
Pathophysiology and Clinical Impacts of Chronic Kidney Disease on Coronary Artery Calcification

Abstract
:1. Introduction
2. Impact of CKD on CAC
2.1. Possible Mechanisms of CKD on Vascular Calcification
2.2. Electrolyte Disorders

{kind=link}
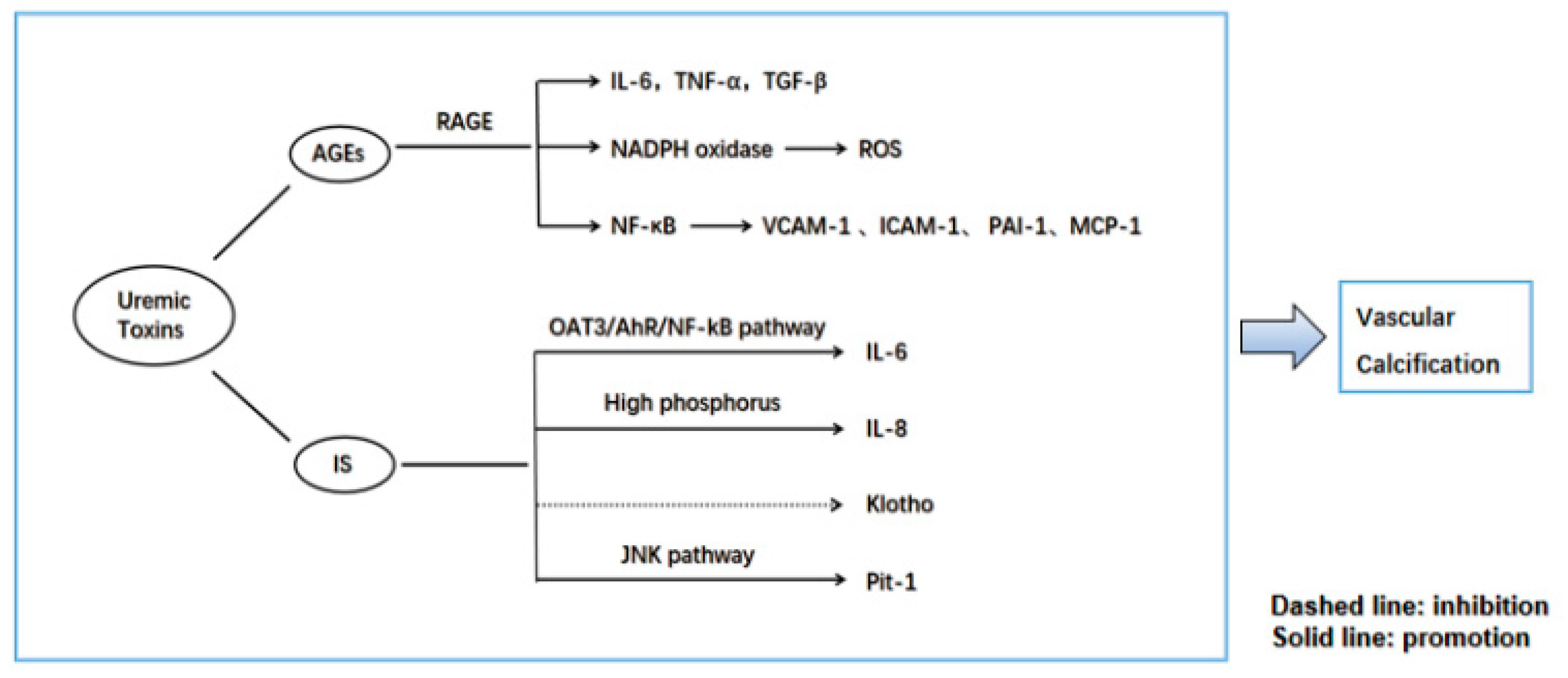{kind=link}
| Electrolyte | Mechanisms | Ref. |
|---|---|---|
| Phosphate | Hyperphosphatemia stimulates the expression of VSMC-related osteogenic transcriptional cytokines and promotes the osteogenic-like transition of VSMCs. | [26] |
| Hyperphosphatemia activates Wnt/β-catenin signaling and increases Pit-1 expression. | [29,30] | |
| Hyperphosphatemia activates the TLR4/NF-κB signaling. | [31] | |
| Hyperphosphatemia induces methylation of the SM22α promoter. | [32] | |
| Calcium | Hypocalcemia combines with high blood phosphorus leading to ectopic calcification and secondary hyperparathyroidism. | [34] |
| Hypocalcemia leads to intracellular calcium overload, mediating intravascular plaque formation. | [35] | |
| Hypercalcemia impairs VSMC function, causing changes in vascular tone. | [36] | |
| Hypercalcemia promotes apoptosis and VSMC matrix vesicle release, providing hydroxyapatite nucleation sites. | [39] | |
| Magnesium | Hypomagnesemia inhibits the conversion of calcium and phosphorus to hydroxyapatite and passively interferes with calcium salt deposition. | [42] |
| Hypomagnesemia inhibits the Wnt/β-catenin signaling pathway and activates the calcium-sensing receptor in VSMCs. | [43,44] | |
| Hypomagnesemia participates in the regulation of oxidative stress and protects endothelial cell function. | [45] |
2.3. Parathyroid Hormone and Vitamin D
2.4. Inflammation
2.5. Uremic Toxins
2.6. Fibroblast Growth Factor 23
2.7. Osteoprotegerin
2.8. Matrix Gla Protein
2.9. Fetuin-A
2.10. Pyrophosphate
2.11. Zinc
2.12. Oxidative Stress
3. Research Progresses
4. Conclusions and Perspective
Author Contributions
Funding
Conflicts of Interest
References
- Lindstrom, M.; DeCleene, N.; Dorsey, H.; Fuster, V.; Johnson, C.O.; LeGrand, K.E.; Mensah, G.A.; Razo, C.; Stark, B.; Varieur Turco, J.; et al. Global Burden of Cardiovascular Diseases and Risks Collaboration, 1990–2021. J. Am. Coll. Cardiol. 2022, 80, 2372–2425. [Google Scholar] [CrossRef] [PubMed]
- Liabeuf, S.; Okazaki, H.; Desjardins, L.; Fliser, D.; Goldsmith, D.; Covic, A.; Wiecek, A.; Ortiz, A.; Martinez-Castelao, A.; Lindholm, B.; et al. Vascular calcification in chronic kidney disease: Are biomarkers useful for probing the pathobiology and the health risks of this process in the clinical scenario? Nephrol. Dial. Transplant. 2014, 29, 1275–1284. [Google Scholar] [CrossRef] [PubMed]
- Vervloet, M.; Cozzolino, M. Vascular calcification in chronic kidney disease: Different bricks in the wall? Kidney Int. 2017, 91, 808–817. [Google Scholar] [CrossRef] [PubMed]
- GBD Chronic Kidney Disease Collaboration. Global, regional, and national burden of chronic kidney disease, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2020, 395, 709–733. [Google Scholar] [CrossRef] [PubMed]
- Durham, A.L.; Speer, M.Y.; Scatena, M.; Giachelli, C.M.; Shanahan, C.M. Role of smooth muscle cells in vascular calcification: Implications in atherosclerosis and arterial stiffness. Cardiovasc. Res. 2018, 114, 590–600. [Google Scholar] [CrossRef]
- Gao, C.; Fu, Y.; Li, Y.; Zhang, X.; Zhang, L.; Yu, F.; Xu, S.S.; Xu, Q.; Zhu, Y.; Guan, Y.; et al. Microsomal Prostaglandin E Synthase-1-Derived PGE2 Inhibits Vascular Smooth Muscle Cell Calcification. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 108–121. [Google Scholar] [CrossRef]
- Nakahara, T.; Dweck, M.R.; Narula, N.; Pisapia, D.; Narula, J.; Strauss, H.W. Coronary Artery Calcification: From Mechanism to Molecular Imaging. JACC Cardiovasc. Imaging 2017, 10, 582–593. [Google Scholar] [CrossRef]
- Lee, S.J.; Lee, I.K.; Jeon, J.H. Vascular Calcification-New Insights into Its Mechanism. Int. J. Mol. Sci. 2020, 21, 2685. [Google Scholar] [CrossRef]
- Chen, N.X.; Moe, S.M. Pathophysiology of Vascular Calcification. Curr. Osteoporos. Rep. 2015, 13, 372–380. [Google Scholar] [CrossRef]
- Detrano, R.; Guerci, A.D.; Carr, J.J.; Bild, D.E.; Burke, G.; Folsom, A.R.; Liu, K.; Shea, S.; Szklo, M.; Bluemke, D.A.; et al. Coronary calcium as a predictor of coronary events in four racial or ethnic groups. N. Engl. J. Med. 2008, 358, 1336–1345. [Google Scholar] [CrossRef]
- Wallin, R.; Wajih, N.; Greenwood, G.T.; Sane, D.C. Arterial calcification: A review of mechanisms, animal models, and the prospects for therapy. Med. Res. Rev. 2001, 21, 274–301. [Google Scholar] [CrossRef] [PubMed]
- Hecht, H.S. Coronary artery calcium scanning: Past, present, and future. JACC Cardiovasc. Imaging 2015, 8, 579–596. [Google Scholar] [CrossRef] [PubMed]
- Kidney Disease: Improving Global Outcomes (KDIGO) Diabetes Work Group. KDIGO 2022 Clinical Practice Guideline for Diabetes Management in Chronic Kidney Disease. Kidney Int. 2022, 102, S1–S127. [Google Scholar] [CrossRef]
- Chen, J.; Budoff, M.J.; Reilly, M.P.; Yang, W.; Rosas, S.E.; Rahman, M.; Zhang, X.; Roy, J.A.; Lustigova, E.; Nessel, L.; et al. Coronary Artery Calcification and Risk of Cardiovascular Disease and Death Among Patients with Chronic Kidney Disease. JAMA Cardiol. 2017, 2, 635–643. [Google Scholar] [CrossRef] [PubMed]
- Chiu, Y.W.; Adler, S.G.; Budoff, M.J.; Takasu, J.; Ashai, J.; Mehrotra, R. Coronary artery calcification and mortality in diabetic patients with proteinuria. Kidney Int. 2010, 77, 1107–1114. [Google Scholar] [CrossRef]
- Xiang, X.; He, J.; Zhang, W.; He, Q.; Liu, Y. Coronary artery calcification in patients with advanced chronic kidney disease. BMC Cardiovasc. Disord. 2022, 22, 453. [Google Scholar] [CrossRef]
- Watanabe, R.; Lemos, M.M.; Manfredi, S.R.; Draibe, S.A.; Canziani, M.E. Impact of cardiovascular calcification in nondialyzed patients after 24 months of follow-up. Clin. J. Am. Soc. Nephrol. 2010, 5, 189–194. [Google Scholar] [CrossRef]
- Mizuiri, S.; Nishizawa, Y.; Yamashita, K.; Mizuno, K.; Ishine, M.; Doi, S.; Masaki, T.; Shigemoto, K. Coronary artery calcification score and common iliac artery calcification score in non-dialysis CKD patients. Nephrology 2018, 23, 837–845. [Google Scholar] [CrossRef]
- Hwang, I.C.; Park, H.E.; Kim, H.L.; Kim, H.M.; Park, J.B.; Yoon, Y.E.; Lee, S.P.; Kim, H.K.; Cho, G.Y.; Sohn, D.W.; et al. Systemic Inflammation Is Associated with Coronary Artery Calcification and All-Cause Mortality in Chronic Kidney Disease. Circ. J. 2016, 80, 1644–1652. [Google Scholar] [CrossRef]
- Yun, H.R.; Joo, Y.S.; Kim, H.W.; Park, J.T.; Chang, T.I.; Son, N.H.; Yoo, T.H.; Kang, S.W.; Sung, S.; Lee, K.B.; et al. Coronary Artery Calcification Score and the Progression of Chronic Kidney Disease. J. Am. Soc. Nephrol. 2022, 33, 1590–1601. [Google Scholar] [CrossRef]
- Lamarche, M.C.; Hopman, W.M.; Garland, J.S.; White, C.A.; Holden, R.M. Relationship of coronary artery calcification with renal function decline and mortality in predialysis chronic kidney disease patients. Nephrol. Dial. Transplant. 2019, 34, 1715–1722. [Google Scholar] [CrossRef] [PubMed]
- Park, K.S.; Chang, J.W.; Kim, T.Y.; Kim, H.W.; Lee, E.K.; Kim, H.S.; Yang, W.S.; Kim, S.B.; Park, S.K.; Lee, S.K.; et al. Lower concentrations of serum phosphorus within the normal range could be associated with less calcification of the coronary artery in Koreans with normal renal function. Am. J. Clin. Nutr. 2011, 94, 1465–1470. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Li, H.; You, L.; Yu, X.; Zhang, M.; Zhu, R.; Hao, C.; Zhang, Z.; Chen, J. Association of serum phosphorus variability with coronary artery calcification among hemodialysis patients. PLoS ONE 2014, 9, e93360. [Google Scholar] [CrossRef] [PubMed]
- Raggi, P.; Boulay, A.; Chasan-Taber, S.; Amin, N.; Dillon, M.; Burke, S.K.; Chertow, G.M. Cardiac calcification in adult hemodialysis patients. A link between end-stage renal disease and cardiovascular disease? J. Am. Coll. Cardiol. 2002, 39, 695–701. [Google Scholar] [CrossRef]
- Adeney, K.L.; Siscovick, D.S.; Ix, J.H.; Seliger, S.L.; Shlipak, M.G.; Jenny, N.S.; Kestenbaum, B.R. Association of serum phosphate with vascular and valvular calcification in moderate CKD. J. Am. Soc. Nephrol. 2009, 20, 381–387. [Google Scholar] [CrossRef]
- Lanzer, P.; Boehm, M.; Sorribas, V.; Thiriet, M.; Janzen, J.; Zeller, T.; St Hilaire, C.; Shanahan, C. Medial vascular calcification revisited: Review and perspectives. Eur. Heart J. 2014, 35, 1515–1525. [Google Scholar] [CrossRef]
- Voelkl, J.; Tuffaha, R.; Luong, T.T.D.; Zickler, D.; Masyout, J.; Feger, M.; Verheyen, N.; Blaschke, F.; Kuro, O.M.; Tomaschitz, A.; et al. Zinc Inhibits Phosphate-Induced Vascular Calcification through TNFAIP3-Mediated Suppression of NF-kappaB. J. Am. Soc. Nephrol. 2018, 29, 1636–1648. [Google Scholar] [CrossRef]
- Bostrom, K.I.; Rajamannan, N.M.; Towler, D.A. The regulation of valvular and vascular sclerosis by osteogenic morphogens. Circ. Res. 2011, 109, 564–577. [Google Scholar] [CrossRef]
- Yao, L.; Sun, Y.T.; Sun, W.; Xu, T.H.; Ren, C.; Fan, X.; Sun, L.; Liu, L.L.; Feng, J.M.; Ma, J.F.; et al. High phosphorus level leads to aortic calcification via beta-catenin in chronic kidney disease. Am. J. Nephrol. 2015, 41, 28–36. [Google Scholar] [CrossRef]
- Leopold, J.A. Vascular calcification: Mechanisms of vascular smooth muscle cell calcification. Trends Cardiovasc. Med. 2015, 25, 267–274. [Google Scholar] [CrossRef]
- Zhang, D.; Bi, X.; Liu, Y.; Huang, Y.; Xiong, J.; Xu, X.; Xiao, T.; Yu, Y.; Jiang, W.; Huang, Y.; et al. High Phosphate-Induced Calcification of Vascular Smooth Muscle Cells is Associated with the TLR4/NF-κb Signaling Pathway. Kidney Blood Press. Res. 2017, 42, 1205–1215. [Google Scholar] [CrossRef]
- Montes de Oca, A.; Madueno, J.A.; Martinez-Moreno, J.M.; Guerrero, F.; Munoz-Castaneda, J.; Rodriguez-Ortiz, M.E.; Mendoza, F.J.; Almaden, Y.; Lopez, I.; Rodriguez, M.; et al. High-phosphate-induced calcification is related to SM22α promoter methylation in vascular smooth muscle cells. J. Bone Miner. Res. 2010, 25, 1996–2005. [Google Scholar] [CrossRef] [PubMed]
- Kovesdy, C.P.; Kuchmak, O.; Lu, J.L.; Kalantar-Zadeh, K. Outcomes associated with serum calcium level in men with non-dialysis-dependent chronic kidney disease. Clin. J. Am. Soc. Nephrol. 2010, 5, 468–476. [Google Scholar] [CrossRef] [PubMed]
- Portillo, M.R.; Rodríguez-Ortiz, M.E. Secondary Hyperparthyroidism: Pathogenesis, Diagnosis, Preventive and Therapeutic Strategies. Rev. Endocr. Metab. Disord. 2017, 18, 79–95. [Google Scholar] [CrossRef] [PubMed]
- Kazmi, A.S.; Wall, B.M. Reversible congestive heart failure related to profound hypocalcemia secondary to hypoparathyroidism. Am. J. Med. Sci. 2007, 333, 226–229. [Google Scholar] [CrossRef]
- Moe, S.M. Calcium as a cardiovascular toxin in CKD-MBD. Bone 2017, 100, 94–99. [Google Scholar] [CrossRef] [PubMed]
- Young, E.W.; Akiba, T.; Albert, J.M.; McCarthy, J.T.; Kerr, P.G.; Mendelssohn, D.C.; Jadoul, M. Magnitude and impact of abnormal mineral metabolism in hemodialysis patients in the Dialysis Outcomes and Practice Patterns Study (DOPPS). Am. J. Kidney Dis. 2004, 44, 34–38. [Google Scholar] [CrossRef]
- Masumoto, A.; Sonou, T.; Ohya, M.; Yashiro, M.; Nakashima, Y.; Okuda, K.; Iwashita, Y.; Mima, T.; Negi, S.; Shigematsu, T. Calcium Overload Accelerates Phosphate-Induced Vascular Calcification Via Pit-1, but not the Calcium-Sensing Receptor. J. Atheroscler. Thromb. 2017, 24, 716–724. [Google Scholar] [CrossRef]
- Shroff, R.; Long, D.A.; Shanahan, C. Mechanistic insights into vascular calcification in CKD. J. Am. Soc. Nephrol. 2013, 24, 179–189. [Google Scholar] [CrossRef]
- Chertow, G.M.; Burke, S.K.; Raggi, P. Sevelamer attenuates the progression of coronary and aortic calcification in hemodialysis patients. Kidney Int. 2002, 62, 245–252. [Google Scholar] [CrossRef]
- Taniguchi, M.; Fukagawa, M.; Fujii, N.; Hamano, T.; Shoji, T.; Yokoyama, K.; Nakai, S.; Shigematsu, T.; Iseki, K.; Tsubakihara, Y.; et al. Serum phosphate and calcium should be primarily and consistently controlled in prevalent hemodialysis patients. Ther. Apher. Dial. 2013, 17, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Ter Braake, A.D.; Shanahan, C.M.; de Baaij, J.H.F. Magnesium Counteracts Vascular Calcification: Passive Interference or Active Modulation? Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1431–1445. [Google Scholar] [CrossRef] [PubMed]
- Floege, J. Magnesium in CKD: More than a calcification inhibitor? J. Nephrol. 2015, 28, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Alesutan, I.; Tuffaha, R.; Auer, T.; Feger, M.; Pieske, B.; Lang, F.; Voelkl, J. Inhibition of osteo/chondrogenic transformation of vascular smooth muscle cells by MgCl2 via calcium-sensing receptor. J. Hypertens. 2017, 35, 523–532. [Google Scholar] [CrossRef]
- Munoz-Castaneda, J.R.; Pendon-Ruiz de Mier, M.V.; Rodriguez, M.; Rodriguez-Ortiz, M.E. Magnesium Replacement to Protect Cardiovascular and Kidney Damage? Lack of Prospective Clinical Trials. Int. J. Mol. Sci. 2018, 19, 664. [Google Scholar] [CrossRef]
- Leenders, N.H.J.; Bos, C.; Hoekstra, T.; Schurgers, L.J.; Vervloet, M.G.; Hoenderop, J.G.J. Dietary magnesium supplementation inhibits abdominal vascular calcification in an experimental animal model of chronic kidney disease. Nephrol. Dial. Transplant. 2022, 37, 1049–1058. [Google Scholar] [CrossRef]
- Sakaguchi, Y.; Hamano, T.; Nakano, C.; Obi, Y.; Matsui, I.; Kusunoki, Y.; Mori, D.; Oka, T.; Hashimoto, N.; Takabatake, Y.; et al. Association between Density of Coronary Artery Calcification and Serum Magnesium Levels among Patients with Chronic Kidney Disease. PLoS ONE 2016, 11, e0163673. [Google Scholar] [CrossRef]
- Molnar, A.O.; Biyani, M.; Hammond, I.; Harmon, J.P.; Lavoie, S.; McCormick, B.; Sood, M.M.; Wagner, J.; Pena, E.; Zimmerman, D.L. Lower serum magnesium is associated with vascular calcification in peritoneal dialysis patients: A cross sectional study. BMC Nephrol. 2017, 18, 129. [Google Scholar] [CrossRef]
- Ter Braake, A.D.; Vervloet, M.G.; de Baaij, J.H.F.; Hoenderop, J.G.J. Magnesium to prevent kidney disease-associated vascular calcification: Crystal clear? Nephrol. Dial. Transplant. 2022, 37, 421–429. [Google Scholar] [CrossRef]
- Sakaguchi, Y.; Hamano, T.; Obi, Y.; Monden, C.; Oka, T.; Yamaguchi, S.; Matsui, I.; Hashimoto, N.; Matsumoto, A.; Shimada, K.; et al. A Randomized Trial of Magnesium Oxide and Oral Carbon Adsorbent for Coronary Artery Calcification in Predialysis CKD. J. Am. Soc. Nephrol. 2019, 30, 1073–1085. [Google Scholar] [CrossRef]
- Cozzolino, M.; Mehmeti, F.; Ciceri, P.; Volpi, E.; Stucchi, A.; Brenna, I.; Cusi, D. The Effect of Paricalcitol on Vascular Calcification and Cardiovascular Disease in Uremia: Beyond PTH Control. Int. J. Nephrol. 2011, 2011, 269060. [Google Scholar] [CrossRef]
- Zhao, F.L.; Zhang, Y.Z.; Tai, G.X.; Wang, Y.; Tong, Q.H.; Fu, L. Serum parathyroid hormone as a potential novel biomarker of coronary heart disease. Genet. Test. Mol. Biomark. 2014, 18, 670–674. [Google Scholar] [CrossRef] [PubMed]
- Talmor-Barkan, Y.; Bernheim, J.; Green, J.; Benchetrit, S.; Rashid, G. Calcitriol counteracts endothelial cell pro-inflammatory processes in a chronic kidney disease-like environment. J. Steroid Biochem. Mol. Biol. 2011, 124, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Jean, G.; Bresson, E.; Lorriaux, C.; Mayor, B.; Hurot, J.M.; Deleaval, P.; Chazot, C. Increased levels of serum parathyroid hormone and fibroblast growth factor-23 are the main factors associated with the progression of vascular calcification in long-hour hemodialysis patients. Nephron Clin. Pract. 2012, 120, c132–c138. [Google Scholar] [CrossRef] [PubMed]
- London, G.M.; Marchais, S.J.; Guerin, A.P.; Boutouyrie, P.; Metivier, F.; de Vernejoul, M.C. Association of bone activity, calcium load, aortic stiffness, and calcifications in ESRD. J. Am. Soc. Nephrol. 2008, 19, 1827–1835. [Google Scholar] [CrossRef]
- Malluche, H.H.; Blomquist, G.; Monier-Faugere, M.C.; Cantor, T.L.; Davenport, D.L. High Parathyroid Hormone Level and Osteoporosis Predict Progression of Coronary Artery Calcification in Patients on Dialysis. J. Am. Soc. Nephrol. 2015, 26, 2534–2544. [Google Scholar] [CrossRef]
- Floege, J.; Kim, J.; Ireland, E.; Chazot, C.; Drueke, T.; de Francisco, A.; Kronenberg, F.; Marcelli, D.; Passlick-Deetjen, J.; Schernthaner, G.; et al. Serum iPTH, calcium and phosphate, and the risk of mortality in a European haemodialysis population. Nephrol. Dial. Transplant. 2011, 26, 1948–1955. [Google Scholar] [CrossRef]
- Mary, A.; Henaut, L.; Boudot, C.; Six, I.; Brazier, M.; Massy, Z.A.; Drueke, T.B.; Kamel, S.; Mentaverri, R. Calcitriol prevents in vitro vascular smooth muscle cell mineralization by regulating calcium-sensing receptor expression. Endocrinology 2015, 156, 1965–1974. [Google Scholar] [CrossRef]
- Razzaque, M.S. The dualistic role of vitamin D in vascular calcifications. Kidney Int. 2011, 79, 708–714. [Google Scholar] [CrossRef]
- Haffner, D.; Hocher, B.; Müller, D.; Simon, K.; König, K.; Richter, C.M.; Eggert, B.; Schwarz, J.; Godes, M.; Nissel, R.; et al. Systemic cardiovascular disease in uremic rats induced by 1,25(OH)2D3. J. Hypertens. 2005, 23, 1067–1075. [Google Scholar] [CrossRef]
- Chung, S.; Kim, M.; Koh, E.S.; Hwang, H.S.; Chang, Y.K.; Park, C.W.; Kim, S.Y.; Chang, Y.S.; Hong, Y.A. Serum 1,25-dihydroxyvitamin D Better Reflects Renal Parameters than 25-hydoxyvitamin D in Patients with Glomerular Diseases. Int. J. Med. Sci. 2017, 14, 1080–1087. [Google Scholar] [CrossRef] [PubMed]
- Samaan, F.; Carvalho, A.B.; Pillar, R.; Rocha, L.A.; Cassiolato, J.L.; Cuppari, L.; Canziani, M.E.F. The Effect of Long-Term Cholecalciferol Supplementation on Vascular Calcification in Chronic Kidney Disease Patients with Hypovitaminosis D. J. Ren. Nutr. 2019, 29, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Briese, S.; Wiesner, S.; Will, J.C.; Lembcke, A.; Opgen-Rhein, B.; Nissel, R.; Wernecke, K.D.; Andreae, J.; Haffner, D.; Querfeld, U. Arterial and cardiac disease in young adults with childhood-onset end-stage renal disease-impact of calcium and vitamin D therapy. Nephrol. Dial. Transplant. 2006, 21, 1906–1914. [Google Scholar] [CrossRef]
- Shroff, R.; Egerton, M.; Bridel, M.; Shah, V.; Donald, A.E.; Cole, T.J.; Hiorns, M.P.; Deanfield, J.E.; Rees, L. A bimodal association of vitamin D levels and vascular disease in children on dialysis. J. Am. Soc. Nephrol. 2008, 19, 1239–1246. [Google Scholar] [CrossRef] [PubMed]
- Pillar, R.; Lopes, M.G.G.; Rocha, L.A.; Cuppari, L.; Carvalho, A.B.; Draibe, S.A.; Canziani, M.E. Severe hypovitaminosis D in chronic kidney disease: Association with blood pressure and coronary artery calcification. Hypertens. Res. 2013, 36, 428–432. [Google Scholar] [CrossRef]
- Liu, J.; Ma, K.L.; Gao, M.; Wang, C.X.; Ni, J.; Zhang, Y.; Zhang, X.L.; Liu, H.; Wang, Y.L.; Liu, B.C. Inflammation disrupts the LDL receptor pathway and accelerates the progression of vascular calcification in ESRD patients. PLoS ONE 2012, 7, e47217. [Google Scholar] [CrossRef] [PubMed]
- London, G.M.; Marchais, S.J.; Guerin, A.P.; Metivier, F.; Adda, H.; Pannier, B. Inflammation, arteriosclerosis, and cardiovascular therapy in hemodialysis patients. Kidney Int. Suppl. 2003, 84, S88–S93. [Google Scholar] [CrossRef]
- Stompor, T.; Pasowicz, M.; Sullowicz, W.; Dembinska-Kiec, A.; Janda, K.; Wojcik, K.; Tracz, W.; Zdzienicka, A.; Klimeczek, P.; Janusz-Grzybowska, E. An association between coronary artery calcification score, lipid profile, and selected markers of chronic inflammation in ESRD patients treated with peritoneal dialysis. Am. J. Kidney Dis. 2003, 41, 203–211. [Google Scholar] [CrossRef]
- Krasniak, A.; Drozdz, M.; Pasowicz, M.; Chmiel, G.; Michalek, M.; Szumilak, D.; Podolec, P.; Klimeczek, P.; Konieczynska, M.; Wicher-Muniak, E.; et al. Factors involved in vascular calcification and atherosclerosis in maintenance haemodialysis patients. Nephrol. Dial. Transplant. 2007, 22, 515–521. [Google Scholar] [CrossRef]
- Jung, H.H.; Kim, S.W.; Han, H. Inflammation, mineral metabolism and progressive coronary artery calcification in patients on haemodialysis. Nephrol. Dial. Transplant. 2006, 21, 1915–1920. [Google Scholar] [CrossRef]
- Shang, D.; Xie, Q.; Shang, B.; Zhang, M.; You, L.; Hao, C.M.; Zhu, T. Hyperphosphatemia and hs-CRP Initiate the Coronary Artery Calcification in Peritoneal Dialysis Patients. BioMed Res. Int. 2017, 2017, 2520510. [Google Scholar] [CrossRef] [PubMed]
- Brancaccio, D.; Tetta, C.; Gallieni, M.; Panichi, V. Inflammation, CRP, calcium overload and a high calcium-phosphate product: A “liaison dangereuse”. Nephrol. Dial. Transplant. 2002, 17, 201–203. [Google Scholar] [CrossRef] [PubMed]
- Voelkl, J.; Cejka, D.; Alesutan, I. An overview of the mechanisms in vascular calcification during chronic kidney disease. Curr. Opin. Nephrol. Hypertens. 2019, 28, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Weiss, M.F.; Erhard, P.; Kader-Attia, F.A.; Wu, Y.C.; Deoreo, P.B.; Araki, A.; Glomb, M.A.; Monnier, V.M. Mechanisms for the formation of glycoxidation products in end-stage renal disease. Kidney Int. 2000, 57, 2571–2585. [Google Scholar] [CrossRef] [PubMed]
- Wei, Q.; Ren, X.; Jiang, Y.; Jin, H.; Liu, N.; Li, J. Advanced glycation end products accelerate rat vascular calcification through RAGE/oxidative stress. BMC Cardiovasc. Disord. 2013, 13, 13. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Luo, W.; Han, J.; Khan, Z.A.; Fang, Q.; Jin, Y.; Chen, X.; Zhang, Y.; Wang, M.; Qian, J.; et al. MD2 activation by direct AGE interaction drives inflammatory diabetic cardiomyopathy. Nat. Commun. 2020, 11, 2148. [Google Scholar] [CrossRef]
- Kosmopoulos, M.; Drekolias, D.; Zavras, P.D.; Piperi, C.; Papavassiliou, A.G. Impact of advanced glycation end products (AGEs) signaling in coronary artery disease. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 611–619. [Google Scholar] [CrossRef]
- Fukami, K.; Yamagishi, S.; Okuda, S. Role of AGEs-RAGE system in cardiovascular disease. Curr. Pharm. Des. 2014, 20, 2395–2402. [Google Scholar] [CrossRef]
- Taki, K.; Takayama, F.; Tsuruta, Y.; Niwa, T. Oxidative stress, advanced glycation end product, and coronary artery calcification in hemodialysis patients. Kidney Int. 2006, 70, 218–224. [Google Scholar] [CrossRef]
- Fonseca, L.F.; Araujo, A.B.; Quadros, K.R.d.S.; Carbonara, C.E.M.; Dertkigil, S.S.J.; Sposito, A.C.; Oliveira, R.B. AGEs accumulation is related to muscle degeneration and vascular calcification in peritoneal dialysis patients. J. Bras. Nefrol. 2021, 43, 191–199. [Google Scholar] [CrossRef]
- Muteliefu, G.; Enomoto, A.; Jiang, P.; Takahashi, M.; Niwa, T. Indoxyl sulphate induces oxidative stress and the expression of osteoblast-specific proteins in vascular smooth muscle cells. Nephrol. Dial. Transplant. 2009, 24, 2051–2058. [Google Scholar] [CrossRef] [PubMed]
- Adelibieke, Y.; Yisireyili, M.; Ng, H.Y.; Saito, S.; Nishijima, F.; Niwa, T. Indoxyl sulfate induces IL-6 expression in vascular endothelial and smooth muscle cells through OAT3-mediated uptake and activation of AhR/NF-kappaB pathway. Nephron Exp. Nephrol. 2014, 128, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Bouabdallah, J.; Zibara, K.; Issa, H.; Lenglet, G.; Kchour, G.; Caus, T.; Six, I.; Choukroun, G.; Kamel, S.; Bennis, Y. Endothelial cells exposed to phosphate and indoxyl sulphate promote vascular calcification through interleukin-8 secretion. Nephrol. Dial. Transplant. 2019, 34, 1125–1134. [Google Scholar] [CrossRef]
- Wu, Y.; Han, X.; Wang, L.; Diao, Z.; Liu, W. Indoxyl sulfate promotes vascular smooth muscle cell calcification via the JNK/Pit-1 pathway. Ren. Fail. 2016, 38, 1702–1710. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Chen, J.; Shen, Z.; Gu, Y.; Xu, L.; Hu, J.; Zhang, X.; Ding, X. Indoxyl sulfate accelerates vascular smooth muscle cell calcification via microRNA-29b dependent regulation of Wnt/beta-catenin signaling. Toxicol. Lett. 2018, 284, 29–36. [Google Scholar] [CrossRef]
- Chen, G.; Liu, Y.; Goetz, R.; Fu, L.; Jayaraman, S.; Hu, M.C.; Moe, O.W.; Liang, G.; Li, X.; Mohammadi, M. alpha-Klotho is a non-enzymatic molecular scaffold for FGF23 hormone signalling. Nature 2018, 553, 461–466. [Google Scholar] [CrossRef]
- Chen, J.; Zhang, X.; Zhang, H.; Liu, T.; Zhang, H.; Teng, J.; Ji, J.; Ding, X. Indoxyl Sulfate Enhance the Hypermethylation of Klotho and Promote the Process of Vascular Calcification in Chronic Kidney Disease. Int. J. Biol. Sci. 2016, 12, 1236–1246. [Google Scholar] [CrossRef]
- Barreto, F.C.; Barreto, D.V.; Liabeuf, S.; Meert, N.; Glorieux, G.; Temmar, M.; Choukroun, G.; Vanholder, R.; Massy, Z.A.; European Uremic Toxin Work Group. Serum Indoxyl sulfate is associated with vascular disease and mortality in chronic kidney disease patients. Clin. J. Am. Soc. Nephrol. 2009, 4, 1551–1558. [Google Scholar] [CrossRef]
- Asami, M.; Tanabe, K.; Ito, S.; Yoshida, E.; Aoki, J.; Tanimoto, S.; Horiuchi, Y.; Yoshida, M. Impact of Indoxyl Sulfate on Coronary Plaques in Patients on Hemodialysis. Int. Heart J. 2018, 59, 489–496. [Google Scholar] [CrossRef]
- Martin, A.; David, V.; Quarles, L.D. Regulation and function of the FGF23/klotho endocrine pathways. Physiol. Rev. 2012, 92, 131–155. [Google Scholar] [CrossRef]
- Di Lullo, L.; Gorini, A.; Bellasi, A.; Morrone, L.F.; Rivera, R.; Russo, L.; Santoboni, A.; Russo, D. Fibroblast growth factor 23 and parathyroid hormone predict extent of aortic valve calcifications in patients with mild to moderate chronic kidney disease. Clin. Kidney J. 2015, 8, 732–736. [Google Scholar] [CrossRef]
- Barthel, T.K.; Mathern, D.R.; Whitfield, G.K.; Haussler, C.A.; Hopper, H.A., 4th; Hsieh, J.C.; Slater, S.A.; Hsieh, G.; Kaczmarska, M.; Jurutka, P.W.; et al. 1,25-Dihydroxyvitamin D3/VDR-mediated induction of FGF23 as well as transcriptional control of other bone anabolic and catabolic genes that orchestrate the regulation of phosphate and calcium mineral metabolism. J. Steroid Biochem. Mol. Biol. 2007, 103, 381–388. [Google Scholar] [CrossRef]
- Silswal, N.; Touchberry, C.D.; Daniel, D.R.; McCarthy, D.L.; Zhang, S.; Andresen, J.; Stubbs, J.R.; Wacker, M.J. FGF23 directly impairs endothelium-dependent vasorelaxation by increasing superoxide levels and reducing nitric oxide bioavailability. Am. J. Physiol. Endocrinol. Metab. 2014, 307, E426–E436. [Google Scholar] [CrossRef]
- Srivaths, P.R.; Goldstein, S.L.; Silverstein, D.M.; Krishnamurthy, R.; Brewer, E.D. Elevated FGF 23 and phosphorus are associated with coronary calcification in hemodialysis patients. Pediatr. Nephrol. 2011, 26, 945–951. [Google Scholar] [CrossRef]
- Desjardins, L.; Liabeuf, S.; Renard, C.; Lenglet, A.; Lemke, H.D.; Choukroun, G.; Drueke, T.B.; Massy, Z.A.; European Uremic Toxin Work Group. FGF23 is independently associated with vascular calcification but not bone mineral density in patients at various CKD stages. Osteoporos. Int. 2012, 23, 2017–2025. [Google Scholar] [CrossRef] [PubMed]
- Nasrallah, M.M.; El-Shehaby, A.R.; Salem, M.M.; Osman, N.A.; El Sheikh, E.; Sharaf El Din, U.A. Fibroblast growth factor-23 (FGF-23) is independently correlated to aortic calcification in haemodialysis patients. Nephrol. Dial. Transplant. 2010, 25, 2679–2685. [Google Scholar] [CrossRef]
- Hu, M.C.; Shi, M.; Zhang, J.; Quinones, H.; Griffith, C.; Kuro-o, M.; Moe, O.W. Klotho deficiency causes vascular calcification in chronic kidney disease. J. Am. Soc. Nephrol. 2011, 22, 124–136. [Google Scholar] [CrossRef] [PubMed]
- Razzaque, M.S. The FGF23-Klotho axis: Endocrine regulation of phosphate homeostasis. Nat. Rev. Endocrinol. 2009, 5, 611–619. [Google Scholar] [CrossRef] [PubMed]
- Yamada, S.; Giachelli, C.M. Vascular calcification in CKD-MBD: Roles for phosphate, FGF23, and Klotho. Bone 2017, 100, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Zununi Vahed, S.; Nikasa, P.; Ardalan, M. Klotho and renal fibrosis. Nephrourol. Mon. 2013, 5, 946–948. [Google Scholar] [CrossRef]
- Yamamoto, M.; Clark, J.D.; Pastor, J.V.; Gurnani, P.; Nandi, A.; Kurosu, H.; Miyoshi, M.; Ogawa, Y.; Castrillon, D.H.; Rosenblatt, K.P.; et al. Regulation of oxidative stress by the anti-aging hormone klotho. J. Biol. Chem. 2005, 280, 38029–38034. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Mao, H.; Chen, C.; Wu, L.; Wang, N.; Zhao, X.; Qian, J.; Xing, C. The Role and Mechanism of alpha-Klotho in the Calcification of Rat Aortic Vascular Smooth Muscle Cells. Biomed. Res. Int. 2015, 2015, 194362. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, O.M.; Januzzi, J.L.; Isakova, T.; Laliberte, K.; Smith, K.; Collerone, G.; Sarwar, A.; Hoffmann, U.; Coglianese, E.; Christenson, R.; et al. Fibroblast growth factor 23 and left ventricular hypertrophy in chronic kidney disease. Circulation 2009, 119, 2545–2552. [Google Scholar] [CrossRef] [PubMed]
- Scialla, J.J.; Lau, W.L.; Reilly, M.P.; Isakova, T.; Yang, H.Y.; Crouthamel, M.H.; Chavkin, N.W.; Rahman, M.; Wahl, P.; Amaral, A.P.; et al. Fibroblast growth factor 23 is not associated with and does not induce arterial calcification. Kidney Int. 2013, 83, 1159–1168. [Google Scholar] [CrossRef] [PubMed]
- Lau, W.L.; Leaf, E.M.; Hu, M.C.; Takeno, M.M.; Kuro-o, M.; Moe, O.W.; Giachelli, C.M. Vitamin D receptor agonists increase klotho and osteopontin while decreasing aortic calcification in mice with chronic kidney disease fed a high phosphate diet. Kidney Int. 2012, 82, 1261–1270. [Google Scholar] [CrossRef]
- Davenport, C.; Harper, E.; Forde, H.; Rochfort, K.D.; Murphy, R.P.; Smith, D.; Cummins, P.M. RANKL promotes osteoblastic activity in vascular smooth muscle cells by upregulating endothelial BMP-2 release. Int. J. Biochem. Cell Biol. 2016, 77, 171–180. [Google Scholar] [CrossRef]
- Panizo, S.; Cardus, A.; Encinas, M.; Parisi, E.; Valcheva, P.; López, S.; Coll, B.; Fernandez, E.; Valdivielso, J.M. RANKL Increases Vascular Smooth Muscle Cell Calcification Through a RANK-BMP4–Dependent Pathway. Circ. Res. 2009, 104, 1041–1048. [Google Scholar] [CrossRef]
- Dellegrottaglie, S.; Sanz, J.; Rajagopalan, S. Molecular determinants of vascular calcification: A bench to bedside view. Curr. Mol. Med. 2006, 6, 515–524. [Google Scholar] [CrossRef]
- Wright, H.L.; McCarthy, H.S.; Middleton, J.; Marshall, M.J. RANK, RANKL and osteoprotegerin in bone biology and disease. Curr. Rev. Musculoskelet. Med. 2009, 2, 56–64. [Google Scholar] [CrossRef]
- Znorko, B.; Oksztulska, E.; Michałowska, M.; Kamiński, T.; Pawlak, K. Does the OPG/RANKL system contribute to the bone-vascular axis in chronic kidney disease? A systematic review. Adv. Med. Sci. 2017, 62, 52–64. [Google Scholar] [CrossRef]
- Di Bartolo, B.A.; Cartland, S.P.; Harith, H.H.; Bobryshev, Y.V.; Schoppet, M.; Kavurma, M.M. TRAIL-deficiency accelerates vascular calcification in atherosclerosis via modulation of RANKL. PLoS ONE 2013, 8, e74211. [Google Scholar] [CrossRef]
- Kazama, J.J.; Shigematsu, T.; Yano, K.; Tsuda, E.; Miura, M.; Iwasaki, Y.; Kawaguchi, Y.; Gejyo, F.; Kurokawa, K.; Fukagawa, M. Increased circulating levels of osteoclastogenesis inhibitory factor (osteoprotegerin) in patients with chronic renal failure. Am. J. Kidney Dis. 2002, 39, 525–532. [Google Scholar] [CrossRef] [PubMed]
- Nitta, K.; Akiba, T.; Uchida, K.; Otsubo, S.; Takei, T.; Yumura, W.; Kabaya, T.; Nihei, H. Serum osteoprotegerin levels and the extent of vascular calcification in haemodialysis patients. Nephrol. Dial. Transplant. 2004, 19, 1886–1889. [Google Scholar] [CrossRef] [PubMed]
- Oštrić, M.; Kukuljan, M.; Markić, D.; Gršković, A.; Ivančić, A.; Bobinac, D.; Španjol, J.; Maroević, J.; Šoša, I.; Ćelić, T. Expression of bone-related proteins in vascular calcification and its serum correlations with coronary artery calcification score. J. Biol. Regul. Homeost. Agents 2019, 33, 29–38. [Google Scholar] [PubMed]
- Barreto, D.V.; Barreto, F.C.; Carvalho, A.B.; Cuppari, L.; Cendoroglo, M.; Draibe, S.A.; Moyses, R.M.; Neves, K.R.; Jorgetti, V.; Blair, A.; et al. Coronary calcification in hemodialysis patients: The contribution of traditional and uremia-related risk factors. Kidney Int. 2005, 67, 1576–1582. [Google Scholar] [CrossRef]
- Mesquita, M.; Demulder, A.; Damry, N.; Melot, C.; Wittersheim, E.; Willems, D.; Dratwa, M.; Bergmann, P. Plasma osteoprotegerin is an independent risk factor for mortality and an early biomarker of coronary vascular calcification in chronic kidney disease. Clin. Chem. Lab. Med. 2009, 47, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Kurnatowska, I.; Grzelak, P.; Kaczmarska, M.; Stefanczyk, L.; Nowicki, M. Serum osteoprotegerin is a predictor of progression of atherosclerosis and coronary calcification in hemodialysis patients. Nephron Clin. Pract. 2011, 117, c297–c304. [Google Scholar] [CrossRef] [PubMed]
- Marques, G.L.; Hayashi, S.; Bjallmark, A.; Larsson, M.; Riella, M.; Olandoski, M.; Lindholm, B.; Nascimento, M.M. Osteoprotegerin is a marker of cardiovascular mortality in patients with chronic kidney disease stages 3–5. Sci. Rep. 2021, 11, 2473. [Google Scholar] [CrossRef]
- Hjelmesaeth, J.; Ueland, T.; Flyvbjerg, A.; Bollerslev, J.; Leivestad, T.; Jenssen, T.; Hansen, T.K.; Thiel, S.; Sagedal, S.; Roislien, J.; et al. Early posttransplant serum osteoprotegerin levels predict long-term (8-year) patient survival and cardiovascular death in renal transplant patients. J. Am. Soc. Nephrol. 2006, 17, 1746–1754. [Google Scholar] [CrossRef] [PubMed]
- Roumeliotis, S.; Dounousi, E.; Eleftheriadis, T.; Liakopoulos, V. Association of the Inactive Circulating Matrix Gla Protein with Vitamin K Intake, Calcification, Mortality, and Cardiovascular Disease: A Review. Int. J. Mol. Sci. 2019, 20, 628. [Google Scholar] [CrossRef]
- Schurgers, L.J.; Spronk, H.M.; Soute, B.A.; Schiffers, P.M.; DeMey, J.G.; Vermeer, C. Regression of warfarin-induced medial elastocalcinosis by high intake of vitamin K in rats. Blood 2007, 109, 2823–2831. [Google Scholar] [CrossRef]
- Wallin, R.; Cain, D.; Hutson, S.M.; Sane, D.C.; Loeser, R. Modulation of the binding of matrix Gla protein (MGP) to bone morphogenetic protein-2 (BMP-2). Thromb. Haemost. 2000, 84, 1039–1044. [Google Scholar]
- Chatrou, M.L.; Cleutjens, J.P.; van der Vusse, G.J.; Roijers, R.B.; Mutsaers, P.H.; Schurgers, L.J. Intra-Section Analysis of Human Coronary Arteries Reveals a Potential Role for Micro-Calcifications in Macrophage Recruitment in the Early Stage of Atherosclerosis. PLoS ONE 2015, 10, e0142335. [Google Scholar] [CrossRef] [PubMed]
- Shanahan, C.M. Mechanisms of vascular calcification in renal disease. Clin. Nephrol. 2005, 63, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Viegas, C.S.; Rafael, M.S.; Enriquez, J.L.; Teixeira, A.; Vitorino, R.; Luis, I.M.; Costa, R.M.; Santos, S.; Cavaco, S.; Neves, J.; et al. Gla-rich protein acts as a calcification inhibitor in the human cardiovascular system. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Roumeliotis, S.; Dounousi, E.; Salmas, M.; Eleftheriadis, T.; Liakopoulos, V. Vascular Calcification in Chronic Kidney Disease: The Role of Vitamin K- Dependent Matrix Gla Protein. Front. Med. 2020, 7, 154. [Google Scholar] [CrossRef] [PubMed]
- Kaesler, N.; Magdeleyns, E.; Herfs, M.; Schettgen, T.; Brandenburg, V.; Fliser, D.; Vermeer, C.; Floege, J.; Schlieper, G.; Kruger, T. Impaired vitamin K recycling in uremia is rescued by vitamin K supplementation. Kidney Int. 2014, 86, 286–293. [Google Scholar] [CrossRef]
- McCabe, K.M.; Booth, S.L.; Fu, X.; Shobeiri, N.; Pang, J.J.; Adams, M.A.; Holden, R.M. Dietary vitamin K and therapeutic warfarin alter the susceptibility to vascular calcification in experimental chronic kidney disease. Kidney Int. 2013, 83, 835–844. [Google Scholar] [CrossRef]
- Kurnatowska, I.; Grzelak, P.; Masajtis-Zagajewska, A.; Kaczmarska, M.; Stefańczyk, L.; Vermeer, C.; Maresz, K.; Nowicki, M. Effect of vitamin K2 on progression of atherosclerosis and vascular calcification in nondialyzed patients with chronic kidney disease stages 3-5. Pol. Arch. Med. Wewn. 2015, 125, 631–640. [Google Scholar] [CrossRef] [PubMed]
- Witham, M.D.; Lees, J.S.; White, M.; Band, M.; Bell, S.; Chantler, D.J.; Ford, I.; Fulton, R.L.; Kennedy, G.; Littleford, R.C.; et al. Vitamin K Supplementation to Improve Vascular Stiffness in CKD: The K4Kidneys Randomized Controlled Trial. J. Am. Soc. Nephrol. 2020, 31, 2434–2445. [Google Scholar] [CrossRef]
- Oikonomaki, T.; Papasotiriou, M.; Ntrinias, T.; Kalogeropoulou, C.; Zabakis, P.; Kalavrizioti, D.; Papadakis, I.; Goumenos, D.S.; Papachristou, E. The effect of vitamin K2 supplementation on vascular calcification in haemodialysis patients: A 1-year follow-up randomized trial. Int. Urol. Nephrol. 2019, 51, 2037–2044. [Google Scholar] [CrossRef] [PubMed]
- Levy-Schousboe, K.; Frimodt-Moller, M.; Hansen, D.; Peters, C.D.; Kjaergaard, K.D.; Jensen, J.D.; Strandhave, C.; Elming, H.; Larsen, C.T.; Sandstrom, H.; et al. Vitamin K supplementation and arterial calcification in dialysis: Results of the double-blind, randomized, placebo-controlled RenaKvit trial. Clin. Kidney J. 2021, 14, 2114–2123. [Google Scholar] [CrossRef] [PubMed]
- Meuwese, C.L.; Olauson, H.; Qureshi, A.R.; Ripsweden, J.; Barany, P.; Vermeer, C.; Drummen, N.; Stenvinkel, P. Associations between Thyroid Hormones, Calcification Inhibitor Levels and Vascular Calcification in End-Stage Renal Disease. PLoS ONE 2015, 10, e0132353. [Google Scholar] [CrossRef] [PubMed]
- Mizuiri, S.; Nishizawa, Y.; Yamashita, K.; Ono, K.; Naito, T.; Tanji, C.; Usui, K.; Doi, S.; Masaki, T.; Shigemoto, K. Relationship of matrix Gla protein and vitamin K with vascular calcification in hemodialysis patients. Ren. Fail. 2019, 41, 770–777. [Google Scholar] [CrossRef]
- Jaminon, A.M.G.; Dai, L.; Qureshi, A.R.; Evenepoel, P.; Ripsweden, J.; Soderberg, M.; Witasp, A.; Olauson, H.; Schurgers, L.J.; Stenvinkel, P. Matrix Gla protein is an independent predictor of both intimal and medial vascular calcification in chronic kidney disease. Sci. Rep. 2020, 10, 6586. [Google Scholar] [CrossRef]
- Schurgers, L.J.; Barreto, D.V.; Barreto, F.C.; Liabeuf, S.; Renard, C.; Magdeleyns, E.J.; Vermeer, C.; Choukroun, G.; Massy, Z.A. The circulating inactive form of matrix gla protein is a surrogate marker for vascular calcification in chronic kidney disease: A preliminary report. Clin. J. Am. Soc. Nephrol. 2010, 5, 568–575. [Google Scholar] [CrossRef]
- Holt, S.G.; Smith, E.R. Fetuin-A-containing calciprotein particles in mineral trafficking and vascular disease. Nephrol. Dial. Transplant. 2016, 31, 1583–1587. [Google Scholar] [CrossRef]
- Jahnen-Dechent, W.; Heiss, A.; Schafer, C.; Ketteler, M. Fetuin-A regulation of calcified matrix metabolism. Circ. Res. 2011, 108, 1494–1509. [Google Scholar] [CrossRef]
- Lyu, B.; Banerjee, T.; Scialla, J.J.; Shafi, T.; Yevzlin, A.S.; Powe, N.R.; Parekh, R.S.; Astor, B.C. Vascular Calcification Markers and Hemodialysis Vascular Access Complications. Am. J. Nephrol. 2018, 48, 330–338. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Mondal, S.A.; Kumar, M.; Dutta, D. Proinflammatory and antiinflammatory attributes of fetuin-a: A novel hepatokine modulating cardiovascular and glycemic outcomes in metabolic syndrome. Endocr. Pract. 2014, 20, 1345–1351. [Google Scholar] [CrossRef]
- Mori, K.; Emoto, M.; Inaba, M. Fetuin-A and the cardiovascular system. Adv. Clin. Chem. 2012, 56, 175–195. [Google Scholar]
- Hermans, M.M.H.; Brandenburg, V.; Ketteler, M.; Kooman, J.P.; van der Sande, F.M.; Boeschoten, E.W.; Leunissen, K.M.L.; Krediet, R.T.; Dekker, F.W. Association of serum fetuin-A levels with mortality in dialysis patients. Kidney Int. 2007, 72, 202–207. [Google Scholar] [CrossRef] [PubMed]
- Caglar, K.; Yilmaz, M.I.; Saglam, M.; Cakir, E.; Acikel, C.; Eyileten, T.; Yenicesu, M.; Oguz, Y.; Vural, A.; Carrero, J.J.; et al. Short-term treatment with sevelamer increases serum fetuin-a concentration and improves endothelial dysfunction in chronic kidney disease stage 4 patients. Clin. J. Am. Soc. Nephrol. 2008, 3, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Turkmen, K.; Gorgulu, N.; Uysal, M.; Ozkok, A.; Sakaci, T.; Unsal, A.; Yildiz, A. Fetuin-A, inflammation, and coronary artery calcification in hemodialysis patients. Indian J. Nephrol. 2011, 21, 90–94. [Google Scholar] [CrossRef]
- Kirkpantur, A.; Altun, B.; Hazirolan, T.; Akata, D.; Arici, M.; Kirazli, S.; Turgan, C. Association among serum fetuin-A level, coronary artery calcification, and bone mineral densitometry in maintenance hemodialysis patients. Artif. Organs 2009, 33, 844–854. [Google Scholar] [CrossRef] [PubMed]
- Mehrotra, R.; Westenfeld, R.; Christenson, P.; Budoff, M.; Ipp, E.; Takasu, J.; Gupta, A.; Norris, K.; Ketteler, M.; Adler, S. Serum fetuin-A in nondialyzed patients with diabetic nephropathy: Relationship with coronary artery calcification. Kidney Int. 2005, 67, 1070–1077. [Google Scholar] [CrossRef]
- Rudloff, S.; Janot, M.; Rodriguez, S.; Dessalle, K.; Jahnen-Dechent, W.; Huynh-Do, U. Fetuin-A is a HIF target that safeguards tissue integrity during hypoxic stress. Nat. Commun. 2021, 12, 549. [Google Scholar] [CrossRef]
- Ossareh, S.; Rayatnia, M.; Vahedi, M.; Jafari, H.; Zebarjadi, M. Association of Serum Fetuin-A with Vascular Calcification in Hemodialysis Patients and Its’ Impact on 3-year Mortality. Iran. J. Kidney Dis. 2020, 14, 500–509. [Google Scholar]
- Mikami, S.; Hamano, T.; Fujii, N.; Nagasawa, Y.; Isaka, Y.; Moriyama, T.; Matsuhisa, M.; Ito, T.; Imai, E.; Hori, M. Serum osteoprotegerin as a screening tool for coronary artery calcification score in diabetic pre-dialysis patients. Hypertens. Res. 2008, 31, 1163–1170. [Google Scholar] [CrossRef]
- Ulutas, O.; Taskapan, M.C.; Dogan, A.; Baysal, T.; Taskapan, H. Vascular calcification is not related to serum fetuin-A and osteopontin levels in hemodialysis patients. Int. Urol. Nephrol. 2018, 50, 137–142. [Google Scholar] [CrossRef]
- Villa-Bellosta, R.; Wang, X.; Millan, J.L.; Dubyak, G.R.; O’Neill, W.C. Extracellular pyrophosphate metabolism and calcification in vascular smooth muscle. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H61–H68. [Google Scholar] [CrossRef]
- Villa-Bellosta, R.; Rivera-Torres, J.; Osorio, F.G.; Acin-Perez, R.; Enriquez, J.A.; Lopez-Otin, C.; Andres, V. Defective extracellular pyrophosphate metabolism promotes vascular calcification in a mouse model of Hutchinson-Gilford progeria syndrome that is ameliorated on pyrophosphate treatment. Circulation 2013, 127, 2442–2451. [Google Scholar] [CrossRef] [PubMed]
- Riser, B.L.; Barreto, F.C.; Rezg, R.; Valaitis, P.W.; Cook, C.S.; White, J.A.; Gass, J.H.; Maizel, J.; Louvet, L.; Drueke, T.B.; et al. Daily peritoneal administration of sodium pyrophosphate in a dialysis solution prevents the development of vascular calcification in a mouse model of uraemia. Nephrol. Dial. Transplant. 2011, 26, 3349–3357. [Google Scholar] [CrossRef]
- Lomashvili, K.A.; Khawandi, W.; O’Neill, W.C. Reduced plasma pyrophosphate levels in hemodialysis patients. J. Am. Soc. Nephrol. 2005, 16, 2495–2500. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, W.C.; Sigrist, M.K.; McIntyre, C.W. Plasma pyrophosphate and vascular calcification in chronic kidney disease. Nephrol. Dial. Transplant. 2010, 25, 187–191. [Google Scholar] [CrossRef]
- Azpiazu, D.; Gonzalez-Parra, E.; Egido, J.; Villa-Bellosta, R. Hydrolysis of Extracellular Pyrophosphate increases in post-hemodialysis plasma. Sci. Rep. 2018, 8, 11089. [Google Scholar] [CrossRef] [PubMed]
- Beattie, J.H.; Gordon, M.J.; Duthie, S.J.; McNeil, C.J.; Horgan, G.W.; Nixon, G.F.; Feldmann, J.; Kwun, I.S. Suboptimal dietary zinc intake promotes vascular inflammation and atherogenesis in a mouse model of atherosclerosis. Mol. Nutr. Food Res. 2012, 56, 1097–1105. [Google Scholar] [CrossRef]
- Ari, E.; Kaya, Y.; Demir, H.; Asicioglu, E.; Keskin, S. The correlation of serum trace elements and heavy metals with carotid artery atherosclerosis in maintenance hemodialysis patients. Biol. Trace Elem. Res. 2011, 144, 351–359. [Google Scholar] [CrossRef]
- Alcantara, E.H.; Lomeda, R.A.; Feldmann, J.; Nixon, G.F.; Beattie, J.H.; Kwun, I.S. Zinc deprivation inhibits extracellular matrix calcification through decreased synthesis of matrix proteins in osteoblasts. Mol. Nutr. Food Res. 2011, 55, 1552–1560. [Google Scholar] [CrossRef]
- Nagy, A.; Petho, D.; Gall, T.; Zavaczki, E.; Nyitrai, M.; Posta, J.; Zarjou, A.; Agarwal, A.; Balla, G.; Balla, J. Zinc Inhibits HIF-Prolyl Hydroxylase Inhibitor-Aggravated VSMC Calcification Induced by High Phosphate. Front. Physiol. 2019, 10, 1584. [Google Scholar] [CrossRef]
- Eshak, E.S.; Iso, H.; Yamagishi, K.; Maruyama, K.; Umesawa, M.; Tamakoshi, A. Associations between copper and zinc intakes from diet and mortality from cardiovascular disease in a large population-based prospective cohort study. J. Nutr. Biochem. 2018, 56, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Bates, C.J.; Hamer, M.; Mishra, G.D. Redox-modulatory vitamins and minerals that prospectively predict mortality in older British people: The National Diet and Nutrition Survey of people aged 65 years and over. Br. J. Nutr. 2011, 105, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zheng, Y.; Wang, L.; Zhong, X.; Qin, D.; Chen, W.; Tan, R.; Liu, Y. Lower Levels of Blood Zinc Associated with Intradialytic Hypertension in Maintenance Hemodialysis Patients. Biol. Trace Elem. Res. 2021, 199, 2514–2522. [Google Scholar] [CrossRef]
- Chen, W.; Eisenberg, R.; Mowrey, W.B.; Wylie-Rosett, J.; Abramowitz, M.K.; Bushinsky, D.A.; Melamed, M.L. Association between dietary zinc intake and abdominal aortic calcification in US adults. Nephrol. Dial. Transplant. 2020, 35, 1171–1178. [Google Scholar] [CrossRef] [PubMed]
- Al-Qaridhi, A.; Ghosh, S.; Luo, D.; Huang, H. Magnesium and Zinc Intake Ratio Mediates the Increase of Coronary Artery Calcification through Upregulating Interleukin 6. Libyan J. Med. 2022, 17, 2028997. [Google Scholar] [CrossRef]
- Massy, Z.A.; Maziere, C.; Kamel, S.; Brazier, M.; Choukroun, G.; Tribouilloy, C.; Slama, M.; Andrejak, M.; Maziere, J.C. Impact of inflammation and oxidative stress on vascular calcifications in chronic kidney disease. Pediatr. Nephrol. 2005, 20, 380–382. [Google Scholar] [CrossRef] [PubMed]
- Small, D.M.; Coombes, J.S.; Bennett, N.; Johnson, D.W.; Gobe, G.C. Oxidative stress, anti-oxidant therapies and chronic kidney disease. Nephrology 2012, 17, 311–321. [Google Scholar] [CrossRef]
- Huang, M.; Zheng, L.; Xu, H.; Tang, D.; Lin, L.; Zhang, J.; Li, C.; Wang, W.; Yuan, Q.; Tao, L.; et al. Oxidative stress contributes to vascular calcification in patients with chronic kidney disease. J. Mol. Cell. Cardiol. 2020, 138, 256–268. [Google Scholar] [CrossRef]
- Byon, C.H.; Javed, A.; Dai, Q.; Kappes, J.C.; Clemens, T.L.; Darley-Usmar, V.M.; McDonald, J.M.; Chen, Y. Oxidative stress induces vascular calcification through modulation of the osteogenic transcription factor Runx2 by AKT signaling. J. Biol. Chem. 2008, 283, 15319–15327. [Google Scholar] [CrossRef]
- Zhao, M.M.; Xu, M.J.; Cai, Y.; Zhao, G.; Guan, Y.; Kong, W.; Tang, C.; Wang, X. Mitochondrial reactive oxygen species promote p65 nuclear translocation mediating high-phosphate-induced vascular calcification in vitro and in vivo. Kidney Int. 2011, 79, 1071–1079. [Google Scholar] [CrossRef]
- Dalfino, G.; Simone, S.; Porreca, S.; Cosola, C.; Balestra, C.; Manno, C.; Schena, F.P.; Grandaliano, G.; Pertosa, G. Bone morphogenetic protein-2 may represent the molecular link between oxidative stress and vascular stiffness in chronic kidney disease. Atherosclerosis 2010, 211, 418–423. [Google Scholar] [CrossRef]
- Moi, P.; Chan, K.; Asunis, I.; Cao, A.; Kan, Y.W. Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region. Proc. Natl. Acad. Sci. USA 1994, 91, 9926–9930. [Google Scholar] [CrossRef]
- Owuor, E.D.; Kong, A.N. Antioxidants and oxidants regulated signal transduction pathways. Biochem. Pharmacol. 2002, 64, 765–770. [Google Scholar] [CrossRef] [PubMed]
- Canning, P.; Sorrell, F.J.; Bullock, A.N. Structural basis of Keap1 interactions with Nrf2. Free Radic. Biol. Med. 2015, 88 Pt B, 101–107. [Google Scholar] [CrossRef]
- Aminzadeh, M.A.; Nicholas, S.B.; Norris, K.C.; Vaziri, N.D. Role of impaired Nrf2 activation in the pathogenesis of oxidative stress and inflammation in chronic tubulo-interstitial nephropathy. Nephrol. Dial. Transplant. 2013, 28, 2038–2045. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Siow, R.C.; Sugden, D.; Gao, L.; Cheng, X.; Mann, G.E. Induction of HO-1 and redox signaling in endothelial cells by advanced glycation end products: A role for Nrf2 in vascular protection in diabetes. Nutr. Metab. Cardiovasc. Dis. 2011, 21, 277–285. [Google Scholar] [CrossRef]
- Liu, Y.; Chan, F.; Sun, H.; Yan, J.; Fan, D.; Zhao, D.; An, J.; Zhou, D. Resveratrol protects human keratinocytes HaCaT cells from UVA-induced oxidative stress damage by downregulating Keap1 expression. Eur. J. Pharmacol. 2011, 650, 130–137. [Google Scholar] [CrossRef]
- Balogh, E.; Chowdhury, A.; Ababneh, H.; Csiki, D.M.; Toth, A.; Jeney, V. Heme-Mediated Activation of the Nrf2/HO-1 Axis Attenuates Calcification of Valve Interstitial Cells. Biomedicines 2021, 9, 427. [Google Scholar] [CrossRef] [PubMed]
- Russo, D.; Bellasi, A.; Pota, A.; Russo, L.; Di Iorio, B. Effects of phosphorus-restricted diet and phosphate-binding therapy on outcomes in patients with chronic kidney disease. J. Nephrol. 2015, 28, 73–80. [Google Scholar] [CrossRef]
- Matias, P.J.; Jorge, C.; Azevedo, A.; Laranjinha, I.; Navarro, D.; Mendes, M.; Amaral, T.; Ferreira, C.; Aires, I.; Gil, C.; et al. Calcium Acetate/Magnesium Carbonate and Cardiovascular Risk Factors in Chronic Hemodialysis Patients. Nephron 2016, 132, 317–326. [Google Scholar] [CrossRef]
- Giger, E.V.; Castagner, B.; Leroux, J.C. Biomedical applications of bisphosphonates. J. Control. Release 2013, 167, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, M.; Yamanaka, S.; Yoshimoto, W.; Shigematsu, T. Alendronate as an effective treatment for bone loss and vascular calcification in kidney transplant recipients. J. Transplant. 2014, 2014, 269613. [Google Scholar] [CrossRef]
- Chen, C.L.; Chen, N.C.; Wu, F.Z.; Wu, M.T. Impact of denosumab on cardiovascular calcification in patients with secondary hyperparathyroidism undergoing dialysis: A pilot study. Osteoporos. Int. 2020, 31, 1507–1516. [Google Scholar] [CrossRef]
- Suzuki, S.; Suzuki, M.; Hanafusa, N.; Tsuchiya, K.; Nitta, K. Denosumab Recovers Aortic Arch Calcification During Long-Term Hemodialysis. Kidney Int. Rep. 2021, 6, 605–612. [Google Scholar] [CrossRef]
- Massy, Z.A.; Henaut, L.; Larsson, T.E.; Vervloet, M.G. Calcium-sensing receptor activation in chronic kidney disease: Effects beyond parathyroid hormone control. Semin. Nephrol. 2014, 34, 648–659. [Google Scholar] [CrossRef] [PubMed]
- Raggi, P.; Chertow, G.M.; Torres, P.U.; Csiky, B.; Naso, A.; Nossuli, K.; Moustafa, M.; Goodman, W.G.; Lopez, N.; Downey, G.; et al. The ADVANCE study: A randomized study to evaluate the effects of cinacalcet plus low-dose vitamin D on vascular calcification in patients on hemodialysis. Nephrol. Dial. Transplant. 2010, 26, 1327–1339. [Google Scholar] [CrossRef]
- Eidman, K.E.; Wetmore, J.B. Treatment of secondary hyperparathyroidism: How do cinacalcet and etelcalcetide differ? Semin. Dial. 2018, 31, 440–444. [Google Scholar] [CrossRef] [PubMed]
- Perello, J.; Gomez, M.; Ferrer, M.D.; Rodriguez, N.Y.; Salcedo, C.; Buades, J.M.; Perez, M.M.; Torregrosa, J.V.; Martin, E.; Maduell, F. SNF472, a novel inhibitor of vascular calcification, could be administered during hemodialysis to attain potentially therapeutic phytate levels. J. Nephrol. 2018, 31, 287–296. [Google Scholar] [CrossRef]
- Raggi, P.; Bellasi, A.; Bushinsky, D.; Bover, J.; Rodriguez, M.; Ketteler, M.; Sinha, S.; Salcedo, C.; Gillotti, K.; Padgett, C.; et al. Slowing Progression of Cardiovascular Calcification with SNF472 in Patients on Hemodialysis: Results of a Randomized Phase 2b Study. Circulation 2020, 141, 728–739. [Google Scholar] [CrossRef] [PubMed]


| Factors | Subjects | Conclusion | Whether Related to CAC | Ref. |
|---|---|---|---|---|
| Phosphorus | 77 HD patients | Blood phosphorus variability was an independent predictor of CAC, and maintaining stable serum phosphorus levels may lead to a lower CACS. | Yes | [23] |
| 205 HD patients | A positive correlation existed between elevated blood phosphorus and CAC severity, and was associated with ischemic CVD. | Yes | [24] | |
| 439 CKD patients in stage CKD 3 | Higher serum phosphate concentrations (within the normal range) were strongly linked with a high incidence of CAC; each 1 mg/dL increase in blood phosphorus concentration was linked with a 21% increase in the incidence of CAC. | Yes | [25] | |
| Calcium, phosphorus | 200 HD patients | When hypercalcemia and hyperphosphatemia co-occur, the development of CAC and aortic calcification is hastened. | Yes | [40] |
| Magnesium | 109 CKD patients | Serum magnesium in ESRD patients was negatively associated with CAC; this association was more pronounced in patients with high serum phosphorus concentrations. | Yes | [47] |
| 324 CKD patients in stage CKD 3–4 | Magnesium oxide treatment was effective in slowing the progression of CAC, but it did not suppress the progression of calcification in the thoracic aorta. | Yes | [50] | |
| PTH | 213 patients in stage CKD-5D | Nine times higher than normal PTH levels were closely associated with CAC progression. | Yes | [56] |
| Vitamin D | 40 CKD patients | Active vitamin D drug use was positively associated with CAC. | Yes | [63] |
| 80 CKD patients | No association was found between the degree of vitamin D deficiency and CAC. | No | [65] | |
| Inflammation | 43 PD patients | IL-6 and CRP levels were significantly higher in patients with a high CACS (>400 points) than in those with low CACS (<10 points) values. | Yes | [68] |
| 73 PD patients | The CACS and the Common Carotid Artery Intima-Medial Thickness Index were positively correlated with CRP and IL-6. | Yes | [69] | |
| 40 HD patients | High CRP levels accelerated the development of CAC. | Yes | [70] | |
| 70 PD patients | CRP was an independent risk factor for the occurrence of CAC. | Yes | [71] | |
| AGEs | 40 HD patients | AGEs and oxidative stress were strongly associated with extensive CAC. | Yes | [79] |
| 27 PD patients | Continued accumulation of AGEs was found to be positively linked to CACS. | Yes | [80] | |
| FGF23 | 16 HD patients | FGF23 levels were independently linked to aortic, peripheral calcification and CAC. | Yes | [94] |
| 142 CKD patients | Patients with elevated FGF23 levels had higher aortic and CACS values than patients with lower FGF23 levels. | Yes | [95] | |
| 1501 patients in CKD stage 2–4 | A correlation between blood FGF23 levels and CAC was not found. | No | [104] | |
| OPG | 185 CKD patients | A high serum OPG concentration favored a high CAC. | Yes | [114] |
| 101 HD patients | A higher OPG level was independently associated with CAC. | Yes | [115] | |
| 77 CKD patients | OPG is an independent risk factor for death in patients with CKD and an early predictor of CAC. | Yes | [116] | |
| MGP | 97 CKD patients | MGP did not correlate with vascular sclerosis and CACS. | No | [133] |
| 112 HD patients | The MGP levels were substantially higher than those in the control groups, but were not related to CACS. | No | [134] | |
| 141 patients in CKD stage 5 | High vascular expression of MGP was associated with higher CAC scores and plasma dp-ucMGP levels. | Yes | [135] | |
| Fetuin-A | 78 HD patients | Serum fetuin-A levels were associated with the total CACS. | Yes | [144] |
| 72 HD patients | The CACS, mass, and volume of plaques in coronary arteries correlated significantly with the serum fetuin-A levels. | Yes | [145] | |
| 88 diabetic nephropathy patients | The fetuin-A levels were significantly higher in diabetic nephropathy patients who had not yet undergone maintenance dialysis than in diabetic control patients, and were directly associated with CAC. | Yes | [146] | |
| 85 diabetic pre-dialysis patients | There was no association between fetuin-A and CACS. | No | [149] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dai, Z.; Zhang, X. Pathophysiology and Clinical Impacts of Chronic Kidney Disease on Coronary Artery Calcification. J. Cardiovasc. Dev. Dis. 2023, 10, 207. https://doi.org/10.3390/jcdd10050207
Dai Z, Zhang X. Pathophysiology and Clinical Impacts of Chronic Kidney Disease on Coronary Artery Calcification. Journal of Cardiovascular Development and Disease. 2023; 10(5):207. https://doi.org/10.3390/jcdd10050207
Chicago/Turabian StyleDai, Zhuoming, and Xiangyu Zhang. 2023. "Pathophysiology and Clinical Impacts of Chronic Kidney Disease on Coronary Artery Calcification" Journal of Cardiovascular Development and Disease 10, no. 5: 207. https://doi.org/10.3390/jcdd10050207
APA StyleDai, Z., & Zhang, X. (2023). Pathophysiology and Clinical Impacts of Chronic Kidney Disease on Coronary Artery Calcification. Journal of Cardiovascular Development and Disease, 10(5), 207. https://doi.org/10.3390/jcdd10050207