
Novel QSAR Models for Molecular Initiating Event Modeling in Two Intersecting Adverse Outcome Pathways Based Pulmonary Fibrosis Prediction for Biocidal Mixtures


Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Sets
2.1.1. Co-Exposure Biocide Data Collection
2.1.2. Receptor and Ligand Data Collection for MD and Pharmacophore Modeling
2.1.3. Data Collection for QSAR Modeling
2.1.4. Selection of Test Substance
2.2. MD Modeling for PPAR-γ
2.2.1. Data Preparation
2.2.2. Calculation of MD and MM-GBSA
2.3. Pharmacophore Modeling for TLR4
2.4. QSAR Modeling for PPAR-γ and TLR4
2.4.1. Data Curation
2.4.2. Calculation of Molecular Descriptors
2.4.3. Model Development and Validation
2.4.4. Y-Randomization
2.5. In Vitro Study
2.5.1. Cell Culture
2.5.2. Western Blot Analysis for Evaluating PPAR-γ Inactivation
2.5.3. Reporter Gene Assay for Evaluating TLR4 Activation
2.5.4. Reporter Gene Assay for Evaluating TGF-β Pathway Activation
2.5.5. Statistical Analysis
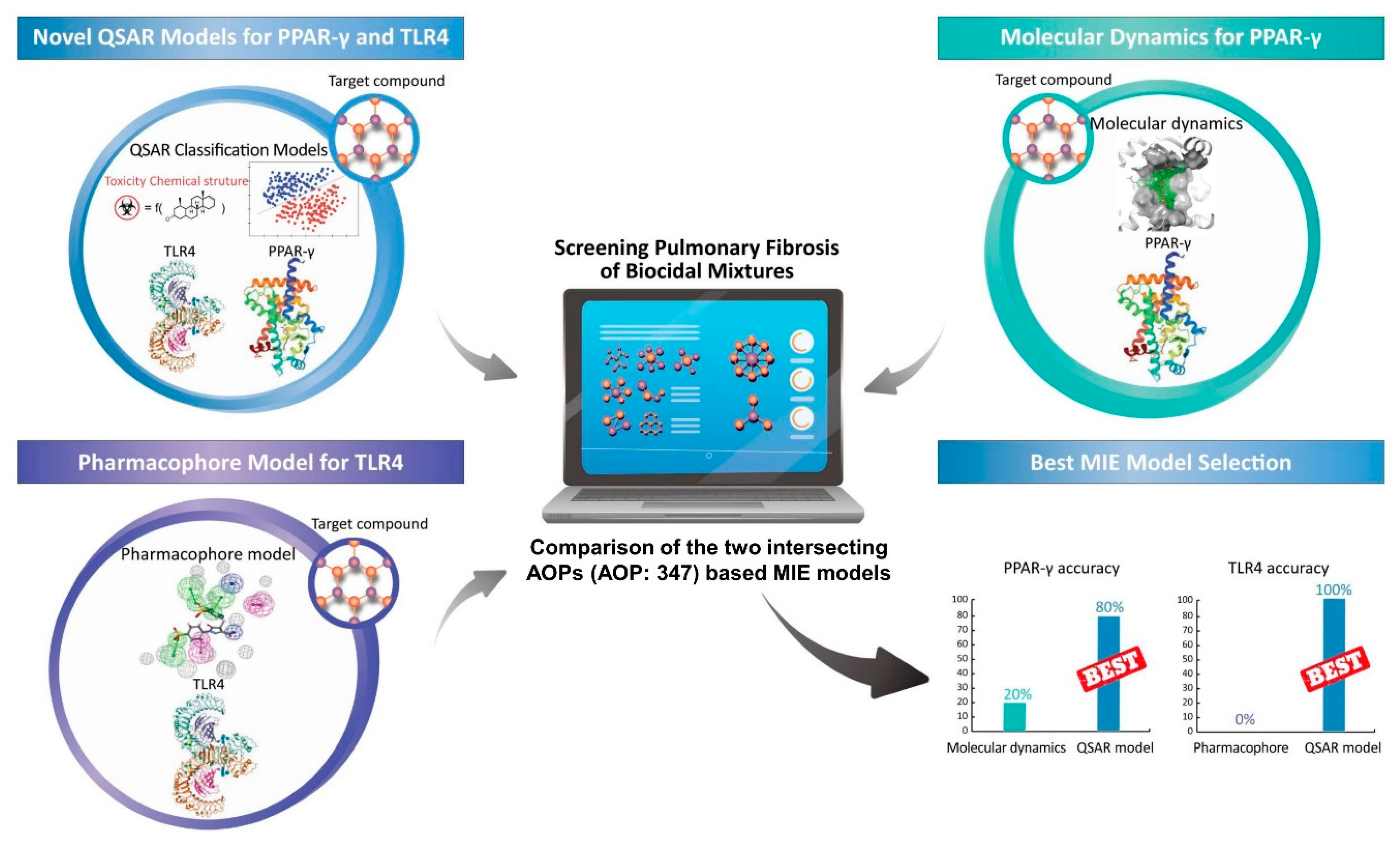
3. Results
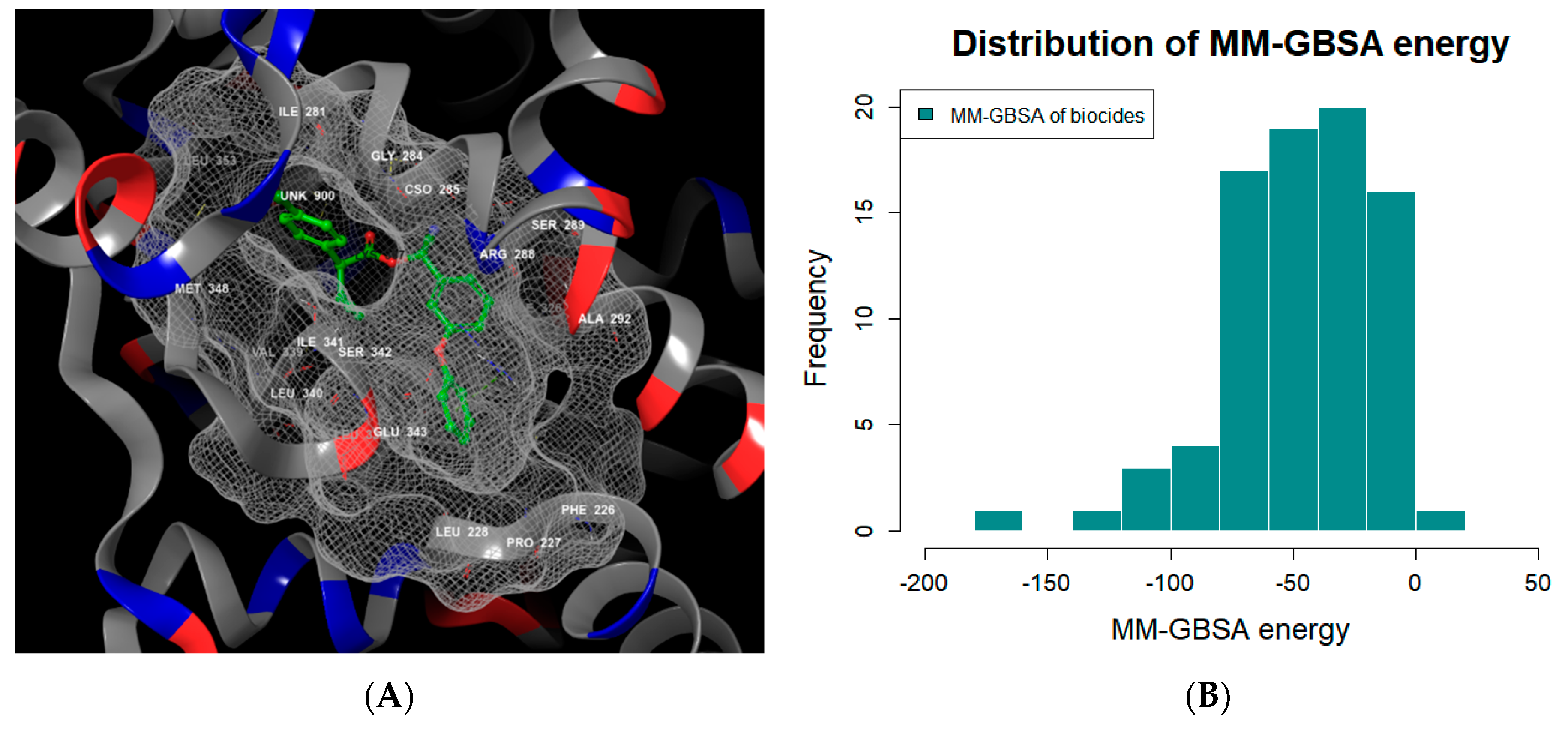
3.1. MD Modeling of PPAR-γ
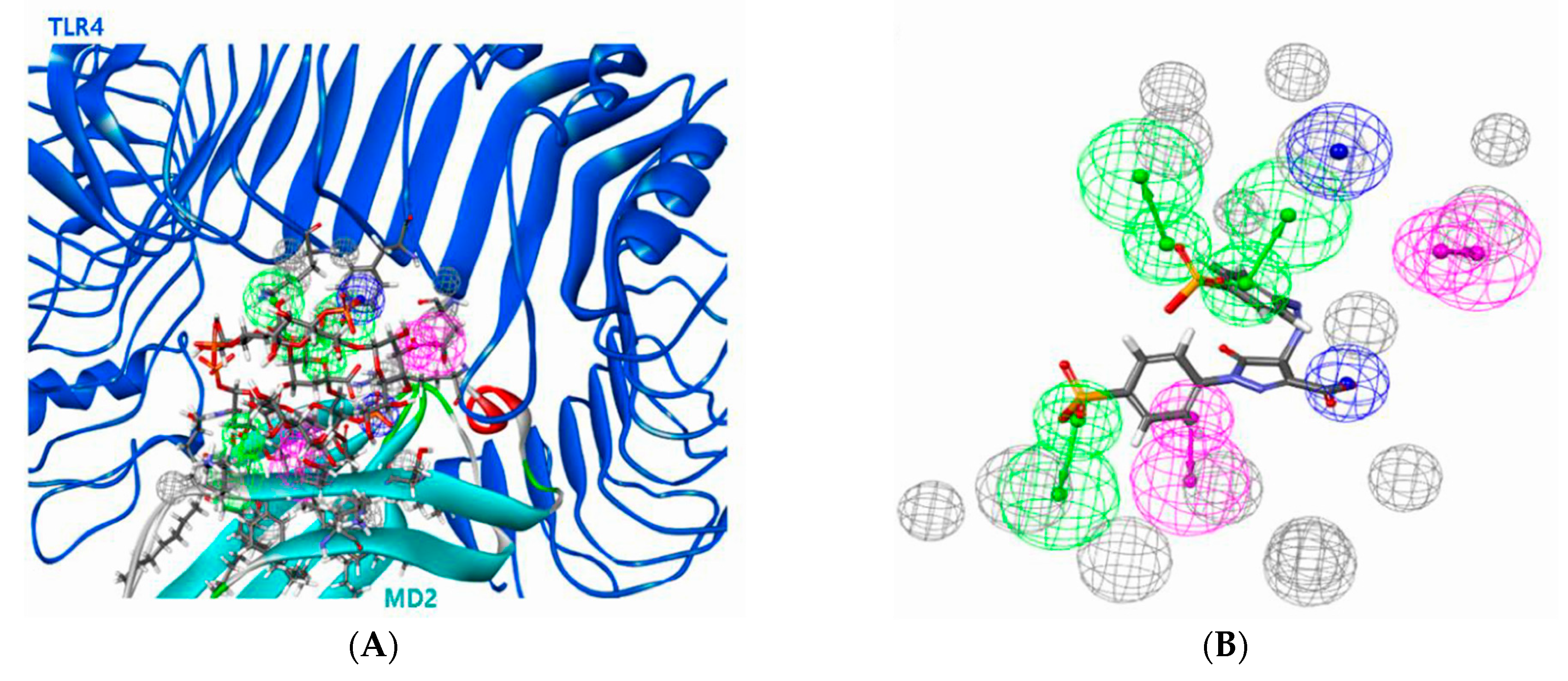
3.2. Pharmacophore Modeling Results for TLR4
3.3. QSAR Modeling Result
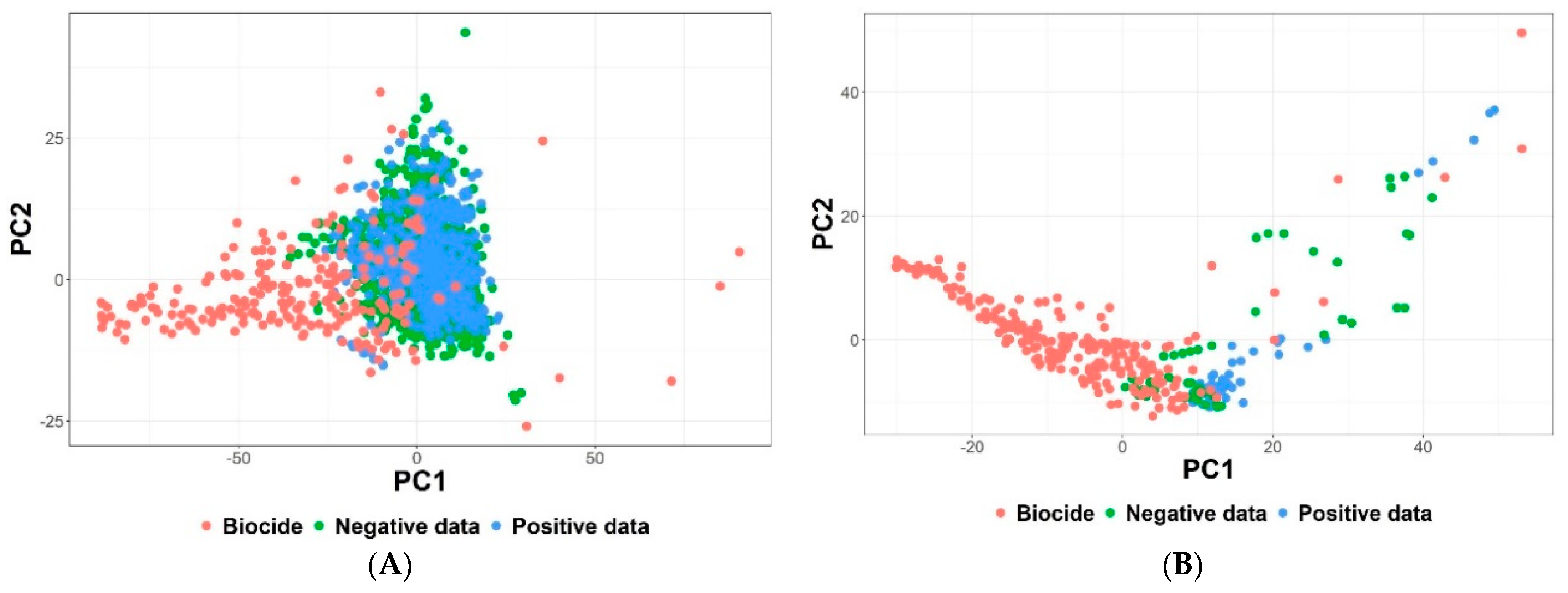
3.3.1. Results of Data Curation
3.3.2. Classification-Based QSAR Models
3.4. Application of MIE Modeling
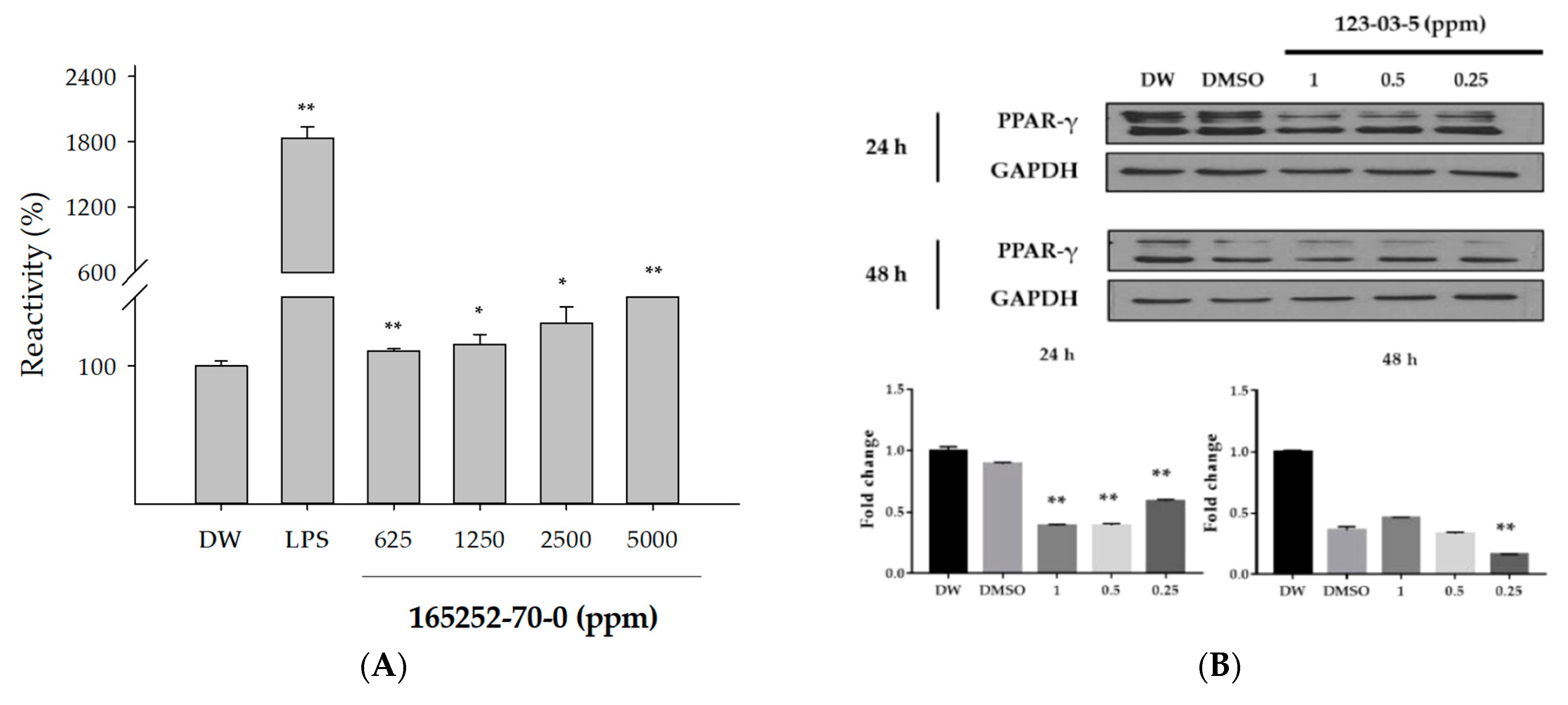
3.4.1. In Vitro Evaluation for MIE Regulation
3.4.2. Comparison between MIE Modeling and In Vitro Testing
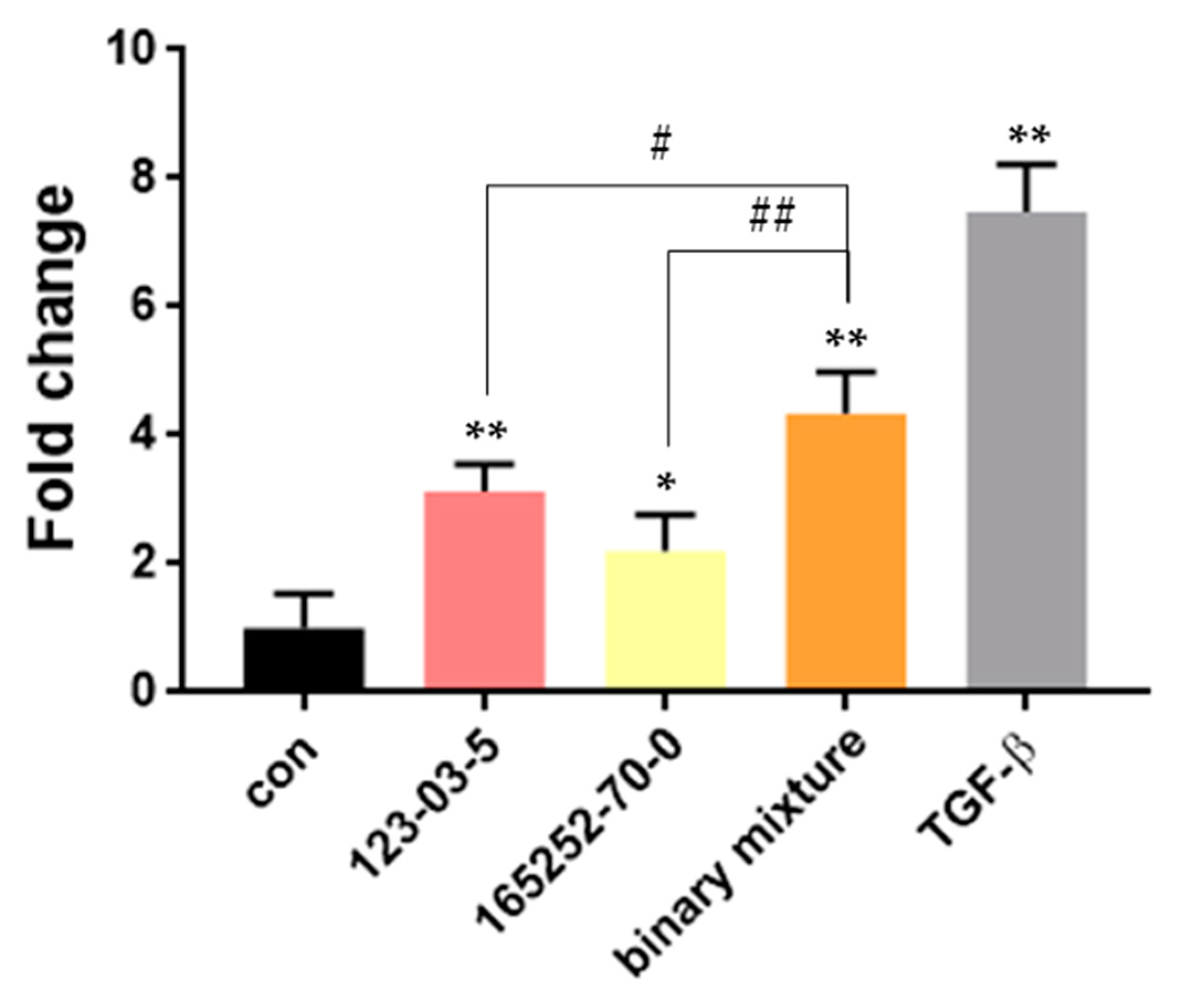
3.4.3. In Vitro Validation of Selected Substances in the AOP Network
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Coors, A.; Vollmar, P.; Heim, J.; Sacher, F.; Kehrer, A. Environmental risk assessment of biocidal products: Identification of relevant components and reliability of a component-based mixture assessment. Environ. Sci. Eur. 2018, 30, 1–15. [Google Scholar] [CrossRef] [PubMed]
- EU Parliament. Regulation (EU) No 528/2012 of the European Parliament and of the Council of 22 May 2012 concerning the making available on the market and use of biocidal products. Off. J. Eur. Union 2012, 55, 1–123. [Google Scholar]
- Korean Ministry of Environment. Act No. 15511 of the Korean Ministry of Environment of the Council of 20 March 2018 Concerning Household Chemical Products and Biocidal Products Safety; Ministry of Environment: Sejong, Korea, 2018. (In Korean) [Google Scholar]
- Villaverde, J.J.; Sevilla-Morán, B.; López-Goti, C.; Alonso-Prados, J.L.; Sandín-España, P. Computational Methodologies for the Risk Assessment of Pesticides in the European Union. J. Agric. Food Chem. 2017, 65, 2017–2018. [Google Scholar] [CrossRef] [PubMed]
- Villaverde, J.; Sevilla-Morán, B.; López-Goti, C.; Alonso-Prados, J.; Sandín-España, P. QSAR/QSPR models based on quantum chemistry for risk assessment of pesticides according to current European legislation. SAR QSAR Environ. Res. 2019, 31, 49–72. [Google Scholar] [CrossRef]
- Kar, S.; Leszczynski, J. Exploration of Computational Approaches to Predict the Toxicity of Chemical Mixtures. Toxics 2019, 7, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Kim, S.; Schaumann, G.E. Reliable predictive computational toxicology methods for mixture toxicity: Toward the development of innovative integrated models for environmental risk assessment. Rev. Environ. Sci. Bio/Technol. 2013, 12, 235–256. [Google Scholar] [CrossRef]
- Ankley, G.T.; Bennett, R.S.; Erickson, R.J.; Hoff, D.J.; Hornung, M.W.; Johnson, R.D.; Mount, D.R.; Nichols, J.W.; Russom, C.L.; Schmieder, P.K.; et al. Adverse outcome pathways: A conceptual framework to support ecotoxicology research and risk assessment. Environ. Toxicol. Chem. 2010, 29, 730–741. [Google Scholar] [CrossRef]
- Villeneuve, D.L.; Crump, D.; Garcia-Reyero, N.; Hecker, M.; Hutchinson, T.H.; Lalone, C.A.; Landesmann, B.; Lettieri, T.; Munn, S.; Nepelska, M.; et al. Adverse Outcome Pathway Development II: Best Practices. Toxicol. Sci. 2014, 142, 321–330. [Google Scholar] [CrossRef] [Green Version]
- Knapen, D.; Vergauwen, L.; Villeneuve, D.L.; Ankley, G.T. The potential of AOP networks for reproductive and developmental toxicity assay development. Reprod. Toxicol. 2015, 56, 52–55. [Google Scholar] [CrossRef]
- Allen, T.E.H.; Liggi, S.; Goodman, J.M.; Gutsell, S.; Russell, P.J. Using Molecular Initiating Events To Generate 2D Structure–Activity Relationships for Toxicity Screening. Chem. Res. Toxicol. 2016, 29, 1611–1627. [Google Scholar] [CrossRef]
- Gadaleta, D.; Manganelli, S.; Roncaglioni, A.; Toma, C.; Benfenati, E.; Mombelli, E. QSAR Modeling of ToxCast Assays Relevant to the Molecular Initiating Events of AOPs Leading to Hepatic Steatosis. J. Chem. Inf. Model. 2018, 58, 1501–1517. [Google Scholar] [CrossRef] [Green Version]
- Mellor, C.L.; Steinmetz, F.P.; Cronin, M.T.D. Using Molecular Initiating Events to Develop a Structural Alert Based Screening Workflow for Nuclear Receptor Ligands Associated with Hepatic Steatosis. Chem. Res. Toxicol. 2016, 29, 203–212. [Google Scholar] [CrossRef] [Green Version]
- Kulkarni, A.A.; Woeller, C.F.; Thatcher, T.H.; Ramon, S.; Phipps, R.P.; Sime, P.J. Emerging PPARγ-Independent Role of PPARγLigands in Lung Diseases. PPAR Res. 2012, 2012, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Jeong, J.; Garcia-Reyero, N.; Burgoon, L.; Perkins, E.J.; Park, T.; Kim, C.; Roh, J.-Y.; Choi, J. Development of Adverse Outcome Pathway for PPARγ Antagonism Leading to Pulmonary Fibrosis and Chemical Selection for Its Validation: ToxCast Database and a Deep Learning Artificial Neural Network Model-Based Approach. Chem. Res. Toxicol. 2019, 32, 1212–1222. [Google Scholar] [CrossRef] [PubMed]
- Toll-Like Receptor 4 Activation and Peroxisome Proliferator-Activated Receptor Gamma Activation Leading to Pulmonary Fibrosis. Available online: https://aopwiki.org/aops/347 (accessed on 15 March 2021).
- Rücker, C.; Scarsi, M.; Meringer, M. 2D QSAR of PPARγ agonist binding and transactivation. Bioorg. Med. Chem. 2006, 14, 5178–5195. [Google Scholar] [CrossRef]
- Rathi, L.; Kashaw, S.K.; Dixit, A.; Pandey, G.; Saxena, A.K. Pharmacophore identification and 3D-QSAR studies in N-(2-benzoyl phenyl)-l-tyrosines as PPARγ agonists. Bioorg. Med. Chem. 2004, 12, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Dolezal, R.; Trejbal, J.; Mesicek, J.; Milanov, A.; Racakova, V.; Krenek, J. Designing QSAR Models for Promising TLR4 Agonists Isolated from Euodia Asteridula by Artificial Neural Networks Enhanced by Optimal Brain Surgeon. Comput. Vis. 2016, 9876, 271–281. [Google Scholar] [CrossRef]
- Kim, S.; Seo, M.; Na, M.; Kim, J. Investigation on Combined Inhalation Exposure Scenarios to Biocidal Mixtures: Biocidal and Household Chemical Products in South Korea. Toxics 2021, 9, 32. [Google Scholar] [CrossRef]
- Evan, E.B.; Yanli, W.; Paul, A.T.; Stephen, H.B. Integrated Platform of Small Molecules and Biological Activities. Annu. Rep. Comput. Chem. 2008, 4, 217–241. [Google Scholar]
- Brusotti, G.; Montanari, R.; Capelli, D.; Cattaneo, G.; Laghezza, A.; Tortorella, P.; Loiodice, F.; Peiretti, F.; Bonardo, B.; Paiardini, A.; et al. Betulinic acid is a PPARγ antagonist that improves glucose uptake, promotes osteogenesis and inhibits adipogenesis. Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marciano, D.P.; Kuruvilla, D.S.; Boregowda, S.V.; Asteian, A.; Hughes, T.S.; Garcia-Ordonez, R.; Corzo, C.A.; Khan, T.M.; Novick, S.J.; Park, H.; et al. Pharmacological repression of PPARγ promotes osteogenesis. Nat. Commun. 2015, 6, 7443. [Google Scholar] [CrossRef] [Green Version]
- Leesnitzer, L.M.; Parks, D.J.; Bledsoe, R.K.; Cobb, J.E.; Collins, J.L.; Consler, T.G.; Davis, R.G.; Hull-Ryde, E.A.; Lenhard, J.M.; Patel, L.; et al. Functional Consequences of Cysteine Modification in the Ligand Binding Sites of Peroxisome Proliferator Activated Receptors by GW9662. Biochemistry 2002, 41, 6640–6650. [Google Scholar] [CrossRef]
- Handeli, S.; Simon, J.A. A small-molecule inhibitor of Tcf/β-catenin signaling down-regulates PPARγ and PPARδ activities. Mol. Cancer Ther. 2008, 7, 521–529. [Google Scholar] [CrossRef] [Green Version]
- Frkic, R.L.; Marshall, A.C.; Blayo, A.-L.; Pukala, T.L.; Kamenecka, T.M.; Griffin, P.R.; Bruning, J.B. PPARγ in Complex with an Antagonist and Inverse Agonist: A Tumble and Trap Mechanism of the Activation Helix. iScience 2018, 5, 69–79. [Google Scholar] [CrossRef]
- Park, B.S.; Song, D.H.; Kim, H.M.; Choi, B.-S.; Lee, H.; Lee, J.-O. The structural basis of lipopolysaccharide recognition by the TLR4–MD-2 complex. Nat. Cell Biol. 2009, 458, 1191–1195. [Google Scholar] [CrossRef] [PubMed]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein−Ligand Complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowers, K.J.; Sacerdoti, F.D.; Salmon, J.K.; Shan, Y.; Shaw, D.E.; Chow, E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; et al. Molecular dynamics-Scalable algorithms for molecular dynamics simulations on commodity clusters. In Proceedings of the SC ’06: Proceedings of the 2006 ACM/IEEE Conference on Supercomputing, Tampa, FL, USA, 11–17 November 2006; p. 84. [Google Scholar]
- Jacobson, M.P.; Pincus, D.L.; Rapp, C.S.; Day, T.J.; Honig, B.; Shaw, D.E.; Friesner, R.A. A hierarchical approach to all-atom protein loop prediction. Proteins Struct. Funct. Bioinform. 2004, 55, 351–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harder, E.; Damm, W.; Maple, J.R.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L.; et al. OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory Comput. 2016, 12, 281–296. [Google Scholar] [CrossRef]
- BIOVIA. Dassault Systems, Discovery Studio, v20.1.0.19; Dassult Systems: San Diego, CA, USA, 2019. [Google Scholar]
- Tomek, I. Two Modification of CNN. IEEE Trans. Syst. Man Commun. 1976, 6, 769–772. [Google Scholar]
- Mauri, A. alvaDesc: A Tool to Calculate and Analyze Molecular Descriptors and Fingerprint. In Ecotoxicological QSARs; Methods in Pharmacology and Toxicology; Roy, K., Ed.; Springer: Berlin, Germany, 2020; pp. 801–820. [Google Scholar]
- Holland, J.H. Adaptation in Natural and Artificial Systems: An Introductory Analysis with Applications to Biology, Control, and Artificial Intelligence; MIT Press: Cambridge, MA, USA, 1975. [Google Scholar]
- Mierswa, I.; Wurst, M.; Klinkenberg, R.; Scholz, M.; Euler, T. YALE. In Proceedings of the 12th ACM SIGKDD International Conference on Knowledge Discovery and Data Mining—KDD ’06, Philadelphia, PA, USA, 20–23 August 2006; Association for Computing Machinery (ACM): New York, NY, USA, 2006; p. 935. [Google Scholar]
- Rücker, C.; Rücker, G.; Meringer, M. y-Randomization and Its Variants in QSPR/QSAR. J. Chem. Inf. Model. 2007, 47, 2345–2357. [Google Scholar] [CrossRef] [PubMed]
- Matthews, B. Comparison of the predicted and observed secondary structure of T4 phage lysozyme. Biochim. Biophys. Acta Protein Struct. 1975, 405, 442–451. [Google Scholar] [CrossRef]
- Tharwat, A. Classification assessment methods. Appl. Comput. Inform. 2020. [Google Scholar] [CrossRef]
- Kanno, S.; Hirano, S.; Kato, H.; Fukuta, M.; Mukai, T.; Aoki, Y. Benzalkonium chloride and cetylpyridinium chloride induce apoptosis in human lung epithelial cells and alter surface activity of pulmonary surfactant monolayers. Chem. Interact. 2020, 317, 108962. [Google Scholar] [CrossRef] [PubMed]
- Kienzler, A.; Berggren, E.; Bessems, J.; Bopp, S.; Van Der Linden, S.; Worth, A. Assessment of Mixtures—Review of Regulation Requirements and Guidance; Publications Office of the European Union: Luxembourg, 2017. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MIE 1 | Substances | CAS No. | Prediction |
|---|---|---|---|
| PPAR-γ inactivation | Cetylpyridinium chloride | 123-03-5 | Positive (Antagonist) |
| Cyfluthrin | 68359-37-5 | Positive (Antagonist) | |
| Deltamethrin | 52918-63-5 | Positive (Antagonist) | |
| Muscalure | 27519-02-4 | Positive (Antagonist) | |
| Piperonyl butoxide | 51-03-6 | Positive (Antagonist) | |
| TLR4 activation | Propetamphos | 31218-83-4 | Positive (Agonist) |
| Prallethrin | 23031-36-9 | Positive (Agonist) | |
| Novaluron | 116714-46-6 | Positive (Agonist) | |
| Imiprothrin | 72963-72-5 | Positive (Agonist) | |
| Dinotefuran | 165252-70-0 | Positive (Agonist) |
| Number | Reference Antagonists | CAS No. | Dock Score 1 (kcal/mol) | MM-GBSA 2 Score (kcal/mol) | Avg. Similarity Distance with Biocides 3 |
|---|---|---|---|---|---|
| 1 | Betulinic acid | 472-15-1 | −6.36 | −134.75 | 1.75 |
| 2 | SR 1664 | 1338259-05-4 | −11.90 | −58.20 | 1.53 |
| 3 | GW9662 | 22978-25-2 | −7.66 | −69.71 | 1.11 |
| 4 | FH535 | 108409-83-2 | −6.50 | −50.70 | 1.38 |
| QSAR Models | Descriptors | Description |
|---|---|---|
| PPAR-γ inactivation | B10[F-F] | Presence/absence of F-F at topological distance 10 |
| B10[S-Cl] | Presence/absence of S-Cl at topological distance 10 | |
| MAXDN | Maximal electrotopological negative variation | |
| NCconj | Number of non-aromatic conjugated C (sp2) | |
| SpMax7_Bh(e) | Largest eigenvalue n. 7 OF Burden matrix weighted by Sanderson electronegativity | |
| SssCH2 | Sum of ssCH2 E-state | |
| TLR4 activation | nR10 | Number of 10-membered rings |
| F02[C-O] | Frequency of C-O at topological distance 2 |
| QSAR Models | Internal Validation (80% of Data Set) | External Validation (20% of Data Set) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ACC 2 | AUC 3 | MCC 4 | Sensitivty 5 | Specificity 6 | BA 7 | ACC | AUC | MCC | Sensitivity | Specificity | BA | |
| PPAR-γ inactivation | 0.82 | 0.87 | 0.63 | 0.83 | 0.80 | 0.81 | 0.82 | 0.88 | 0.64 | 0.83 | 0.81 | 0.82 |
| TLR4 activation | 0.98 | 1.00 | 0.97 | 0.99 | 0.97 | 0.98 | 0.97 | 1.00 | 0.97 | 0.96 | 0.94 | 0.95 |
| MIE 2 | Substance | CAS No. | In Vitro | MD 3/Pharmacophore | QSAR 4 |
|---|---|---|---|---|---|
| PPAR-γ inactivation | Cetylpyridinium chloride | 123-03-5 | Positive | Positive | Negative |
| Cyfluthrin | 68359-37-5 | Negative | Positive | Negative | |
| Deltamethrin | 52918-63-5 | Negative | Positive | Negative | |
| Muscalure | 27519-02-4 | Negative | Positive | Negative | |
| Piperonyl butoxide | 51-03-6 | Negative | Positive | Negative | |
| TLR4 activation | Propetamphos | 31218-83-4 | Negative | Positive | Negative |
| Prallethrin | 23031-36-9 | Negative | Positive | Negative | |
| Novaluron | 116714-46-6 | Negative | Positive | Negative | |
| Imiprothrin | 72963-72-5 | Negative | Positive | Negative | |
| Dinotefuran | 165252-70-0 | Positive | Negative | Positive |
| Performance Indices | PPAR-γ Inactivation | TLR4 Activation | ||
|---|---|---|---|---|
| MD 2 | QSAR 3 | Pharmacophore | QSAR | |
| Accuracy | 0.20 | 0.80 | 0.00 | 1.00 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seo, M.; Chae, C.H.; Lee, Y.; Kim, H.R.; Kim, J. Novel QSAR Models for Molecular Initiating Event Modeling in Two Intersecting Adverse Outcome Pathways Based Pulmonary Fibrosis Prediction for Biocidal Mixtures. Toxics 2021, 9, 59. https://doi.org/10.3390/toxics9030059
Seo M, Chae CH, Lee Y, Kim HR, Kim J. Novel QSAR Models for Molecular Initiating Event Modeling in Two Intersecting Adverse Outcome Pathways Based Pulmonary Fibrosis Prediction for Biocidal Mixtures. Toxics. 2021; 9(3):59. https://doi.org/10.3390/toxics9030059
Chicago/Turabian StyleSeo, Myungwon, Chong Hak Chae, Yuno Lee, Ha Ryong Kim, and Jongwoon Kim. 2021. "Novel QSAR Models for Molecular Initiating Event Modeling in Two Intersecting Adverse Outcome Pathways Based Pulmonary Fibrosis Prediction for Biocidal Mixtures" Toxics 9, no. 3: 59. https://doi.org/10.3390/toxics9030059
APA StyleSeo, M., Chae, C. H., Lee, Y., Kim, H. R., & Kim, J. (2021). Novel QSAR Models for Molecular Initiating Event Modeling in Two Intersecting Adverse Outcome Pathways Based Pulmonary Fibrosis Prediction for Biocidal Mixtures. Toxics, 9(3), 59. https://doi.org/10.3390/toxics9030059