
A Toolbox for the Determination of Nitroaromatic Explosives in Marine Water, Sediment, and Biota Samples on Femtogram Levels by GC-MS/MS

 , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials and Chemicals
2.2. Water Sample Preparation
2.3. Sediment Sample Preparation
2.4. Mussel Sample Preparation
2.5. GC-MS/MS Analysis
2.6. External and Internal Standards
2.7. Matrix Standards
3. Results
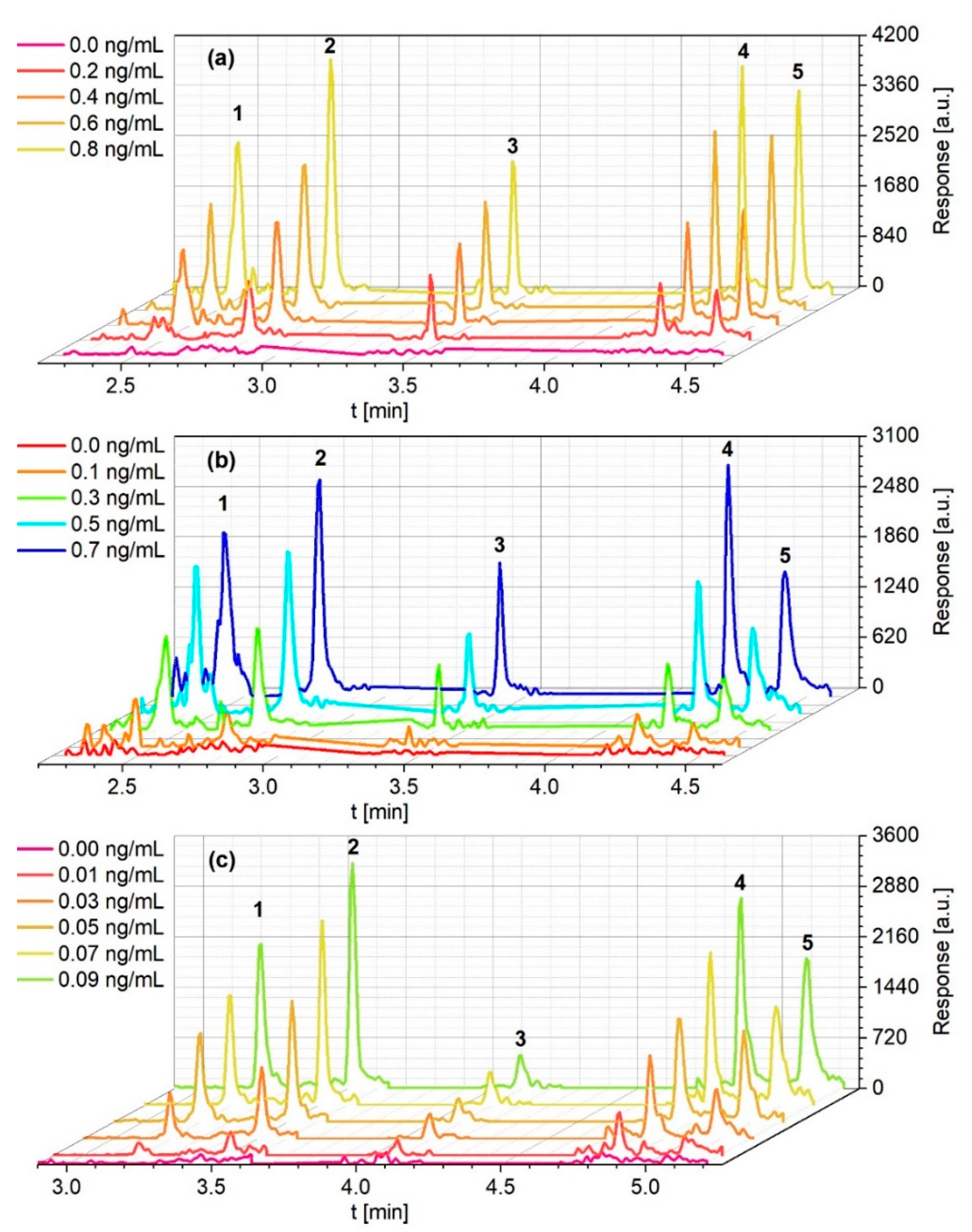
3.1. GC-MS/MS Method Development and Optimization
3.1.1. Splitless Injection Methods
3.1.2. PTV Large Volume Injection Method
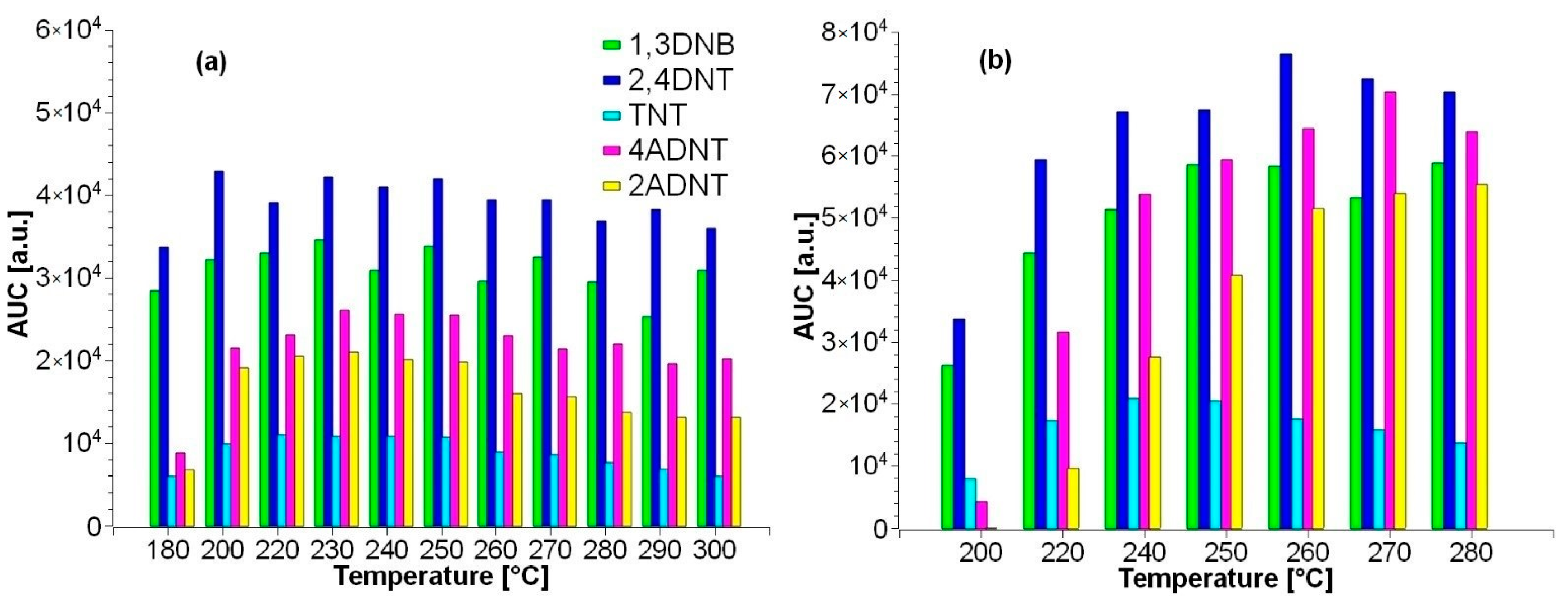
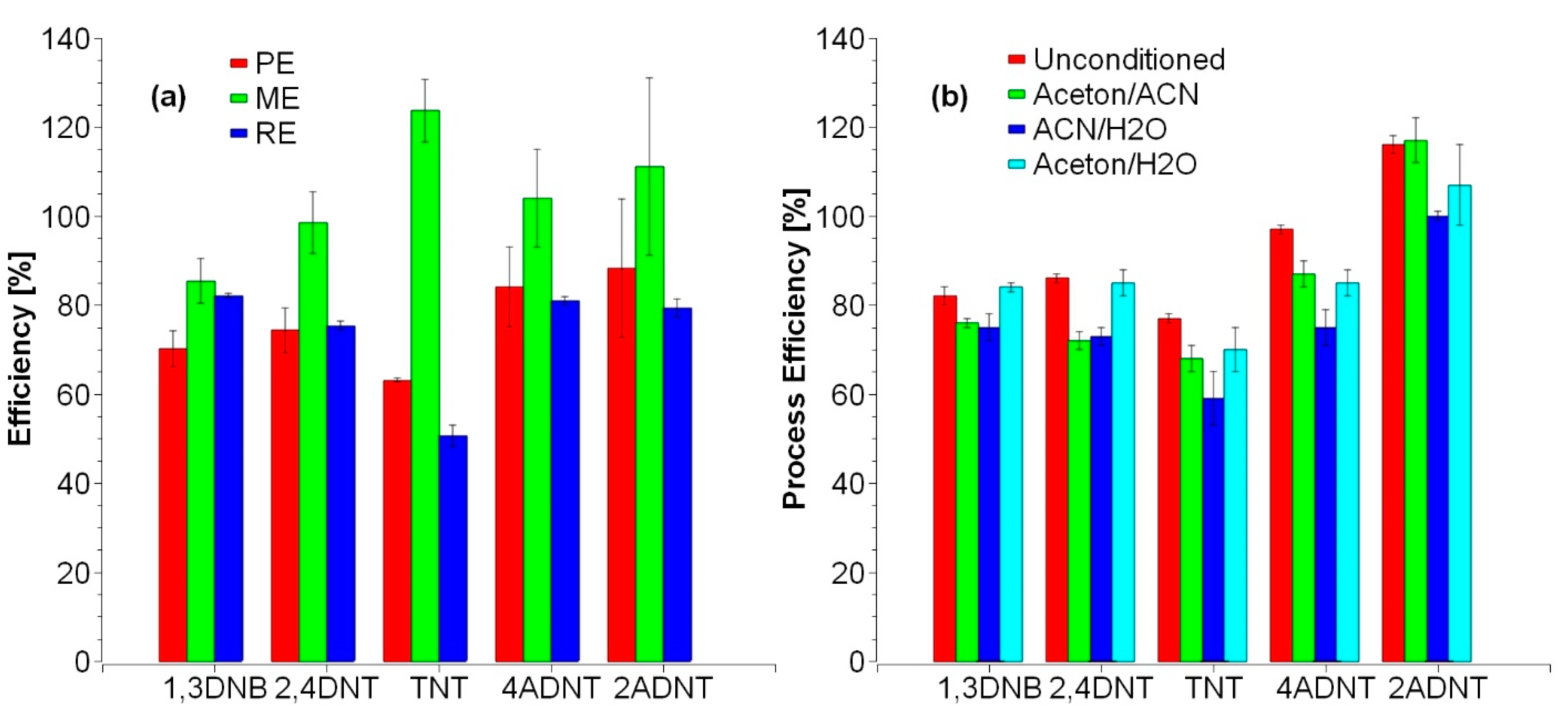
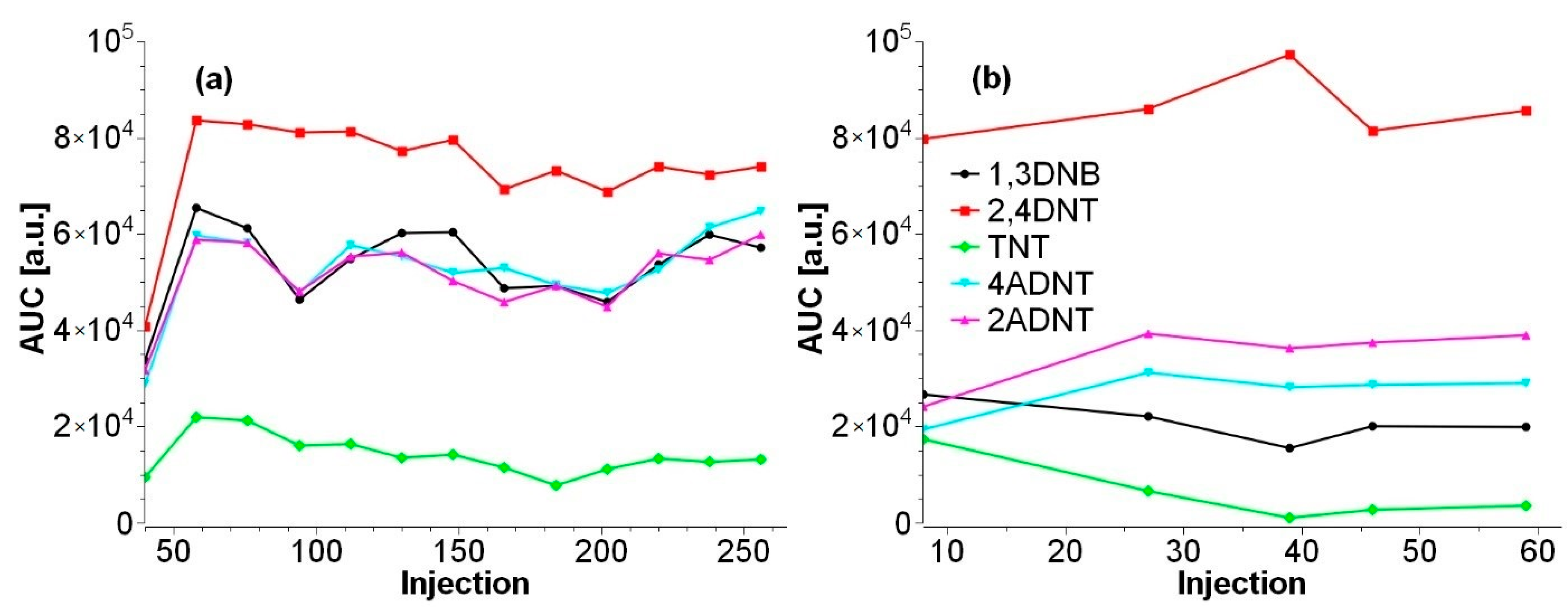
3.2. Improvement of Sample Preparation
3.2.1. Water Samples
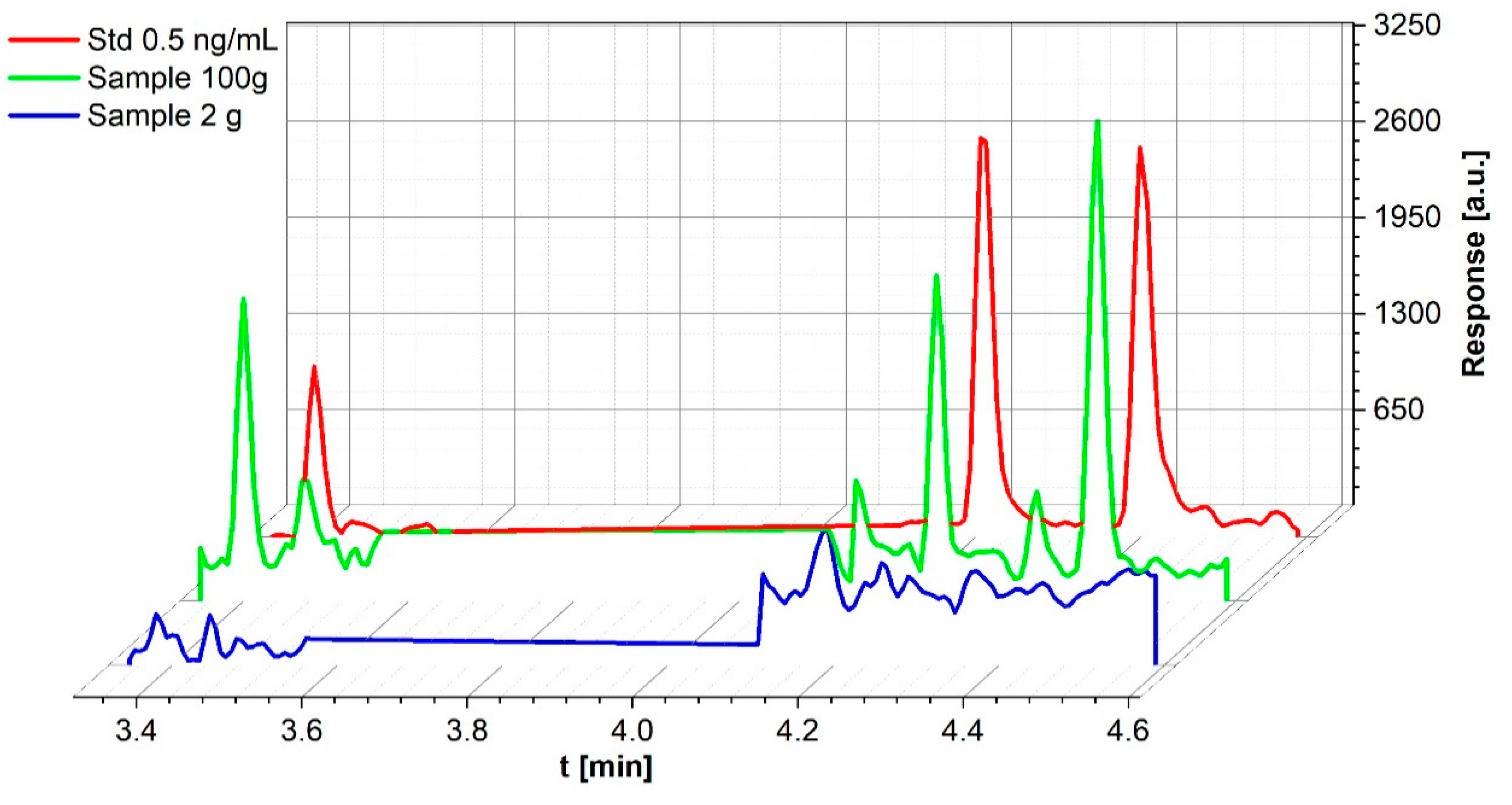
3.2.2. Sediment Samples
3.2.3. Mussel Samples
3.2.4. Limits of Detection/-Quantification and Linear Range
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bełdowski, J.; Brenner, M.; Lehtonen, K.K. Contaminated by War: A Brief History of Sea-Dumping of Munitions. Mar. Environ. Res. 2020, 162, 105189. [Google Scholar] [CrossRef]
- Böttcher, C.; Knobloch, T.; Rühl, N.-P.; Sternheim, J.; Wichert, U.; Wöhler, J. Munitionsbelastung der Deutschen Meeresgewässer—Bestandsaufnahme und Empfehlungen; Bundesamt für Seeschifffahrt und Hydrographie (BSH), Sekretariat Bund/Länder-Messprogramm für die Meeresumwelt von Nord-und Ostsee (BMLP): Hamburg, Germany, 2011; p. 174. [Google Scholar]
- Strehse, J.S.; Appel, D.; Geist, C.; Martin, H.-J.; Maser, E. Biomonitoring of 2,4,6-Trinitrotoluene and Degradation Products in the Marine Environment with Transplanted Blue Mussels (m. Edulis). Toxicology 2017, 390, 117–123. [Google Scholar] [CrossRef]
- Appel, D.; Strehse, J.S.; Martin, H.-J.; Maser, E. Bioaccumulation of 2,4,6-Trinitrotoluene (TNT) and Its Metabolites Leaking from Corroded Munition in Transplanted Blue Mussels (m. Edulis). Mar. Pollut. Bull. 2018, 135, 1072–1078. [Google Scholar] [CrossRef] [PubMed]
- Maser, E.; Strehse, J.S. “Don’t Blast”: Blast-in-Place (BiP) Operations of Dumped World War Munitions in the Oceans Signifi-cantly Increase Hazards to the Environment and the Human Seafood Consumer. Arch. Toxicol. 2020, 94, 1941–1953. [Google Scholar] [CrossRef]
- Strehse, J.S.; Maser, E. Marine Bivalves as Bioindicators for Environmental Pollutants with Focus on Dumped Munitions in the Sea: A Review. Mar. Environ. Res. 2020, 158, 105006. [Google Scholar] [CrossRef]
- Koske, D.; Straumer, K.; Goldenstein, N.I.; Hanel, R.; Lang, T.; Kammann, U. First Evidence of Explosives and Their Degra-dation Products in Dab (Limanda Limanda L.) from a Munition Dumpsite in the Baltic Sea. Mar. Pollut. Bull. 2020, 155, 111131. [Google Scholar] [CrossRef]
- Koske, D.; Goldenstein, N.I.; Rosenberger, T.; Machulik, U.; Hanel, R.; Kammann, U. Dumped Munitions: New Insights into the Metabolization of 2,4,6-Trinitrotoluene in Baltic Flatfish. Mar. Environ. Res. 2020, 160, 104992. [Google Scholar] [CrossRef]
- Williams, A.F.; Murray, W.J. Determination of Traces of Ethyleneglycol Dinitrate (and Nitroglycerine) in Blood and Urine. Nature 1966, 210, 816–817. [Google Scholar] [CrossRef]
- Alley, B.J.; Dykes, H.W. Gas-Liquid Chromatographic Determination of Nitroglycerine in Pharmaceutical Preparations. J. Chromatogr. 1972, 72, 182–186. [Google Scholar] [CrossRef]
- Patterson, D.G.; Welch, S.M.; Turner, W.E.; Sjödin, A.; Focant, J.-F. Cryogenic Zone Compression for the Measurement of Dioxins in Human Serum by Isotope Dilution at the Attogram Level Using Modulated Gas Chromatography Coupled to High Resolution Magnetic Sector Mass Spectrometry. J. Chromatogr. A 2011, 1218, 3274–3281. [Google Scholar] [CrossRef] [Green Version]
- Munch, J.W. Method 529 Determination of Explosives and Related Compounds in Drinking Water by Solid Phaseextraction and Capillary Column Gaschromatography/Mass Spectrometry (GC/MS); National Exposure Research Laboratory, Office of Research and Development, US Environmental Protection Agency: Cincinnati, OH, USA, 2002. [Google Scholar]
- US Environmental Protection Agency. Method 8330B Nitroaromatics, Nitramines, and Nitrate Esters by Highperformance Liquid Chroma-Tography (HPLC); US Environmental Protection Agency: Washington, DC, USA, 2006.
- US Environmental Protection Agency. Method 8095 Explosives by Gas Chromatography; US Environmental Protection Agency: Washington, DC, USA, 2007.
- Preslan, J.E.; Hatrel, B.B.; Emerson, M.; White, L.; George, W.J. An Improved Method for Analysis of 2,4,6-Trinitrotoluene and Its Metabolites from Compost and Contaminated Soils. J. Hazard. Mater. 1993, 33, 329–337. [Google Scholar] [CrossRef]
- Staples, E.J.; Viswanathan, S. Ultrahigh-Speed Chromatography and Virtual Chemical Sensors for Detecting Explosives and Chemical Warfare Agents. IEEE Sens. J. 2005, 5, 622–631. [Google Scholar] [CrossRef]
- Sigman, M.E.; Ma, C.-Y. Detection Limits for GC/MS Analysis of Organic Explosives. J. Forensic Sci. 2001, 46, 6–11. [Google Scholar] [CrossRef]
- Barshick, S.-A.; Griest, W.H. Trace Analysis of Explosives in Seawater Using Solid-Phase Microextraction and Gas Chroma-tography/Ion Trap Mass Spectrometry. Anal. Chem. 1998, 70, 3015–3020. [Google Scholar] [CrossRef]
- Gledhill, M.; Beck, A.J.; Stamer, B.; Schlosser, C.; Achterberg, E.P. Quantification of Munition Compounds in the Marine Environment by Solid Phase Extraction—Ultra High Performance Liquid Chromatography with Detection by Electrospray Ionisation—Mass Spectrometry. Talanta 2019, 200, 366–372. [Google Scholar] [CrossRef]
- Gordon, D.; Nawała, J.; Szala, M.; Dziedzic, D.; Dawidziuk, B.; Popiel, S. Development of Analytical Methods Used for the Study of 2,4,6-Trinitrotoluene Degradation Kinetics in Simulated Sediment Samples from the Baltic Sea. Mar. Pollut. Bull. 2018, 135, 397–410. [Google Scholar] [CrossRef]
- Emmrich, M.; Lahrz, T.; Spyra, W. Influence of Solvents and Gas Chromatographic Injector Conditions on the Detectability of Nitroaromatic Compounds. J. Chromatogr. A 2001, 918, 121–126. [Google Scholar] [CrossRef]
- Marder, D.; Tzanani, N.; Prihed, H.; Gura, S. Trace Detection of Explosives with a Unique Large Volume Injection Gas Chromatography-Mass Spectrometry (LVI-GC-MS) Method. Anal. Methods 2018, 10, 2712–2721. [Google Scholar] [CrossRef]
- Guidry, R.M.; Davis, L.P. Thermochemical Decomposition of Explosives. I. TNT Kinetic Parameters Determined from ESR Investigation. Thermochim. Acta 1979, 32, 1–18. [Google Scholar] [CrossRef]
- Ahmad, M.F.; Hussain, A.; Malik, A.Q. Thermal decomposition and kinetic evaluation of decanted 2,4,6-trinitrotoluene (TNT) for reutilization as composite material. In IOP Conference Series: Materials Science and Engineering, Proceedings of the 14th International Symposium on Advanced Materials, National Centre for Physics, Islamabad, Pakistan, 12–16 October 2015; IOP Publishing Ltd.: Bristol, UK, 2016; Volume 146, p. 012032. [Google Scholar] [CrossRef] [Green Version]
- Beck, A.J.; van der Lee, E.M.; Eggert, A.; Stamer, B.; Gledhill, M.; Schlosser, C.; Achterberg, E.P. In situ measurements of explosive compound dissolution fluxes from exposed munition material in the Baltic Sea. Environ. Sci. Technol. 2019, 53, 5652–5660. [Google Scholar] [CrossRef]
- Kampmeier, M.; Greinert, J.; Strehse, J.; Beck, A.J.; Wichert, U.; Diller, N.; Kurbjuhn, T.; Wenzlaff, E.; Schröder, J. RV Poseidon POS530Cruise Report “MineMoni 2018”. Available online: https://oceanrep.geomar.de/47567/1/CruiseReport_POS530_v05_final.pdf (accessed on 26 February 2021).
- Greinert, J.; Appel, D.; Beck, A.; Eggert, A.; Gräwe, U.; Kampmeier, M.; Martin, H.-J.; Maser, E.; Schlosser, C.; Song, Y.; et al. Practical Guide for Environmental Monitoring of Conventional Munitions in the Seas. Results from the BMBF Funded Pro-ject UDEMM “Umweltmonitoring Für Die Delaboration von Munition Im Meer”; GEOMAR Report, N. Ser. 054; GEOMAR Helmholtz Centre for Ocean Research: Kiel, Germany, 2019. [Google Scholar]
- Bae, B.; Autenrieth, R.; Bonner, J.S. Baseline separation of 2,4,6-Trinitrotoluene (TNT) and its Biotrans-Formation products using HPLC: Precautions for analytes loss. Environ. Eng. Res. 1999, 4, 135–142. [Google Scholar]
- European Commission, Joint Research Centre. Guidance Document on the Estimation of LOD and LOQ for Measurements in the Field of Contaminants in Feed and Food; Publications Office of the EU: Luxembourg, 2016. [Google Scholar]
- Dawidziuk, B.; Nawała, J.; Dziedzic, D.; Gordon, D.; Popiel, S. Development, Validation and Comparison of Three Methods of Sample Preparation Used for Identification and Quantification of 2,4,6-Trinitrotoluene and Products of Its Degradation in Sediments by GC-MS/MS. Anal. Methods 2018, 10, 5188–5196. [Google Scholar] [CrossRef]
- Kirchner, M.; Matisová, E.; Hrouzková, S.; Húšková, R. Fast GC and GC-MS analysis of explosives. Petroleum Coal 2007, 49, 2–79. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Splitless | Large Volume Injection | |
|---|---|---|---|
| Injector | Split-/splitless | Programmable temp. vaporization | |
| Inlet liner | Quartz wool | CarboFrit | Quartz wool |
| Injection volume | 1 µL | 5 µL | |
| Injection temperature | 230 °C | 270 °C | 70 °C, (0.18 min, 50 mL × min−1) 5 °C/s to 240 °C (1.5 min, no split) 240 °C (5 min, 200 mL/min−1) |
| Column flow | 1.5 mL × min−1 | 1.2 mL × min−1 | |
| Oven temp. | 100 °C (0.20 min), 30 °C/min to 220 °C (0.30 min), 80 °C to 280 °C (1 min) | 100 °C (1 min), 35 °C/min to 220 °C (0.7 min), 70 °C to 280 °C (1 min) | |
| Total run time | 6.25 min | 6.99 min | |
| Transfer line temp. | 250 °C | ||
| Ion source temp. | 300 °C | ||
| Ionization method | EI | ||
| Compound | Rt SL [min] | Rt LVI [min] | Molecular Mass [g × mol−1] | Transition [m/z] | CE [eV] | |
|---|---|---|---|---|---|---|
| 1,3-Dinitrobenzene | 2.43 | 3.20 | 168.11 | Q | 122.0 > 75.0 | 12 |
| q | 168.0 > 75.0 | 20 | ||||
| q | 168.0 > 122.0 | 8 | ||||
| 2,4-Dinitrotoluene | 2.77 | 3.52 | 182.13 | Q | 165.0 > 63.1 | 22 |
| q | 165.0 > 90.1 | 16 | ||||
| q | 165.0 > 118.1 | 8 | ||||
| Trinitrotoluene | 3.41 | 4.09 | 227.13 | Q | 210.0 > 164.1 | 6 |
| q | 164.0 > 90.1 | 10 | ||||
| q | 108.0 > 76.1 | 12 | ||||
| 13C15N-Trinitrotoluene | 3.41 | 4.09 | 237.06 | Q | 220.1 > 173.1 | 6 |
| q | 220.1 > 203.1 | 8 | ||||
| q | 189.1 > 82.1 | 10 | ||||
| 4-Amino-2,6-dinitrotoluene | 4.22 | 4.85 | 197.15 | Q | 197.0 > 180.1 | 6 |
| q | 180.0 > 163.1 | 8 | ||||
| q | 163.0 > 78.0 | 14 | ||||
| 2-Amino-4,6-dinitrotoluene | 4.42 | 5.07 | 197.15 | Q | 197.0 > 180.1 | 6 |
| q | 180.0 > 133.0 | 6 | ||||
| q | 180.0 > 67.0 | 12 | ||||
| Compound | SL QW | Splitless, CarboFrit Liner | PTV-LVI | ||||||
|---|---|---|---|---|---|---|---|---|---|
| LOD LOQ | R2 | LOD LOQ | R2 | LOD LOQ | R2 | ||||
| fg/µL | fg/µL | fg/µL | |||||||
| 1,3-DNB | 333 | 1099 | 0.9444 | 330 | 1089 | 0.9664 | 32 | 105 | 0.9644 |
| 2,4-DNT | 77 | 254 | 0.9968 | 86 | 284 | 0.9951 | 10 | 33 | 0.9934 |
| TNT | 152 | 502 | 0.9879 | 150 | 495 | 0.9852 | 47 | 155 | 0.9878 |
| 4-ADNT | 95 | 314 | 0.9951 | 88 | 289 | 0.9896 | 8 | 26 | 0.9959 |
| 2-ADNT | 103 | 341 | 0.9943 | 60 | 210 | 0.9944 | 11 | 37 | 0.9919 |
| Compound | Water | Sediment | Mussels by Strehse et al. 2017 [3] | Freeze Dried Mussels by Solid Phase Extraction | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SL QW | PTV LVI | ||||||||||||||
| LOD LOQ ng/L | R2 | LOD LOQ ng/g d.w. | R2 | LOD LOQ ng/g w.w. | R2 | LOD LOQ ng/g d.w. | R2 | LOD LOQ ng/g d.w. | R2 | ||||||
| 1,3DNB | 0.20 | 0.66 | 0.978 | 0.10 | 0.33 | 0.944 | 2.2 | 7.2 | 0.975 | 0.27 | 0.88 | 0.977 | 0.03 | 0.10 | 0.985 |
| 2,4DNT | 0.05 | 0.15 | 0.998 | 0.02 | 0.08 | 0.996 | 2.2 | 7.3 | 0.975 | 0.23 | 0.75 | 0.983 | 0.04 | 0.12 | 0.990 |
| TNT | 0.09 | 0.30 | 0.981 | 0.05 | 0.15 | 0.988 | 3.5 | 11.5 | 0.940 | 0.47 | 1.60 | 0.964 | 0.20 | 0.68 | 0.986 |
| 4ADNT | 0.06 | 0.19 | 0.994 | 0.03 | 0.09 | 0.995 | 1.5 | 5.1 | 0.987 | 0.27 | 0.90 | 0.988 | 0.05 | 0.17 | 0.973 |
| 2ADNT | 0.06 | 0.20 | 0.992 | 0.03 | 0.10 | 0.994 | 1.2 | 4.0 | 0.992 | 0.10 | 0.32 | 0.999 | 0.04 | 0.14 | 0.980 |
| Present Study | Gledhill et al. 2019 [19] | Gordon et al. 2018 [20] | Dawidziuk et al. 2018 [30] | Kirchner et al. 2007 [31] | ||
|---|---|---|---|---|---|---|
| Method | SL QW | PTV LVI | LC-Orbitrap | GC-MS/MS | GC-MS/MS | GC-MS |
| pg/µL | ||||||
| 1,3-DNB | 0.333 | 0.032 | 0.15 | 3.6 | 4.8 | |
| 2,4-DNT | 0.077 | 0.010 | 1.05 | 2.7 | 4.3 | 0.063 |
| TNT | 0.152 | 0.047 | 0.036 | 560.4 | 19.9 | 0.029 |
| 4-ADNT | 0.095 | 0.008 | 0.050 | 47 | ||
| 2-ADNT | 0.103 | 0.011 | 0.030 | 13.1 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bünning, T.H.; Strehse, J.S.; Hollmann, A.C.; Bötticher, T.; Maser, E. A Toolbox for the Determination of Nitroaromatic Explosives in Marine Water, Sediment, and Biota Samples on Femtogram Levels by GC-MS/MS. Toxics 2021, 9, 60. https://doi.org/10.3390/toxics9030060
Bünning TH, Strehse JS, Hollmann AC, Bötticher T, Maser E. A Toolbox for the Determination of Nitroaromatic Explosives in Marine Water, Sediment, and Biota Samples on Femtogram Levels by GC-MS/MS. Toxics. 2021; 9(3):60. https://doi.org/10.3390/toxics9030060
Chicago/Turabian StyleBünning, Tobias Hartwig, Jennifer Susanne Strehse, Ann Christin Hollmann, Tom Bötticher, and Edmund Maser. 2021. "A Toolbox for the Determination of Nitroaromatic Explosives in Marine Water, Sediment, and Biota Samples on Femtogram Levels by GC-MS/MS" Toxics 9, no. 3: 60. https://doi.org/10.3390/toxics9030060
APA StyleBünning, T. H., Strehse, J. S., Hollmann, A. C., Bötticher, T., & Maser, E. (2021). A Toolbox for the Determination of Nitroaromatic Explosives in Marine Water, Sediment, and Biota Samples on Femtogram Levels by GC-MS/MS. Toxics, 9(3), 60. https://doi.org/10.3390/toxics9030060