M. Neuroprotective effect of morin on lead acetate- induced apoptosis by preventing cytochrome c translocation via regulation of Bax/Bcl-2 ratio. Neurotoxicol. Teratol. 2018, 66, 35–45. [Google Scholar] [CrossRef]
- Bazrgar, M.; Goudarzi, I.; Lashkarbolouki, T.; Elahdadi Salmani, M. Melatonin ameliorates oxidative damage induced by maternal lead exposure in rat pups. Physiol. Behav. 2015, 151, 178–188. [Google Scholar] [CrossRef]
- Chintapanti, S.; Reddy, K.P.; Reddy, P.S. Behavioral and neurochemical consequences of perinatal exposure to lead in adult male Wistar rats: Protective effect by Centella asiatica. Environ. Sci. Pollut. Res. Int. 2018, 25, 13173–13185. [Google Scholar] [CrossRef]
- Gottipolu, R.R.; Davuljigari, C.B. Perinatal exposure to lead: Reduction in alterations of brain mitochondrial antioxidant system with calcium supplement. Biol. Trace Elem. Res. 2014, 162, 270–277. [Google Scholar] [CrossRef]
- Nam, S.M.; Chang, B.J.; Kim, J.H.; Nahm, S.S.; Lee, J.H. Ascorbic acid ameliorates lead-induced apoptosis in the cerebellar cortex of developing rats. Brain Res. 2018, 1686, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Nam, S.M.; Cho, I.S.; Seo, J.S.; Go, T.H.; Kim, J.H.; Nahm, S.S.; Chang, B.J.; Lee, J.H. Ascorbic Acid Attenuates Lead-Induced Alterations in the Synapses in the Developing Rat Cerebellum. Biol. Trace Elem. Res. 2019, 187, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Yousef, A.O.S.; Fahad, A.F.; Abdel Moneim, A.E.; Metwally, D.M.; El-Khadragy, M.F.; Kassab, R.B. The Neuroprotective Role of Coenzyme Q10 Against Lead Acetate-Induced Neurotoxicity Is Mediated by Antioxidant, Anti-Inflammatory and Anti-Apoptotic Activities. Int. J. Environ. Res. Public Health 2019, 16, 2895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karamian, R.; Komaki, A.; Salehi, I.; Tahmasebi, L.; Komaki, H.; Shahidi, S.; Sarihi, A. Vitamin C reverses lead-induced deficits in hippocampal synaptic plasticity in rats. Brain Res. Bull. 2015, 116, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Xue, W.Z.; Yang, Q.Q.; Chen, Y.; Zou, R.X.; Xing, D.; Xu, Y.; Liu, Y.S.; Wang, H.L. Kiwifruit Alleviates Learning and Memory Deficits Induced by Pb through Antioxidation and Inhibition of Microglia Activation In Vitro and In Vivo. Oxid. Med. Cell. Longev. 2017, 2017, 5645324. [Google Scholar] [CrossRef]
- Su, P.; Zhang, J.; Wang, S.; Aschner, M.; Cao, Z.; Zhao, F.; Wang, D.; Chen, J.; Luo, W. Genistein alleviates lead-induced neurotoxicity in vitro and in vivo: Involvement of multiple signaling pathways. Neurotoxicology 2016, 53, 153–164. [Google Scholar] [CrossRef]
- Cai, S.; Liu, J.; Shi, X.; Hu, S.; Zhao, L. Allicin alleviated learning and memory deficits caused by lead exposure at developmental stage. Life Sci. 2019, 231, 116532. [Google Scholar] [CrossRef]
- Li, W.H.; Shi, Y.C.; Tseng, I.L.; Liao, V.H. Protective efficacy of selenite against lead-induced neurotoxicity in Caenorhabditis elegans. PLoS ONE 2013, 8, e62387. [Google Scholar] [CrossRef] [Green Version]
- Maiti, A.K.; Saha, N.C.; More, S.S.; Panigrahi, A.K.; Paul, G. Neuroprotective Efficacy of Mitochondrial Antioxidant MitoQ in Suppressing Peroxynitrite-Mediated Mitochondrial Dysfunction Inflicted by Lead Toxicity in the Rat Brain. Neurotox. Res. 2017, 31, 358–372. [Google Scholar] [CrossRef]
- Hu, P.; Wang, M.; Chen, W.H.; Liu, J.; Chen, L.; Yin, S.T.; Yong, W.; Chen, J.T.; Wang, H.L.; Ruan, D.Y. Quercetin relieves chronic lead exposure-induced impairment of synaptic plasticity in rat dentate gyrus in vivo. Naunyn Schmiedebergs Arch. Pharmacol. 2008, 378, 43–51. [Google Scholar] [CrossRef]
- Ebuehi, O.A.; Ayinde, O.C. Neurobehavioural and neurotoxic effects of L-ascorbic acid and L-tryptophan in lead exposed rats. Niger. Q. J. Hosp. Med. 2012, 22, 240–244. [Google Scholar]
- Phyu, M.P.; Tangpong, J. Neuroprotective effects of xanthone derivative of Garcinia mangostana against lead-induced acetylcholinesterase dysfunction and cognitive impairment. Food Chem. Toxicol. 2014, 70, 151–156. [Google Scholar] [CrossRef] [PubMed]
- She, J.Q.; Wang, M.; Zhu, D.M.; Tang, M.; Chen, J.T.; Wang, L.; Ruan, D.Y. Monosialoanglioside (GM1) prevents lead-induced neurotoxicity on long-term potentiation, SOD activity, MDA levels, and intracellular calcium levels of hippocampus in rats. Naunyn Schmiedebergs Arch. Pharmacol. 2009, 379, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.M.; Wang, L.X.; Wang, Z.Y.; Sun, L.F.; Pan, X.D.; Pan, G.Q. Tanshinone IIA ameliorates lead (Pb)-induced cognitive deficits and oxidative stress in a rat pup model. Bratisl. Lek. Listy 2017, 118, 196–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phyu, M.P.; Tangpong, J. Protective effect of Thunbergia laurifolia (Linn.) on lead induced acetylcholinesterase dysfunction and cognitive impairment in mice. Biomed. Res. Int. 2013, 2013, 186098. [Google Scholar] [CrossRef] [PubMed]
- El-Sokkary, G.H.; Kamel, E.S.; Reiter, R.J. Prophylactic effect of melatonin in reducing lead-induced neurotoxicity in the rat. Cell. Mol. Biol. Lett. 2003, 8, 461–470. [Google Scholar]



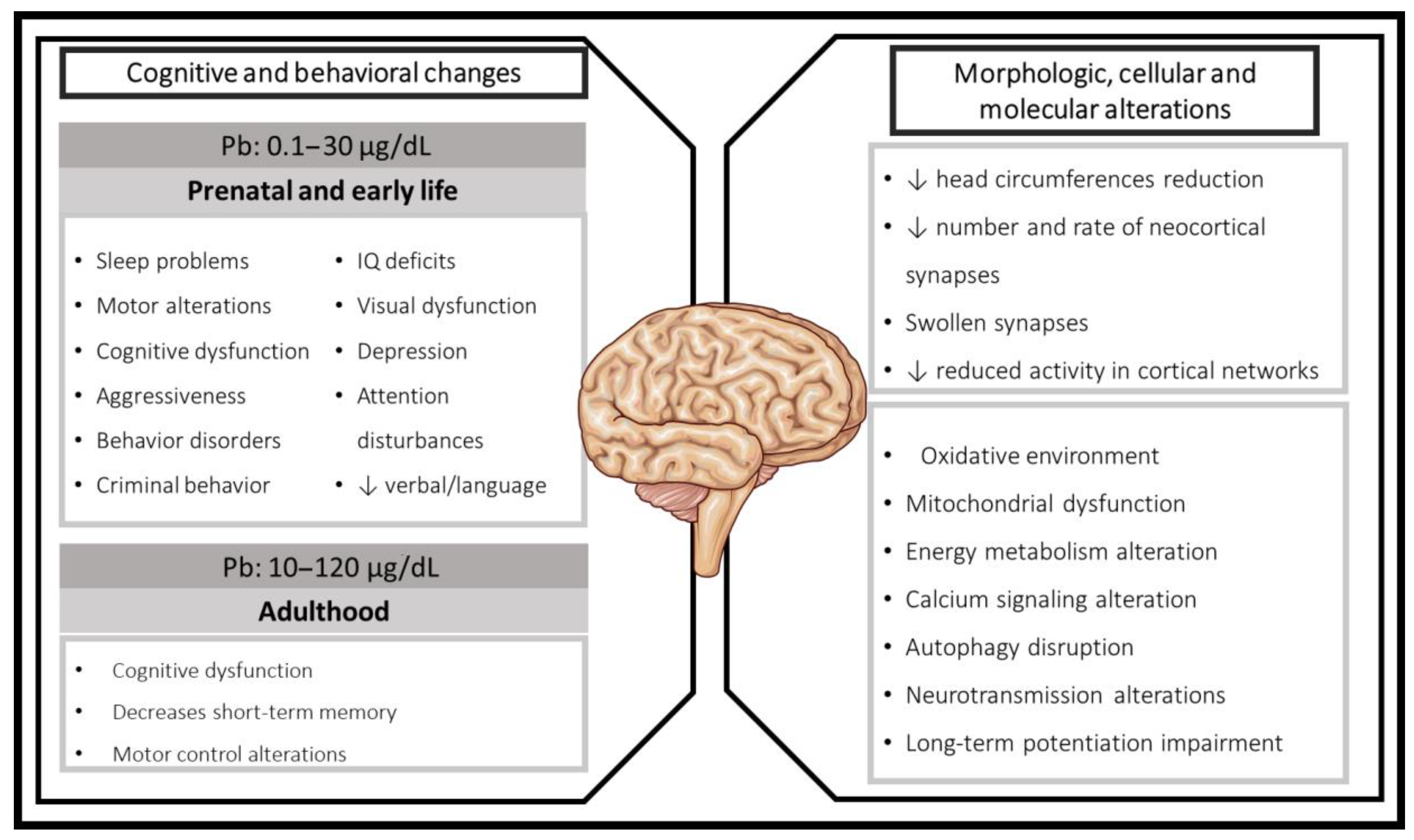{kind=link}
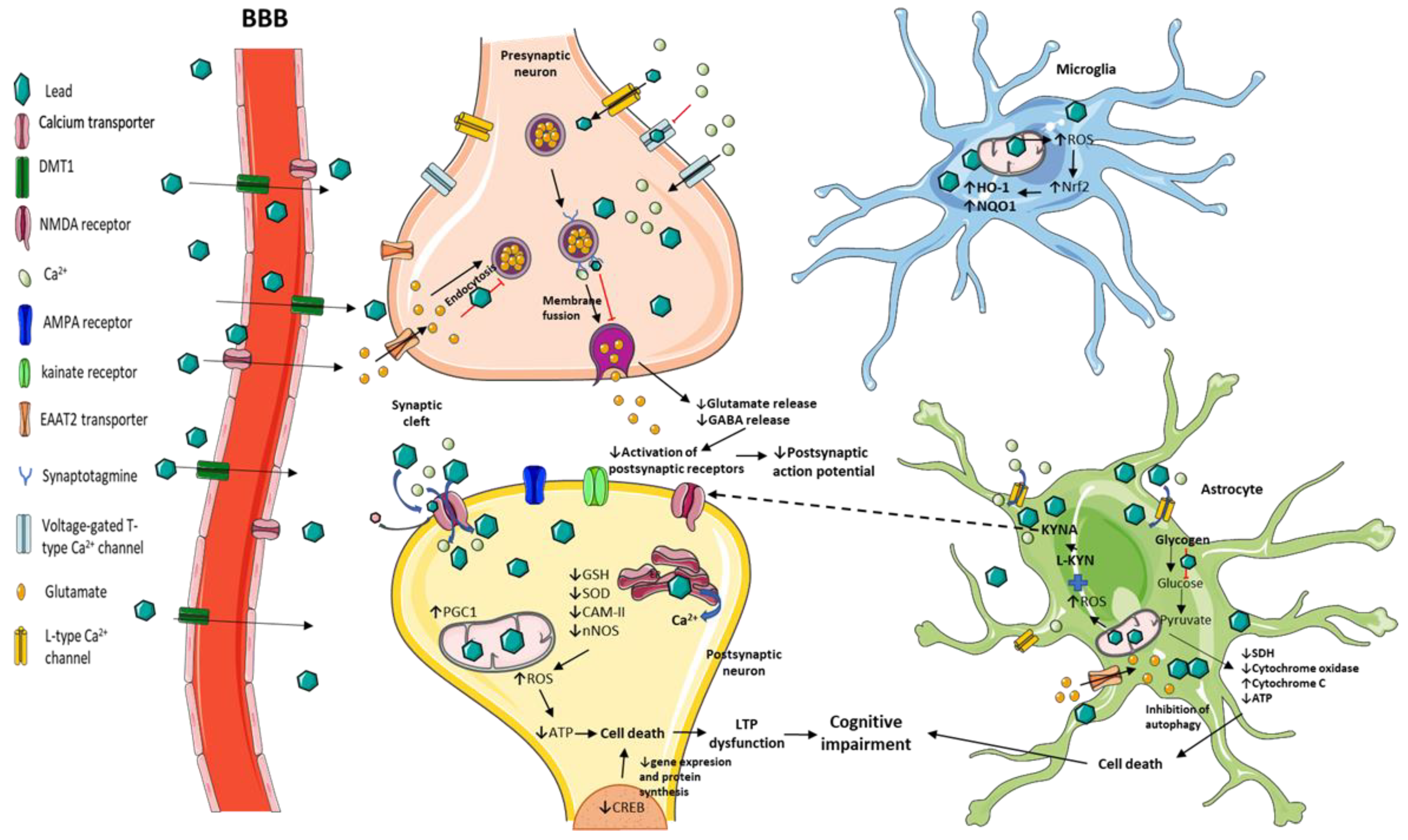{kind=link}
| Population (Age Range) | Study Design and Subjects | Location | Lead Levels | Outcomes | Reference |
|---|---|---|---|---|---|
| Children | |||||
| 2–4 years | Cross-sectional study; 76 children. Gesell Developmental Scale: t-test comparisons. The Achenbach Child Behavior Checklist (CBCL): χ2 test. Scores on each behavior factor and the total behavioral score were analyzed by the rank-sum test. | Xi’an, China | Blood: 4–246 µg/L |
| [11] |
| 2–5 years | Cross-sectional study; 201 children. Child Behavior Checklist (CBCL): χ2 test and t-test comparisons. Total Behavior Problem Score (TBPS) based upon the percentile ranking of raw scores and in the clinical range (90th percentile). Multiple regression analysis for the likelihood ratio and influential factor on the TBPS was measured from the mothers using the Center for Epidemiologic Studies Depression Scale (CES-D). | Baltimore, United States | Blood: 2–30 µg/dL |
| [12] |
| 8–9 years | Cross-sectional study; 167 children. Wechsler Intelligence Scale for Children (WISC), Göttinger Formreproduktions-Test (GFT), Bender Gestalt-Test (German version), Benton Test, Diagnostics for Cerebral Damage Test and Wiener Reaction Device. Duisburg study t tests for correlating samples. Stolberg sample associations were tested using of stepwise multiple regression analysis. | Duisburg city and Stolberg city, Germany | Baby teeth: 1.4–38.5 µg/g Blood: 6.8–34 µg/100 mL |
| [13] |
| 6–12 years | 100 children Wechsler Intelligence Scale for Children, 3rd edition (WISC-III) Spearman’s rank correlation, logistic and linear regressions to test independent predictors for impairment of cognitive function and the relationship between blood lead levels and cognitive function. Receiver operating characteristic (ROC) curve was used to calculate the best cut-off value of blood lead levels (based on the highest sensitivity with the lowest false-positive results) above which the majority of the children have cognitive dysfunction. | Cairo, Egypt | Blood: 3–28 µg/dL |
| [14] |
| Adults | |||||
| 50–60 years | Cross-sectional study; 53 adults. Wechsler adult intelligence scale-revised (WAIS-R), Wechsler memory scale (WMS), a test of attention and visuomotor tracking (trail making), test of verbal fluency (FAS), test of non-verbal reasoning (Raven progressive matrices), test of motor speed (finger tapping) and inventory of current mood (POMS). Wilcoxon signed ranks test, used to compare matched pairs of subjects exposed to lead and controls; and the Mann–Whitney test, used to compare entire groups. χ2 test was used to evaluate the distribution of categorical frequencies. | Boston, United States | Blood: 60–120 µg/dL |
| [8] |
| 50–70 years | Cross-sectional study; 1033 adults. A battery of 20 cognitive test results was standardized and collapsed into 7 cognitive domain scores. All 7 domain scores were standardized for direction so that a negative regression coefficient indicated worse performance. χ2 test for interaction between tibia lead, Neighborhood Psychosocial Hazards Scale (NPH). NPH scale and the 7 domains of Cognitive function in the Baltimore Memory study. Multilevel regression models were used to account for the nesting of persons within neighborhoods. | Baltimore, USA | Tibia 18.8 ± 11.6 µg/g |
| [1] |
| Male workers | |||||
| 22 years | Case report | India | Blood: 128.3 µg/dL |
| [15] |
| 39–50 years | Cross-sectional study 47 adults exposed to Pb for 11.7 ± 9 years. Modified version of the Wisconsin card sorting test, the block design test, the visual recognition test, choice reaction, simple reaction, and digit symbol substitution. One-tailed t-test for independent samples was used for the following tasks: block design and visual recognition tests, simple reaction time, and digit symbol substitution. The results of the choice reaction were analyzed by multivariate analysis of variance. Since scores of the Wisconsin test were not distributed normally, they were analyzed by one-tailed Mann– Whitney test. Because of multiple univariate testing, Bonferroni correction was applied. | Germany | Blood: 30.8 + 11.2 µg/100 mL |
| [16] |
| 25–67 years | Cross-sectional study: 100 adults exposed to Pb for 1–7 years. WHO neurobehavioral core test battery. Multiple linear regression of neurobehavioral function in workers and lead exposure indices. Correlation coefficients (Pearson r) between blood lead concentration and covariates Analysis of covariance for dichotomous exposure variables. | Venezuela | Blood: 9–60 µg/dL |
| [17] |
| 71 years | Case report Twins | Boston, United States | Blood: 15–125 µg/dL Patella: 119–343 µg/g Tibia: 79–189 µg/g |
| [18] |
| Female workers | |||||
| 55–65 years | Cross-sectional study: 31 adults exposed to Pb for 1.4–20.7 years. N-back working memory paradigm. Mean values of continuous variables were compared using the Student’s t-test. Pearson correlation analyses between mean percentage changes of activated brain regions and working memory performance. The effects of blood lead on percent signal change by multiple regression analysis. | Korea | Blood: 0.88–13.5 µg/dL |
| [19] |
| Population | Study Design and Subjects | Study Period and Location | Sample | Lead Levels | Reference |
|---|---|---|---|---|---|
| Pregnant woman | |||||
| Cross-sectional study 53 female patients 29.11 ± 4.77 years | 2007–2008 Szczecin, Poland | Blood Milk | 1.290 ± 0.578 μg/dL 0.174 ± 1.15 μg/dL | [41] | |
| Cross-sectional study 91 female patients | Five months Ontario, Canada | Blood Milk | 22.9 ± 12.5 μg/L 2.08 ± 1.67 μg/L | [42] | |
| Cross-sectional study 156 female patients 29–34 years | November 2015–December 2016 Beijing, China | Blood Cord blood | 23.1 ± 21.2 μg/L 14.2 ± 7.6 μg/L | [43] | |
| Prospective case-control study 21 pregnant woman 26.5 ± 5.5 years | October 2016 to April 2017 Ankara, Turkey | Blood | 12.3 μg/L | [44] | |
| Cross-sectional study 41 patients (29 ± 6 years) | 2003–2004 Oroya City, Peru | Blood Cord blood | 27.4 ± 15.6 μg/L 19.0 ± 12.6 μg/L 319 ± 215 µg/100 g | [45] | |
| Newborns | |||||
| Cross-sectional study 53 newborns | 2007–2008 Szczecin, Poland | Blood | ~1 ug/dL | [41] | |
| Cross-sectional study 91 infants (0–4 months) | Five months Ontario, Canada | Cord Blood | 20.8 ± 16.7 μg/L 16.7 ± 10.4 μg/L | [42,46] | |
| Birth cohort study 79 newborns | September, October 2008 Terai, Nepal | Cord blood | 31.7 + 35.36 μg/L | [47] | |
| Children | |||||
| Cross-sectional study 1–5 years | 2011–2016 South Carolina, United States | Blood | 0.27–20.4 μg/dL | [48] | |
| Cross-sectional study 120 children Age 1–36 months | 2010–2011 Shandong, China | Blood | 42.18 ± 12.13 μg/L | [49] | |
| Cross-sectional study 2397 children 1–4 years | 2013 Sao Paolo, Brazil | Blood | 6.3–8.1 μg/dL | [50] | |
| Cross-sectional study 130 children 0.33-5.8 years | 2003–2013 Ecuador | Blood | 29.4 ± 24.3 µg/dL | [51] | |
| Cross-sectional study 561 children 3 months–9 years | July–August 2017 Kabwe, Zambia | Blood | 5–100 μg/dL | [52] | |
| Cross-sectional study 301 children 6 months–16 years | 2003–2016 Refugees in the United States | Blood | 1–27 μg/dL | [53] | |
| Adults | |||||
| Case control study 454 adults (35–65 years) | 2009 Florence | Blood | 86.1 μg/L | [54] | |
| Cross-sectional study 15,123 residents (≥16 years). | 2009–2015 Missouri | Blood | 1.5–≥25 μg/dL | [55] | |
| Cross-sectional study 30 participants 70.4 ± 9.1 years | 2009–2010 Shanghai, China | Blood Bone | 1.0–23.2 μg/dL 0.9–15 μg/g | [56] | |
| Cross-sectional, case control 80 adults (male and female; 15–47 years) | January 2017–July 2018 India | Blood | 38.02 ± 19.92 μg/dL | [57] | |
| Cross-sectional study 171 male adults 40.9 ± 8.25 years | 2007–2008 Poland | Blood | 42.9 ± 6.3 μg/dL | [58] | |
| Cross-sectional study 52 male adults (39 ± 9 years) | 2019 Tunisia | Blood Urine Hair | 101–535.3 μg/dL 15.8–72.0 μg/dL 2.2 ± 0.1 μg/g | [59] | |
| Cross sectional study 139 adults (male and female; 16–67 years) | 2018 Iraq | Blood | 5.77 μg/dL | [60] |
| Animal Model. | Doses | Behavioral Alterations/Cognitive Impairment | Biochemical and Morphological Alterations | Therapy | Reference |
|---|---|---|---|---|---|
| Male Wistar rats | 20 mg PbAc/kg i.p. for 14 days | ↓ Rotarod activity ↓ Time in open field test ↑ Time in adhesive removal test ↑ Escape latency time in Morris water maze ↑ Immobility time in the forced swim test ↓ Grip strength time in string test | ↑ Lipid peroxidation, nitric oxide, and protein carbonile ↓ Superoxide dismutase, catalase, glutathione peroxidase activity, glutathione reductase, and glutathione-S-transferase activity ↓ Glutathione, vitamin C and E ↓ Na+/K+ ATPase activity ↓ Acetyl cholinesterase ↓ Survival cell number ↑ Bax/Bcl-2 ratio ↓ Mitochondrial cytochrome c ↑ Cytosolic cytochrome c | Morin (40 mg/kg) orally 2 h after the administration of PbAc for 14 days). Attenuates all the behavioral and biochemical alterations induced by PbAc | [187] |
| Pregnant Wistar rats | 0.2% PbAc, daily from the 5th day of gestation until weaning (PD21). | ↓ Rotarod activity ↓ Number of rearing | ↑ TBARS ↓ SOD activity ↓ GPx activity ↓ Cerebellar Purkinje cell number | Melatonin (10 mg/kg) once daily through oral gavage during the gestational and lactational period. Attenuates the effect on behavioral and biochemical alterations induced by Pb. | [188] |
| Wistar rats | 0.015% PbAc in drinking water from gestation until PD21 | ↓ Exploratory, locomotory, cognitive impairment ↑ Analgesic reaction time | ↑ Lipid peroxidation ↓ GSH levels ↓ The activity of SOD, catalase, GPx and glutathione reductase | Extract of Centella asiatica (200 mg of crude/kg body weight/day) from PD21 to PD60. Prevents the behavioral and brain redox alterations induced by Pb. | [189] |
| Wistar rats | 0.2% PbAc in drinking water from gestational day 6 to PD21 | ↓ Activity of serum ceruloplasmin oxidase (Cp), Mn-SOD, Cu/Zn-SOD, GPx, CAT, and xanthine oxidase ↑ Malondialdehyde (MDA) levels increased in the cerebellum and hippocampus | Calcium supplement (0.02% in Pb–water) Reversed Pb toxicity | [190] | |
| Sprague Dawley rats | 0.2% PbAc in drinking water during the gestational period | ↑ Apoptotic cell deaths ↓ The number of Purkinje cells in the cerebellum ↓ Synaptophysin and NMDAr subtype 1 density | Ascorbic acid (100 mg/kg) Reduces Bax and apoptotic neuronal death and prevents the impairment in cerebellar synaptic proteins. | [191,192] | |
| Wistar rats | 20 mg PbAc/kg i.p. for 7 days | ↑ Cortical lipid peroxidation, nitrate/nitrite levels, and inducible nitric oxide synthase expression ↓ Glutathione content, superoxide dismutase, catalase, glutathione peroxidase, glutathione reductase activity and mRNA expression ↓ Nuclear factor erythroid 2–related factor 2 (Nrf2) and hemoxygenase-1 (HO-1) expression. ↑ The cortical levels of serotonin, dopamine, norepinephrine, GABA, and glutamate, ↓ The level of ATP | Coenzyme Q10 (10 mg/kg i.p./7 days) Restores the balance between oxidants and antioxidants, inhibiting the apoptotic cascade, and modulating cortical neurotransmission and energy metabolism. | [193] | |
| Male Wistar rats | 0.2% PbAc daily for 3 months | ↑ Malondialdehyde and total oxidant status in plasma ↓ The excitatory postsynaptic potentials slope and the population spike amplitude | Vitamin C (150 mg/kg, daily for 3 months) Increases total antioxidant capacity inhibiting the effects of Pb | [194] | |
| Sprague-Dawley (SD) rats | 0.2% PbAc during the gestational period to PD43 | Induces working memory deficits | ↓ Dendritic spine density ↓ SOD and GPx activity and expression in the hippocampus. | Kiwi fruit (12 mg/kg daily from 7 to 9 weeks old) Alleviates cognitive deficits and restores the antioxidant environment | [195] |
| Male Sprague–Dawley rats | 200 ppm PbAc for 8 weeks | Impaired spatial reference memory | ↑ Apoptotic cell death and the expression of Bax ↓ Bcl-2 protein expression | Genistein (1 mg/kg/day) Diminishes impairment in cognitive function and protects neurons from Pb toxicity. N-acetyl-l-cysteine (NAC; 1 mg/kg/day) Prevents cognitive impairment and reduced cell death. | [196] |
| Wistar rats | 0.2% PbAc from gestation to PD21 | Induces learning and memory deficits | ↓ GSH content ↓ SOD activity ↑ MDA | Allicin (30 mg/kg allicin twice a day for 21 days) Alleviates learning and memory deficits and reverts biochemical parameters altered by Pb. | [197] |
| Caenorhabditis elegans | 100 µM of PbAc for 24 h | Decline of locomotion behaviors (frequencies of body bends, head thrashes, and reversal) | ↑ Intracellular ROS production | Se(IV) (0.01 µM) Ameliorates locomotion behavioral alterations induced by Pb. Decreases intracellular ROS and protects sensory neurons from Pb. | [198] |
| Sprague Dawley rats | 100 and 400 ppm PbAc for 15 days | ↑ Synaptosomal lipid peroxidation, protein carbonylation and 3-nitrotyrosine levels ↓ Thiol content Inhibits complexes II, III and IV of the mitochondrial respiratory and decreases ATP and transmembrane potential | MitoQ (500 μM for 15 days). Prevents the oxidative damage induced by Pb. Alleviates mitochondrial dysfunction | [199] | |
| Wistar rats | 0.2% PbAc in drinking water | Impairment of synaptic plasticity | Quercetin (30 mg/kg, for 7 days) Prevent Pb alterations. | [200] | |
| Male Sprague Dawley rats | 75 mg/kg PbAc for 4 weeks | Anxiety and Aggression | ↓ Serotonin ↓ Reduced glutathione levels, antioxidant enzyme activity ↑ Lipid peroxidation and brain protein contents | Ascorbic acid (40 mg/kg) Attenuates oxidative stress and abnormalities in behavior induced by Pb. Tryptophan (20 mg/kg) Ameliorates altered neurobehavior induced by Pb, with no significant effect on Pb induced oxidative stress in the brain. | [201] |
| ICR mice | 1% PbAc in drinking water for 38 days | Depression Memory loss | ↓ AChE activity ↑ Malondialdehyde levels | Xanthone derivative of Garcinia mangostana (100 and 200 mg/kg) Increases AChE activity and decreases lipid peroxidation Ameliorates depression-like effect and memory loss induced by Pb | [202] |
| Wistar rats | 0.2% PbAc solution was injected intraperitoneally | Impaired synaptic transmission and plasticity in the hippocampus ↑ SOD activity and malondialdehyde ↑ Intracellular calcium | Monosialoganglioside (100 μg/mL microinjection into hippocampus). Prevents the impairments of synaptic plasticity, antioxidant system function, and intracellular calcium levels | [203] | |
| Wistar rats | 0.2% PbAc for 4 weeks | Affects spatial reference memory ability. Increases in the scape latency | ↓ Body weight ↓ Decrease of antioxidant activities and BDNF content | Tanshinone IIA (4 and 8 mg/kg) Improves antioxidant activities by increasing SOD activity and GSH and decreasing MDA levels Prevents the impairment in the spatial reference memory. | [204] |
| ICR mice | drinking water (1 g/L), 38 days | Impairment memory | ↓ AchE activity | Thunbergia laurifolia (100 and 200 mg/kg/day). Attenuates cognitive impairment and increased AchE activity. | [205] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramírez Ortega, D.; González Esquivel, D.F.; Blanco Ayala, T.; Pineda, B.; Gómez Manzo, S.; Marcial Quino, J.; Carrillo Mora, P.; Pérez de la Cruz, V. Cognitive Impairment Induced by Lead Exposure during Lifespan: Mechanisms of Lead Neurotoxicity. Toxics 2021, 9, 23. https://doi.org/10.3390/toxics9020023
Ramírez Ortega D, González Esquivel DF, Blanco Ayala T, Pineda B, Gómez Manzo S, Marcial Quino J, Carrillo Mora P, Pérez de la Cruz V. Cognitive Impairment Induced by Lead Exposure during Lifespan: Mechanisms of Lead Neurotoxicity. Toxics. 2021; 9(2):23. https://doi.org/10.3390/toxics9020023
Chicago/Turabian StyleRamírez Ortega, Daniela, Dinora F. González Esquivel, Tonali Blanco Ayala, Benjamín Pineda, Saul Gómez Manzo, Jaime Marcial Quino, Paul Carrillo Mora, and Verónica Pérez de la Cruz. 2021. "Cognitive Impairment Induced by Lead Exposure during Lifespan: Mechanisms of Lead Neurotoxicity" Toxics 9, no. 2: 23. https://doi.org/10.3390/toxics9020023
APA StyleRamírez Ortega, D., González Esquivel, D. F., Blanco Ayala, T., Pineda, B., Gómez Manzo, S., Marcial Quino, J., Carrillo Mora, P., & Pérez de la Cruz, V. (2021). Cognitive Impairment Induced by Lead Exposure during Lifespan: Mechanisms of Lead Neurotoxicity. Toxics, 9(2), 23. https://doi.org/10.3390/toxics9020023