


The Impacts of Animal-Based Diets in Cardiovascular Disease Development: A Cellular and Physiological Overview


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
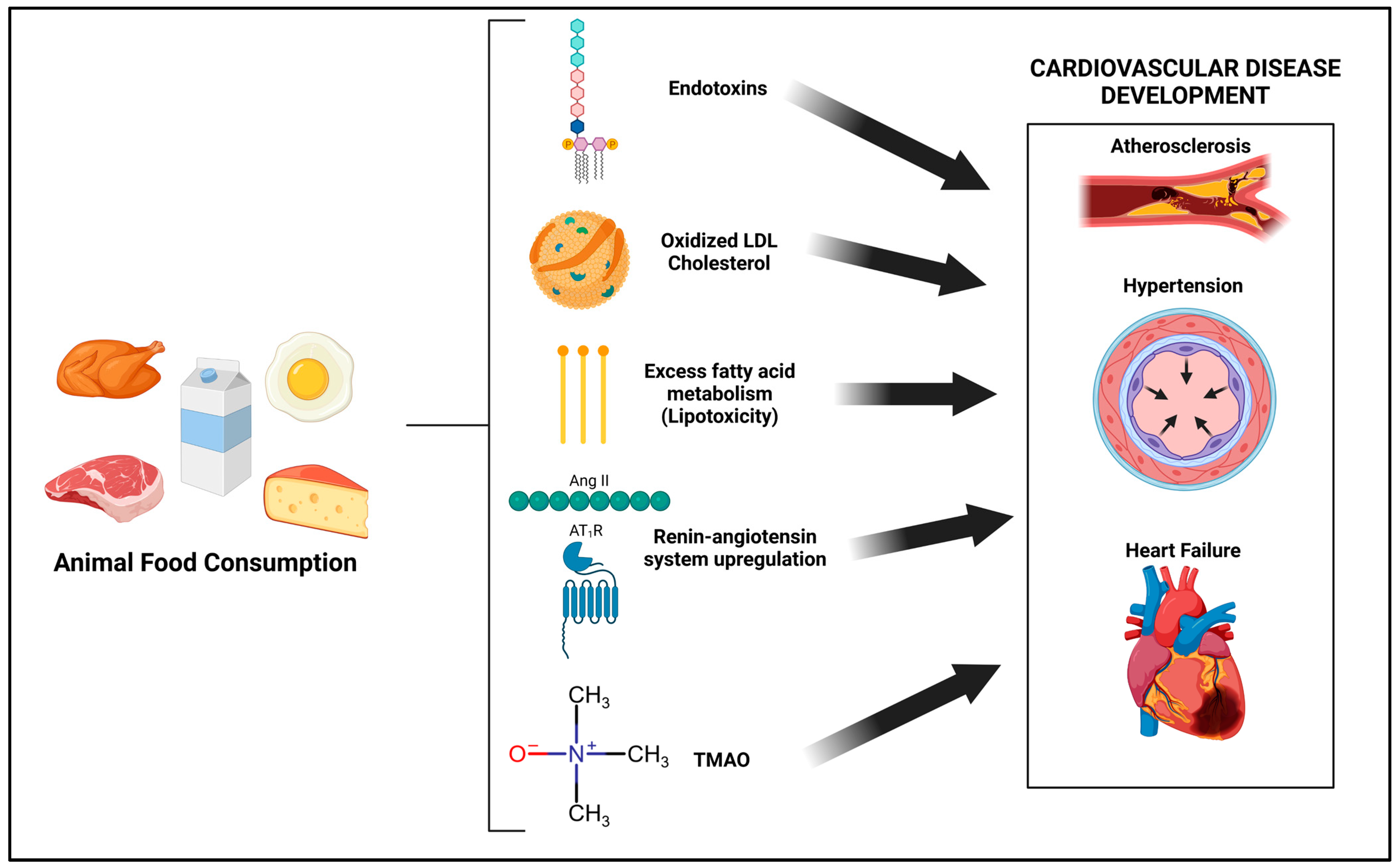
:1. Introduction
Animal Food-Based Diets: Are They Health Promoting?
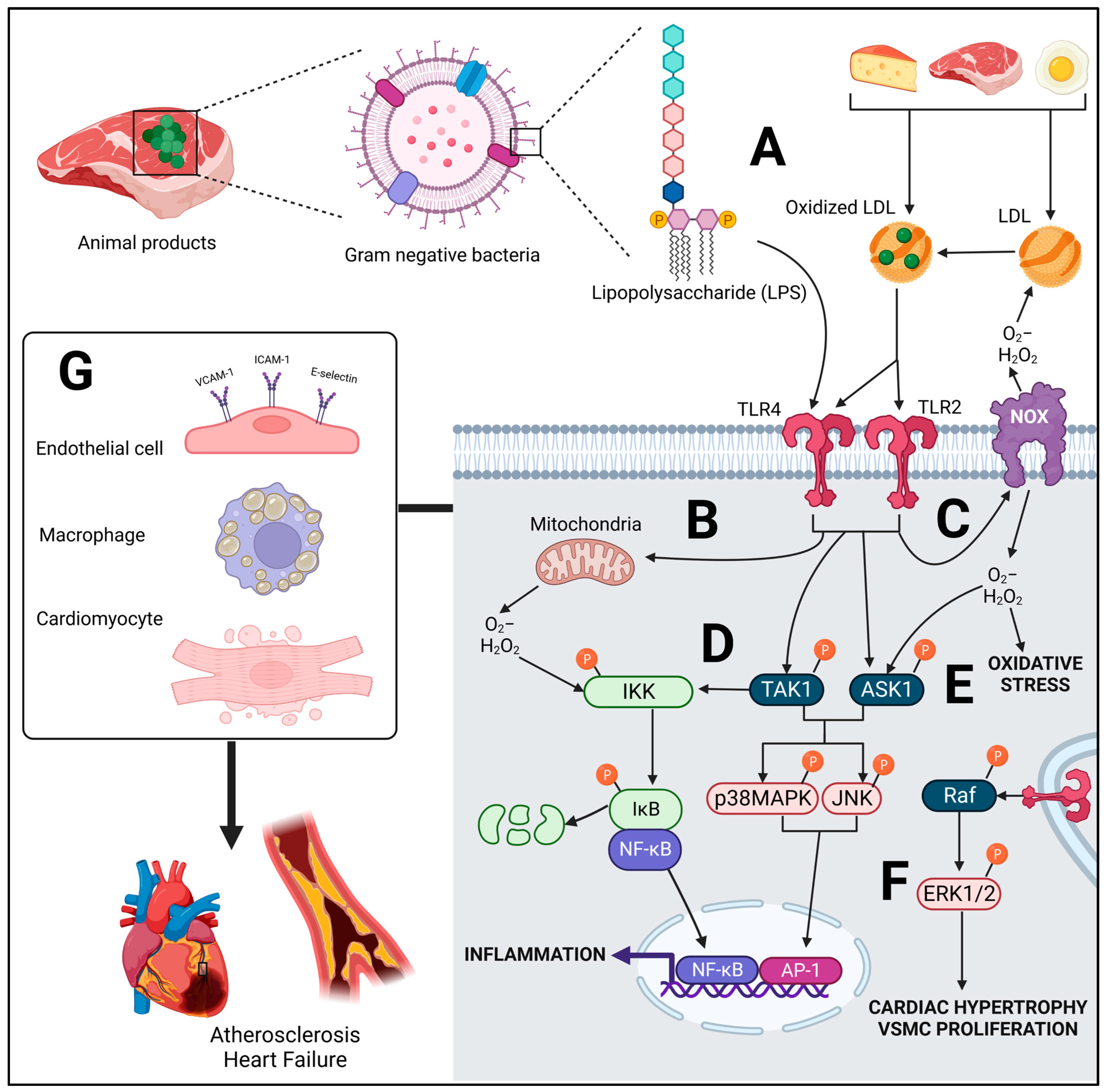
2. Diet-Mediated Toll-Like Receptor (TLR) Activation
2.1. Molecular Signaling of TLR
2.2. Role of TLRs in CVD
2.3. Potential Role of Diet-Derived Endotoxins from Animal Foods in CVD Development
2.4. Oxidized LDL from Diet: TLR-Mediated Effects
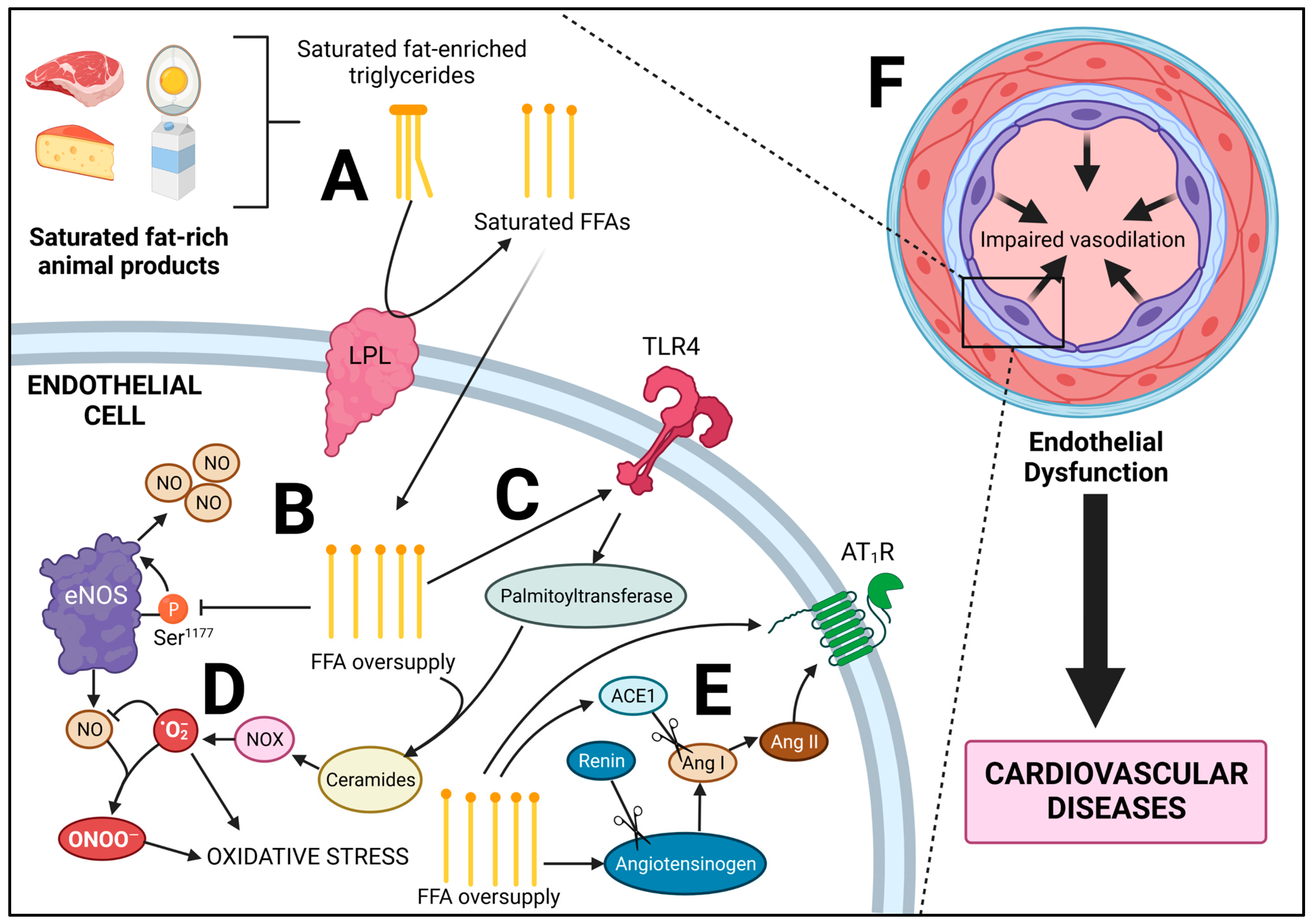
3. Saturated Fat from Animal Foods: Molecular Consequences beyond Increased LDL Cholesterol
3.1. Lipotoxicity
3.2. Lipotoxicity of the Endothelium
3.3. Consumption of Animal Foods and Saturated Fat: A Link to the Renin-Angiotensin System
4. Animal Products and the CVD-Promoting Trimethylamine-N-Oxide Molecule
5. Considerations for Fish Consumption
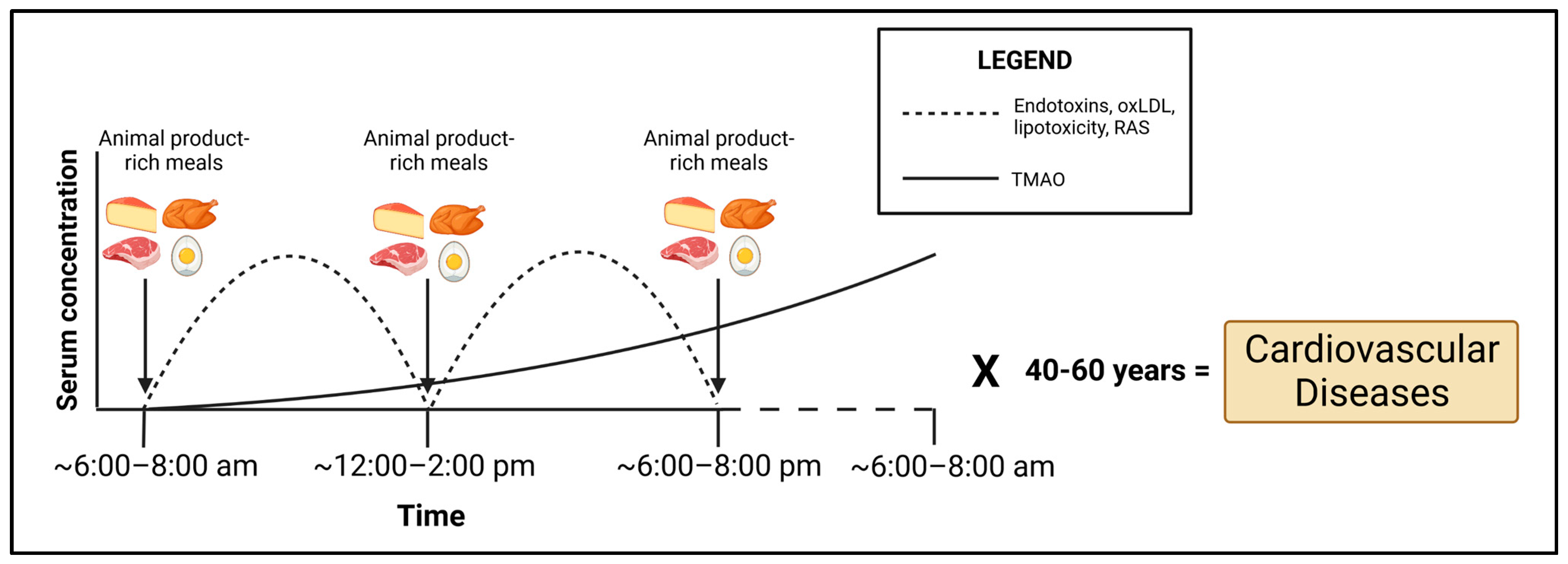
6. Implications and Perspectives
7. Conclusions
Funding
Data Availability Statement
Conflicts of Interest
References
- Anton, S.D.; Hida, A.; Heekin, K.; Sowalsky, K.; Karabetian, C.; Mutchie, H.; Leeuwenburgh, C.; Manini, T.M.; Barnett, T.E. Effects of Popular Diets without Specific Calorie Targets on Weight Loss Outcomes: Systematic Review of Findings from Clinical Trials. Nutrients 2017, 9, 822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roehl, K.; Sewak, S.L. Practice Paper of the Academy of Nutrition and Dietetics: Classic and Modified Ketogenic Diets for Treatment of Epilepsy. J. Acad. Nutr. Diet. 2017, 117, 1279–1292. [Google Scholar] [CrossRef] [PubMed]
- Tuso, P.J.; Ismail, M.H.; Ha, B.P.; Bartolotto, C. Nutritional update for physicians: Plant-based diets. Perm. J. 2013, 17, 61–66. [Google Scholar] [CrossRef] [Green Version]
- Buettner, D.; Skemp, S. Blue Zones: Lessons From the World’s Longest Lived. Am. J. Lifestyle Med. 2016, 10, 318–321. [Google Scholar] [CrossRef] [PubMed]
- Willcox, B.J.; Willcox, D.C.; Todoriki, H.; Fujiyoshi, A.; Yano, K.; He, Q.; Curb, J.D.; Suzuki, M. Caloric restriction, the traditional Okinawan diet, and healthy aging: The diet of the world’s longest-lived people and its potential impact on morbidity and life span. Ann. N. Y. Acad. Sci. 2007, 1114, 434–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraser, G.E.; Shavlik, D.J. Ten years of life: Is it a matter of choice? Arch. Intern. Med. 2001, 161, 1645–1652. [Google Scholar] [CrossRef] [Green Version]
- Le, L.T.; Sabate, J. Beyond meatless, the health effects of vegan diets: Findings from the Adventist cohorts. Nutrients 2014, 6, 2131–2147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kagawa, Y. Impact of Westernization on the nutrition of Japanese: Changes in physique, cancer, longevity and centenarians. Prev. Med. 1978, 7, 205–217. [Google Scholar] [CrossRef]
- Carter, C.; McGee, D.; Yano, K. Morbidity and mortality rates in Okinawan Japanese vs. mainland Japanese: The Honolulu Heart Program. Hum. Biol. 1984, 56, 339–353. [Google Scholar]
- Mizushima, S.; Moriguchi, E.H.; Nakada, Y.; Biosca, M.D.G.; Nara, Y.; Murakami, K.; Horie, R.; Moriguchi, Y.; Mimura, G.; Yamori, Y. The relationship of dietary factors to cardiovascular diseases among Japanese in Okinawa and Japanese immigrants, originally from Okinawa, in Brazil. Hypertens. Res. 1992, 15, 45–55. [Google Scholar] [CrossRef]
- Bang, H.O.; Dyerberg, J.; Hjoorne, N. The composition of food consumed by Greenland Eskimos. Acta Med. Scand. 1976, 200, 69–73. [Google Scholar] [CrossRef] [PubMed]
- Fodor, J.G.; Helis, E.; Yazdekhasti, N.; Vohnout, B. “Fishing” for the origins of the “Eskimos and heart disease” story: Facts or wishful thinking? Can. J. Cardiol. 2014, 30, 864–868. [Google Scholar] [CrossRef] [PubMed]
- Larsson, S.C.; Akesson, A.; Wolk, A. Primary prevention of stroke by a healthy lifestyle in a high-risk group. Neurology 2015, 84, 2224–2228. [Google Scholar] [CrossRef] [Green Version]
- Lakkur, S.; Judd, S.E. Diet and Stroke: Recent Evidence Supporting a Mediterranean-Style Diet and Food in the Primary Prevention of Stroke. Stroke 2015, 46, 2007–2011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reynolds, A.; Mann, J.; Cummings, J.; Winter, N.; Mete, E.; Te Morenga, L. Carbohydrate quality and human health: A series of systematic reviews and meta-analyses. Lancet 2019, 393, 434–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seidelmann, S.B.; Claggett, B.; Cheng, S.; Henglin, M.; Shah, A.; Steffen, L.M.; Folsom, A.R.; Rimm, E.B.; Willett, W.C.; Solomon, S.D. Dietary carbohydrate intake and mortality: A prospective cohort study and meta-analysis. Lancet Public Health 2018, 3, e419–e428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benisi-Kohansal, S.; Saneei, P.; Salehi-Marzijarani, M.; Larijani, B.; Esmaillzadeh, A. Whole-Grain Intake and Mortality from All Causes, Cardiovascular Disease, and Cancer: A Systematic Review and Dose-Response Meta-Analysis of Prospective Cohort Studies. Adv. Nutr. 2016, 7, 1052–1065. [Google Scholar] [CrossRef] [Green Version]
- Huang, T.; Xu, M.; Lee, A.; Cho, S.; Qi, L. Consumption of whole grains and cereal fiber and total and cause-specific mortality: Prospective analysis of 367,442 individuals. BMC Med. 2015, 13, 59. [Google Scholar] [CrossRef] [Green Version]
- Aune, D.; Keum, N.; Giovannucci, E.; Fadnes, L.T.; Boffetta, P.; Greenwood, D.C.; Tonstad, S.; Vatten, L.J.; Riboli, E.; Norat, T. Whole grain consumption and risk of cardiovascular disease, cancer, and all cause and cause specific mortality: Systematic review and dose-response meta-analysis of prospective studies. BMJ 2016, 353, i2716. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.D.; Li, Y.; Bhupathiraju, S.N.; Rosner, B.A.; Sun, Q.; Giovannucci, E.L.; Rimm, E.B.; Manson, J.E.; Willett, W.C.; Stampfer, M.J.; et al. Fruit and Vegetable Intake and Mortality: Results From 2 Prospective Cohort Studies of US Men and Women and a Meta-Analysis of 26 Cohort Studies. Circulation 2021, 143, 1642–1654. [Google Scholar] [CrossRef]
- Quek, J.; Lim, G.; Lim, W.H.; Ng, C.H.; So, W.Z.; Toh, J.; Pan, X.H.; Chin, Y.H.; Muthiah, M.D.; Chan, S.P.; et al. The Association of Plant-Based Diet With Cardiovascular Disease and Mortality: A Meta-Analysis and Systematic Review of Prospect Cohort Studies. Front. Cardiovasc. Med. 2021, 8, 756810. [Google Scholar] [CrossRef] [PubMed]
- Najjar, R.S.; Schwartz, A.M.; Wong, B.J.; Mehta, P.K.; Feresin, R.G. Berries and Their Polyphenols as a Potential Therapy for Coronary Microvascular Dysfunction: A Mini-Review. Int. J. Mol. Sci. 2021, 22, 3373. [Google Scholar] [CrossRef]
- Najjar, R.S.; Feresin, R.G. Protective Role of Polyphenols in Heart Failure: Molecular Targets and Cellular Mechanisms Underlying Their Therapeutic Potential. Int. J. Mol. Sci. 2021, 22, 1668. [Google Scholar] [CrossRef] [PubMed]
- Najjar, R.S.; Turner, C.G.; Wong, B.J.; Feresin, R.G. Berry-Derived Polyphenols in Cardiovascular Pathologies: Mechanisms of Disease and the Role of Diet and Sex. Nutrients 2021, 13, 387. [Google Scholar] [CrossRef] [PubMed]
- Najjar, R.S.; Moore, C.E.; Montgomery, B.D. A defined, plant-based diet utilized in an outpatient cardiovascular clinic effectively treats hypercholesterolemia and hypertension and reduces medications. Clin. Cardiol. 2018, 41, 307–313. [Google Scholar] [CrossRef] [Green Version]
- Najjar, R.S.; Moore, C.E.; Montgomery, B.D. Consumption of a defined, plant-based diet reduces lipoprotein(a), inflammation, and other atherogenic lipoproteins and particles within 4 weeks. Clin. Cardiol. 2018, 41, 1062–1068. [Google Scholar] [CrossRef] [Green Version]
- Najjar, R.S.; Montgomery, B.D. A defined, plant-based diet as a potential therapeutic approach in the treatment of heart failure: A clinical case series. Complement. Ther. Med. 2019, 45, 211–214. [Google Scholar] [CrossRef]
- Jenkins, D.J.; Kendall, C.W.; Popovich, D.G.; Vidgen, E.; Mehling, C.C.; Vuksan, V.; Ransom, T.P.; Rao, A.V.; Rosenberg-Zand, R.; Tariq, N.; et al. Effect of a very-high-fiber vegetable, fruit, and nut diet on serum lipids and colonic function. Metabolism 2001, 50, 494–503. [Google Scholar] [CrossRef]
- Nawrocki, J.W.; Weiss, S.R.; Davidson, M.H.; Sprecher, D.L.; Schwartz, S.L.; Lupien, P.J.; Jones, P.H.; Haber, H.E.; Black, D.M. Reduction of LDL cholesterol by 25% to 60% in patients with primary hypercholesterolemia by atorvastatin, a new HMG-CoA reductase inhibitor. Arterioscler. Thromb. Vasc. Biol. 1995, 15, 678–682. [Google Scholar] [CrossRef]
- Esselstyn, C.B., Jr.; Gendy, G.; Doyle, J.; Golubic, M.; Roizen, M.F. A way to reverse CAD? J. Fam. Pract. 2014, 63, 356–364b. [Google Scholar]
- Ornish, D.; Brown, S.E.; Scherwitz, L.W.; Billings, J.H.; Armstrong, W.T.; Ports, T.A.; McLanahan, S.M.; Kirkeeide, R.L.; Brand, R.J.; Gould, K.L. Can lifestyle changes reverse coronary heart disease? The Lifestyle Heart Trial. Lancet 1990, 336, 129–133. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.W.; Loh, H.C.; Ching, S.M.; Devaraj, N.K.; Hoo, F.K. Effects of Vegetarian Diets on Blood Pressure Lowering: A Systematic Review with Meta-Analysis and Trial Sequential Analysis. Nutrients 2020, 12, 1604. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, Y.; Levin, S.M.; Barnard, N.D. Association between plant-based diets and plasma lipids: A systematic review and meta-analysis. Nutr. Rev. 2017, 75, 683–698. [Google Scholar] [CrossRef] [Green Version]
- Fung, T.T.; van Dam, R.M.; Hankinson, S.E.; Stampfer, M.; Willett, W.C.; Hu, F.B. Low-carbohydrate diets and all-cause and cause-specific mortality: Two cohort studies. Ann. Intern. Med. 2010, 153, 289–298. [Google Scholar] [CrossRef]
- Jenkins, D.J.; Wong, J.M.; Kendall, C.W.; Esfahani, A.; Ng, V.W.; Leong, T.C.; Faulkner, D.A.; Vidgen, E.; Greaves, K.A.; Paul, G.; et al. The effect of a plant-based low-carbohydrate (“Eco-Atkins”) diet on body weight and blood lipid concentrations in hyperlipidemic subjects. Arch. Intern. Med. 2009, 169, 1046–1054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenkins, D.J.; Wong, J.M.; Kendall, C.W.; Esfahani, A.; Ng, V.W.; Leong, T.C.; Faulkner, D.A.; Vidgen, E.; Paul, G.; Mukherjea, R.; et al. Effect of a 6-month vegan low-carbohydrate (‘Eco-Atkins’) diet on cardiovascular risk factors and body weight in hyperlipidaemic adults: A randomised controlled trial. BMJ Open 2014, 4, e003505. [Google Scholar] [CrossRef] [PubMed]
- Sacks, F.M.; Lichtenstein, A.H.; Wu, J.H.Y.; Appel, L.J.; Creager, M.A.; Kris-Etherton, P.M.; Miller, M.; Rimm, E.B.; Rudel, L.L.; Robinson, J.G.; et al. Dietary Fats and Cardiovascular Disease: A Presidential Advisory From the American Heart Association. Circulation 2017, 136, e1–e23. [Google Scholar] [CrossRef]
- Gardner, C.D.; Vadiveloo, M.K.; Petersen, K.S.; Anderson, C.A.M.; Springfield, S.; Van Horn, L.; Khera, A.; Lamendola, C.; Mayo, S.M.; Joseph, J.J.; et al. Popular Dietary Patterns: Alignment With American Heart Association 2021 Dietary Guidance: A Scientific Statement From the American Heart Association. Circulation 2023, 147, 1715–1730. [Google Scholar] [CrossRef]
- Dong, T.; Guo, M.; Zhang, P.; Sun, G.; Chen, B. The effects of low-carbohydrate diets on cardiovascular risk factors: A meta-analysis. PLoS ONE 2020, 15, e0225348. [Google Scholar] [CrossRef]
- Kirkpatrick, C.F.; Bolick, J.P.; Kris-Etherton, P.M.; Sikand, G.; Aspry, K.E.; Soffer, D.E.; Willard, K.E.; Maki, K.C. Review of current evidence and clinical recommendations on the effects of low-carbohydrate and very-low-carbohydrate (including ketogenic) diets for the management of body weight and other cardiometabolic risk factors: A scientific statement from the National Lipid Association Nutrition and Lifestyle Task Force. J. Clin. Lipidol. 2019, 13, 689–711.e681. [Google Scholar] [CrossRef] [Green Version]
- FoodData Central. Available online: https://fdc.nal.usda.gov/ (accessed on 3 March 2023).
- Satija, A.; Bhupathiraju, S.N.; Spiegelman, D.; Chiuve, S.E.; Manson, J.E.; Willett, W.; Rexrode, K.M.; Rimm, E.B.; Hu, F.B. Healthful and Unhealthful Plant-Based Diets and the Risk of Coronary Heart Disease in U.S. Adults. J. Am. Coll. Cardiol. 2017, 70, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Merino, J.; Kones, R.; Ferre, R.; Plana, N.; Girona, J.; Aragones, G.; Ibarretxe, D.; Heras, M.; Masana, L. Negative effect of a low-carbohydrate, high-protein, high-fat diet on small peripheral artery reactivity in patients with increased cardiovascular risk. Br. J. Nutr. 2013, 109, 1241–1247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwingshackl, L.; Hoffmann, G. Low-carbohydrate diets impair flow-mediated dilatation: Evidence from a systematic review and meta-analysis. Br. J. Nutr. 2013, 110, 969–970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenbaum, M.; Hall, K.D.; Guo, J.; Ravussin, E.; Mayer, L.S.; Reitman, M.L.; Smith, S.R.; Walsh, B.T.; Leibel, R.L. Glucose and Lipid Homeostasis and Inflammation in Humans Following an Isocaloric Ketogenic Diet. Obesity 2019, 27, 971–981. [Google Scholar] [CrossRef]
- Fleming, R.M. The effect of high-protein diets on coronary blood flow. Angiology 2000, 51, 817–826. [Google Scholar] [CrossRef]
- Kawasaki, T.; Kawai, T. Toll-like receptor signaling pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef] [Green Version]
- De Kleijn, D.; Pasterkamp, G. Toll-like receptors in cardiovascular diseases. Cardiovasc. Res. 2003, 60, 58–67. [Google Scholar] [CrossRef] [Green Version]
- Hambleton, J.; Weinstein, S.L.; Lem, L.; DeFranco, A.L. Activation of c-Jun N-terminal kinase in bacterial lipopolysaccharide-stimulated macrophages. Proc. Natl. Acad. Sci. USA 1996, 93, 2774–2778. [Google Scholar] [CrossRef] [Green Version]
- Lyte, J.M.; Gabler, N.K.; Hollis, J.H. Postprandial serum endotoxin in healthy humans is modulated by dietary fat in a randomized, controlled, cross-over study. Lipids Health Dis. 2016, 15, 186. [Google Scholar] [CrossRef] [Green Version]
- Ghanim, H.; Abuaysheh, S.; Sia, C.L.; Korzeniewski, K.; Chaudhuri, A.; Fernandez-Real, J.M.; Dandona, P. Increase in plasma endotoxin concentrations and the expression of Toll-like receptors and suppressor of cytokine signaling-3 in mononuclear cells after a high-fat, high-carbohydrate meal: Implications for insulin resistance. Diabetes Care 2009, 32, 2281–2287. [Google Scholar] [CrossRef] [Green Version]
- Zarember, K.A.; Godowski, P.J. Tissue expression of human Toll-like receptors and differential regulation of Toll-like receptor mRNAs in leukocytes in response to microbes, their products, and cytokines. J. Immunol. 2002, 168, 554–561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frantz, S.; Kobzik, L.; Kim, Y.D.; Fukazawa, R.; Medzhitov, R.; Lee, R.T.; Kelly, R.A. Toll4 (TLR4) expression in cardiac myocytes in normal and failing myocardium. J. Clin. Investig. 1999, 104, 271–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Z.; Li, Y.; Jin, J.; Zhang, X.; Lopes-Virella, M.F.; Huang, Y. Toll-like receptor 4 activation in microvascular endothelial cells triggers a robust inflammatory response and cross talk with mononuclear cells via interleukin-6. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1696–1706. [Google Scholar] [CrossRef] [Green Version]
- Hodgkinson, C.P.; Pratt, R.E.; Kirste, I.; Dal-Pra, S.; Cooke, J.P.; Dzau, V.J. Cardiomyocyte Maturation Requires TLR3 Activated Nuclear Factor Kappa, B. Stem Cells 2018, 36, 1198–1209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimmer, S.; Steinmetz, M.; Asdonk, T.; Motz, I.; Coch, C.; Hartmann, E.; Barchet, W.; Wassmann, S.; Hartmann, G.; Nickenig, G. Activation of endothelial toll-like receptor 3 impairs endothelial function. Circ. Res. 2011, 108, 1358–1366. [Google Scholar] [CrossRef] [Green Version]
- Spirig, R.; Tsui, J.; Shaw, S. The Emerging Role of TLR and Innate Immunity in Cardiovascular Disease. Cardiol. Res. Pract. 2012, 2012, 181394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erridge, C. The roles of Toll-like receptors in atherosclerosis. J. Innate Immun. 2009, 1, 340–349. [Google Scholar] [CrossRef]
- Yu, L.; Wang, L.; Chen, S. Endogenous toll-like receptor ligands and their biological significance. J. Cell. Mol. Med. 2010, 14, 2592–2603. [Google Scholar] [CrossRef] [Green Version]
- Yu, L.; Wang, L.; Chen, S. Exogenous or endogenous Toll-like receptor ligands: Which is the MVP in tumorigenesis? Cell. Mol. Life Sci. 2012, 69, 935–949. [Google Scholar] [CrossRef]
- Erridge, C. Endogenous ligands of TLR2 and TLR4: Agonists or assistants? J. Leukoc. Biol. 2010, 87, 989–999. [Google Scholar] [CrossRef]
- Chavez-Sanchez, L.; Garza-Reyes, M.G.; Espinosa-Luna, J.E.; Chavez-Rueda, K.; Legorreta-Haquet, M.V.; Blanco-Favela, F. The role of TLR2, TLR4 and CD36 in macrophage activation and foam cell formation in response to oxLDL in humans. Hum. Immunol. 2014, 75, 322–329. [Google Scholar] [CrossRef] [PubMed]
- Holvoet, P.; Davey, P.C.; De Keyzer, D.; Doukoure, M.; Deridder, E.; Bochaton-Piallat, M.L.; Gabbiani, G.; Beaufort, E.; Bishay, K.; Andrieux, N.; et al. Oxidized low-density lipoprotein correlates positively with toll-like receptor 2 and interferon regulatory factor-1 and inversely with superoxide dismutase-1 expression: Studies in hypercholesterolemic swine and THP-1 cells. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1558–1565. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Zou, C.; Mei, L.; Zhang, Y.; Qian, Y.; You, S.; Pan, Y.; Xu, Z.; Bai, B.; Huang, W.; et al. MD2 mediates angiotensin II-induced cardiac inflammation and remodeling via directly binding to Ang II and activating TLR4/NF-kappaB signaling pathway. Basic. Res. Cardiol. 2017, 112, 9. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, T.; Umemoto, S.; Yoshimura, K.; Matsuda, S.; Itoh, S.; Murata, T.; Fukai, T.; Matsuzaki, M. TLR4 is a critical regulator of angiotensin II-induced vascular remodeling: The roles of extracellular SOD and NADPH oxidase. Hypertens. Res. 2015, 38, 649–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.J.; Choi, Y.; Choi, Y.H.; Park, T. Obesity activates toll-like receptor-mediated proinflammatory signaling cascades in the adipose tissue of mice. J. Nutr. Biochem. 2012, 23, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, R.; Kochumon, S.; Thomas, R.; Atizado, V.; Sindhu, S. Increased adipose tissue expression of TLR8 in obese individuals with or without type-2 diabetes: Significance in metabolic inflammation. J. Inflamm. 2016, 13, 38. [Google Scholar] [CrossRef] [Green Version]
- Cavassani, K.A.; Ishii, M.; Wen, H.; Schaller, M.A.; Lincoln, P.M.; Lukacs, N.W.; Hogaboam, C.M.; Kunkel, S.L. TLR3 is an endogenous sensor of tissue necrosis during acute inflammatory events. J. Exp. Med. 2008, 205, 2609–2621. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Feng, Y.; Zou, L.; Wang, L.; Chen, H.H.; Cai, J.Y.; Xu, J.M.; Sosnovik, D.E.; Chao, W. Role of extracellular RNA and TLR3-Trif signaling in myocardial ischemia-reperfusion injury. J. Am. Heart Assoc. 2014, 3, e000683. [Google Scholar] [CrossRef] [Green Version]
- Hong, C.P.; Yun, C.H.; Lee, G.W.; Park, A.; Kim, Y.M.; Jang, M.H. TLR9 regulates adipose tissue inflammation and obesity-related metabolic disorders. Obesity 2015, 23, 2199–2206. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.; Rutkowsky, J.M.; Snodgrass, R.G.; Ono-Moore, K.D.; Schneider, D.A.; Newman, J.W.; Adams, S.H.; Hwang, D.H. Saturated fatty acids activate TLR-mediated proinflammatory signaling pathways. J. Lipid Res. 2012, 53, 2002–2013. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Singh, V.; Tiwari, R.L.; Chandra, T.; Kumar, A.; Dikshit, M.; Barthwal, M.K. The IRAK-ERK-p67phox-Nox-2 axis mediates TLR4, 2-induced ROS production for IL-1beta transcription and processing in monocytes. Cell. Mol. Immunol. 2016, 13, 745–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, H.S.; Jung, H.Y.; Park, E.Y.; Kim, J.; Lee, W.J.; Bae, Y.S. Cutting edge: Direct interaction of TLR4 with NAD(P)H oxidase 4 isozyme is essential for lipopolysaccharide-induced production of reactive oxygen species and activation of NF-kappa B. J. Immunol. 2004, 173, 3589–3593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, C.S.; Kim, J.J.; Lee, S.J.; Hwang, J.H.; Lee, C.H.; Lee, M.S.; Jo, E.K. TLR3-triggered reactive oxygen species contribute to inflammatory responses by activating signal transducer and activator of transcription-1. J. Immunol. 2013, 190, 6368–6377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.H.; Joo, J.H.; Kim, J.; Lim, H.J.; Kim, S.; Curtiss, L.; Seong, J.K.; Cui, W.; Yabe-Nishimura, C.; Bae, Y.S. Interaction of NADPH oxidase 1 with Toll-like receptor 2 induces migration of smooth muscle cells. Cardiovasc. Res. 2013, 99, 483–493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katare, P.B.; Nizami, H.L.; Paramesha, B.; Dinda, A.K.; Banerjee, S.K. Activation of toll like receptor 4 (TLR4) promotes cardiomyocyte apoptosis through SIRT2 dependent p53 deacetylation. Sci. Rep. 2020, 10, 19232. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-kappaB signaling in inflammation. Signal. Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [Green Version]
- Hayakawa, T.; Matsuzawa, A.; Noguchi, T.; Takeda, K.; Ichijo, H. The ASK1-MAP kinase pathways in immune and stress responses. Microbes Infect. 2006, 8, 1098–1107. [Google Scholar] [CrossRef]
- Wi, S.M.; Moon, G.; Kim, J.; Kim, S.T.; Shim, J.H.; Chun, E.; Lee, K.Y. TAK1-ECSIT-TRAF6 complex plays a key role in the TLR4 signal to activate NF-kappaB. J. Biol. Chem. 2014, 289, 35205–35214. [Google Scholar] [CrossRef] [Green Version]
- Comalada, M.; Xaus, J.; Valledor, A.F.; Lopez-Lopez, C.; Pennington, D.J.; Celada, A. PKC epsilon is involved in JNK activation that mediates LPS-induced TNF-alpha, which induces apoptosis in macrophages. Am. J. Physiol. Cell. Physiol. 2003, 285, C1235–C1245. [Google Scholar] [CrossRef] [Green Version]
- Satta, N.; Kruithof, E.K.; Reber, G.; de Moerloose, P. Induction of TLR2 expression by inflammatory stimuli is required for endothelial cell responses to lipopeptides. Mol. Immunol. 2008, 46, 145–157. [Google Scholar] [CrossRef]
- Jiang, Z.; Zamanian-Daryoush, M.; Nie, H.; Silva, A.M.; Williams, B.R.; Li, X. Poly(I-C)-induced Toll-like receptor 3 (TLR3)-mediated activation of NFkappa B and MAP kinase is through an interleukin-1 receptor-associated kinase (IRAK)-independent pathway employing the signaling components TLR3-TRAF6-TAK1-TAB2-PKR. J. Biol. Chem. 2003, 278, 16713–16719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Deng, L.; Hong, M.; Akkaraju, G.R.; Inoue, J.; Chen, Z.J. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature 2001, 412, 346–351. [Google Scholar] [CrossRef] [PubMed]
- Moriguchi, T.; Kuroyanagi, N.; Yamaguchi, K.; Gotoh, Y.; Irie, K.; Kano, T.; Shirakabe, K.; Muro, Y.; Shibuya, H.; Matsumoto, K.; et al. A novel kinase cascade mediated by mitogen-activated protein kinase kinase 6 and MKK3. J. Biol. Chem. 1996, 271, 13675–13679. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Li, M.; Chen, L.; Yang, K.; Shan, Y.; Zhu, L.; Sun, S.; Li, L.; Wang, C. The TAK1-JNK cascade is required for IRF3 function in the innate immune response. Cell Res. 2009, 19, 412–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, M.J.; Liu, Z.G. Crosstalk of reactive oxygen species and NF-kappaB signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef] [Green Version]
- Meijles, D.N.; Cull, J.J.; Markou, T.; Cooper, S.T.E.; Haines, Z.H.R.; Fuller, S.J.; O’Gara, P.; Sheppard, M.N.; Harding, S.E.; Sugden, P.H.; et al. Redox Regulation of Cardiac ASK1 (Apoptosis Signal-Regulating Kinase 1) Controls p38-MAPK (Mitogen-Activated Protein Kinase) and Orchestrates Cardiac Remodeling to Hypertension. Hypertension 2020, 76, 1208–1218. [Google Scholar] [CrossRef]
- Torres, M.; Forman, H.J. Redox signaling and the MAP kinase pathways. Biofactors 2003, 17, 287–296. [Google Scholar] [CrossRef]
- Dong, L.H.; Wen, J.K.; Liu, G.; McNutt, M.A.; Miao, S.B.; Gao, R.; Zheng, B.; Zhang, H.; Han, M. Blockade of the Ras-extracellular signal-regulated kinase 1/2 pathway is involved in smooth muscle 22 alpha-mediated suppression of vascular smooth muscle cell proliferation and neointima hyperplasia. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 683–691. [Google Scholar] [CrossRef]
- Hu, Y.; Dietrich, H.; Metzler, B.; Wick, G.; Xu, Q. Hyperexpression and activation of extracellular signal-regulated kinases (ERK1/2) in atherosclerotic lesions of cholesterol-fed rabbits. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 18–26. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Elimban, V.; Nijjar, M.S.; Gupta, S.K.; Dhalla, N.S. Role of mitogen-activated protein kinase in cardiac hypertrophy and heart failure. Exp. Clin. Cardiol. 2003, 8, 173–183. [Google Scholar]
- Muslin, A.J. MAPK signalling in cardiovascular health and disease: Molecular mechanisms and therapeutic targets. Clin. Sci. 2008, 115, 203–218. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Yin, Z.; Guo, X.; Hajjar, D.P.; Han, J. Inhibition of ERK1/2 and activation of liver X receptor synergistically induce macrophage ABCA1 expression and cholesterol efflux. J. Biol. Chem. 2010, 285, 6316–6326. [Google Scholar] [CrossRef] [Green Version]
- Amini, N.; Boyle, J.J.; Moers, B.; Warboys, C.M.; Malik, T.H.; Zakkar, M.; Francis, S.E.; Mason, J.C.; Haskard, D.O.; Evans, P.C. Requirement of JNK1 for endothelial cell injury in atherogenesis. Atherosclerosis 2014, 235, 613–618. [Google Scholar] [CrossRef] [Green Version]
- Jagavelu, K.; Tietge, U.J.; Gaestel, M.; Drexler, H.; Schieffer, B.; Bavendiek, U. Systemic deficiency of the MAP kinase-activated protein kinase 2 reduces atherosclerosis in hypercholesterolemic mice. Circ. Res. 2007, 101, 1104–1112. [Google Scholar] [CrossRef] [Green Version]
- Udalova, I.A.; Kwiatkowski, D. Interaction of AP-1 with a cluster of NF-kappa B binding elements in the human TNF promoter region. Biochem. Biophys. Res. Commun. 2001, 289, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Martin, T.; Cardarelli, P.M.; Parry, G.C.; Felts, K.A.; Cobb, R.R. Cytokine induction of monocyte chemoattractant protein-1 gene expression in human endothelial cells depends on the cooperative action of NF-kappa B and AP-1. Eur. J. Immunol. 1997, 27, 1091–1097. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Mackman, N.; Edgington, T.S.; Fan, S.T. Lipopolysaccharide induction of the tumor necrosis factor-alpha promoter in human monocytic cells. Regulation by Egr-1, c-Jun, and NF-kappaB transcription factors. J. Biol. Chem. 1997, 272, 17795–17801. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Wang, Y.; Cao, Z.Y.; Wang, M.M.; Liu, X.M.; Gao, T.; Hu, Q.K.; Yuan, W.J.; Lin, L. Up-regulated TLR4 in cardiomyocytes exacerbates heart failure after long-term myocardial infarction. J. Cell. Mol. Med. 2015, 19, 2728–2740. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Z.; Kong, B.; Yang, H.; Dai, C.; Fang, J.; Qin, T.; Huang, H. Key Player in Cardiac Hypertrophy, Emphasizing the Role of Toll-Like Receptor 4. Front. Cardiovasc. Med. 2020, 7, 579036. [Google Scholar] [CrossRef] [PubMed]
- Anand, A.R.; Bradley, R.; Ganju, R.K. LPS-induced MCP-1 expression in human microvascular endothelial cells is mediated by the tyrosine kinase, Pyk2 via the p38 MAPK/NF-kappaB-dependent pathway. Mol. Immunol. 2009, 46, 962–968. [Google Scholar] [CrossRef] [Green Version]
- Shi, Q.; Cox, L.A.; Glenn, J.; Tejero, M.E.; Hondara, V.; Vandeberg, J.L.; Wang, X.L. Molecular pathways mediating differential responses to lipopolysaccharide between human and baboon arterial endothelial cells. Clin. Exp. Pharmacol. Physiol. 2010, 37, 178–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawa, Y.; Ueki, T.; Hata, M.; Iwasawa, K.; Tsuruga, E.; Kojima, H.; Ishikawa, H.; Yoshida, S. LPS-induced IL-6, IL-8, VCAM-1, and ICAM-1 expression in human lymphatic endothelium. J. Histochem. Cytochem. 2008, 56, 97–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pober, J.S.; Sessa, W.C. Evolving functions of endothelial cells in inflammation. Nat. Rev. Immunol. 2007, 7, 803–815. [Google Scholar] [CrossRef]
- Howell, K.W.; Meng, X.; Fullerton, D.A.; Jin, C.; Reece, T.B.; Cleveland, J.C., Jr. Toll-like receptor 4 mediates oxidized LDL-induced macrophage differentiation to foam cells. J. Surg. Res. 2011, 171, e27–e31. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.K.; Haka, A.S.; Asmal, A.; Barbosa-Lorenzi, V.C.; Grosheva, I.; Chin, H.F.; Xiong, Y.; Hla, T.; Maxfield, F.R. TLR4 (Toll-Like Receptor 4)-Dependent Signaling Drives Extracellular Catabolism of LDL (Low-Density Lipoprotein) Aggregates. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 86–102. [Google Scholar] [CrossRef]
- Riad, A.; Jager, S.; Sobirey, M.; Escher, F.; Yaulema-Riss, A.; Westermann, D.; Karatas, A.; Heimesaat, M.M.; Bereswill, S.; Dragun, D.; et al. Toll-like receptor-4 modulates survival by induction of left ventricular remodeling after myocardial infarction in mice. J. Immunol. 2008, 180, 6954–6961. [Google Scholar] [CrossRef] [Green Version]
- Michelsen, K.S.; Wong, M.H.; Shah, P.K.; Zhang, W.; Yano, J.; Doherty, T.M.; Akira, S.; Rajavashisth, T.B.; Arditi, M. Lack of Toll-like receptor 4 or myeloid differentiation factor 88 reduces atherosclerosis and alters plaque phenotype in mice deficient in apolipoprotein E. Proc. Natl. Acad. Sci. USA 2004, 101, 10679–10684. [Google Scholar] [CrossRef] [Green Version]
- Hu, N.; Zhang, Y. TLR4 knockout attenuated high fat diet-induced cardiac dysfunction via NF-kappaB/JNK-dependent activation of autophagy. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 2001–2011. [Google Scholar] [CrossRef]
- Bjorkbacka, H.; Kunjathoor, V.V.; Moore, K.J.; Koehn, S.; Ordija, C.M.; Lee, M.A.; Means, T.; Halmen, K.; Luster, A.D.; Golenbock, D.T.; et al. Reduced atherosclerosis in MyD88-null mice links elevated serum cholesterol levels to activation of innate immunity signaling pathways. Nat. Med. 2004, 10, 416–421. [Google Scholar] [CrossRef]
- Mullick, A.E.; Tobias, P.S.; Curtiss, L.K. Modulation of atherosclerosis in mice by Toll-like receptor 2. J. Clin. Invest. 2005, 115, 3149–3156. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Ukai, T.; Yumoto, H.; Davey, M.; Goswami, S.; Gibson, F.C., 3rd; Genco, C.A. Toll-like receptor 2 plays a critical role in the progression of atherosclerosis that is independent of dietary lipids. Atherosclerosis 2008, 196, 146–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madan, M.; Amar, S. Toll-like receptor-2 mediates diet and/or pathogen associated atherosclerosis: Proteomic findings. PLoS ONE 2008, 3, e3204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shishido, T.; Nozaki, N.; Yamaguchi, S.; Shibata, Y.; Nitobe, J.; Miyamoto, T.; Takahashi, H.; Arimoto, T.; Maeda, K.; Yamakawa, M.; et al. Toll-like receptor-2 modulates ventricular remodeling after myocardial infarction. Circulation 2003, 108, 2905–2910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arslan, F.; Smeets, M.B.; O’Neill, L.A.; Keogh, B.; McGuirk, P.; Timmers, L.; Tersteeg, C.; Hoefer, I.E.; Doevendans, P.A.; Pasterkamp, G.; et al. Myocardial ischemia/reperfusion injury is mediated by leukocytic toll-like receptor-2 and reduced by systemic administration of a novel anti-toll-like receptor-2 antibody. Circulation 2010, 121, 80–90. [Google Scholar] [CrossRef] [Green Version]
- Ahola, A.J.; Lassenius, M.I.; Forsblom, C.; Harjutsalo, V.; Lehto, M.; Groop, P.H. Dietary patterns reflecting healthy food choices are associated with lower serum LPS activity. Sci. Rep. 2017, 7, 6511. [Google Scholar] [CrossRef] [Green Version]
- Jay, J.M.; Margitic, S.; Shereda, A.L.; Covington, H.V. Determining endotoxin content of ground beef by the Limulus amoebocyte lysate test as a rapid indicator of microbial quality. Appl. Environ. Microbiol. 1979, 38, 885–890. [Google Scholar] [CrossRef] [Green Version]
- Erridge, C. The capacity of foodstuffs to induce innate immune activation of human monocytes in vitro is dependent on food content of stimulants of Toll-like receptors 2 and 4. Br. J. Nutr. 2011, 105, 15–23. [Google Scholar] [CrossRef] [Green Version]
- Erridge, C.; Attina, T.; Spickett, C.M.; Webb, D.J. A high-fat meal induces low-grade endotoxemia: Evidence of a novel mechanism of postprandial inflammation. Am. J. Clin. Nutr. 2007, 86, 1286–1292. [Google Scholar] [CrossRef] [Green Version]
- Deopurkar, R.; Ghanim, H.; Friedman, J.; Abuaysheh, S.; Sia, C.L.; Mohanty, P.; Viswanathan, P.; Chaudhuri, A.; Dandona, P. Differential effects of cream, glucose, and orange juice on inflammation, endotoxin, and the expression of Toll-like receptor-4 and suppressor of cytokine signaling-3. Diabetes Care 2010, 33, 991–997. [Google Scholar] [CrossRef] [Green Version]
- Sipka, S.; Beres, A.; Bertok, L.; Varga, T.; Bruckner, G. Comparison of endotoxin levels in cow’s milk samples derived from farms and shops. Innate Immun. 2015, 21, 531–536. [Google Scholar] [CrossRef]
- Clemente-Postigo, M.; Queipo-Ortuno, M.I.; Murri, M.; Boto-Ordonez, M.; Perez-Martinez, P.; Andres-Lacueva, C.; Cardona, F.; Tinahones, F.J. Endotoxin increase after fat overload is related to postprandial hypertriglyceridemia in morbidly obese patients. J. Lipid Res. 2012, 53, 973–978. [Google Scholar] [CrossRef] [Green Version]
- Harte, A.L.; Varma, M.C.; Tripathi, G.; McGee, K.C.; Al-Daghri, N.M.; Al-Attas, O.S.; Sabico, S.; O’Hare, J.P.; Ceriello, A.; Saravanan, P.; et al. High fat intake leads to acute postprandial exposure to circulating endotoxin in type 2 diabetic subjects. Diabetes Care 2012, 35, 375–382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez-Moreno, J.; Garcia-Carpintero, S.; Jimenez-Lucena, R.; Haro, C.; Rangel-Zuniga, O.A.; Blanco-Rojo, R.; Yubero-Serrano, E.M.; Tinahones, F.J.; Delgado-Lista, J.; Perez-Martinez, P.; et al. Effect of Dietary Lipids on Endotoxemia Influences Postprandial Inflammatory Response. J. Agric. Food Chem. 2017, 65, 7756–7763. [Google Scholar] [CrossRef] [PubMed]
- Ghoshal, S.; Witta, J.; Zhong, J.; de Villiers, W.; Eckhardt, E. Chylomicrons promote intestinal absorption of lipopolysaccharides. J. Lipid Res. 2009, 50, 90–97. [Google Scholar] [CrossRef] [Green Version]
- Hussain, M.; Umair Ijaz, M.; Ahmad, M.I.; Khan, I.A.; Bukhary, S.U.F.; Khan, W.; Hussain, S.; Hashmi, M.S.; Li, C. Gut inflammation exacerbates hepatic injury in C57BL/6J mice via gut-vascular barrier dysfunction with high-fat-incorporated meat protein diets. Food Funct. 2020, 11, 9168–9176. [Google Scholar] [CrossRef]
- McDaniel, J.; Askew, W.; Bennett, D.; Mihalopoulos, J.; Anantharaman, S.; Fjeldstad, A.S.; Rule, D.C.; Nanjee, N.M.; Harris, R.A.; Richardson, R.S. Bison meat has a lower atherogenic risk than beef in healthy men. Nutr. Res. 2013, 33, 293–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arya, F.; Egger, S.; Colquhoun, D.; Sullivan, D.; Pal, S.; Egger, G. Differences in postprandial inflammatory responses to a ‘modern’ v. traditional meat meal: A preliminary study. Br. J. Nutr. 2010, 104, 724–728. [Google Scholar] [CrossRef] [Green Version]
- Iqbal, J.; Hussain, M.M. Intestinal lipid absorption. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E1183–E1194. [Google Scholar] [CrossRef] [Green Version]
- Berg, R.D. The indigenous gastrointestinal microflora. Trends Microbiol. 1996, 4, 430–435. [Google Scholar] [CrossRef]
- Rozentsvit, A.; Vinokur, K.; Samuel, S.; Li, Y.; Gerdes, A.M.; Carrillo-Sepulveda, M.A. Ellagic Acid Reduces High Glucose-Induced Vascular Oxidative Stress Through ERK1/2/NOX4 Signaling Pathway. Cell. Physiol. Biochem. 2017, 44, 1174–1187. [Google Scholar] [CrossRef]
- Patel, H.; Chen, J.; Das, K.C.; Kavdia, M. Hyperglycemia induces differential change in oxidative stress at gene expression and functional levels in HUVEC and HMVEC. Cardiovasc. Diabetol. 2013, 12, 142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zgheib, C.; Hodges, M.M.; Hu, J.; Liechty, K.W.; Xu, J. Long non-coding RNA Lethe regulates hyperglycemia-induced reactive oxygen species production in macrophages. PLoS ONE 2017, 12, e0177453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghanim, H.; Sia, C.L.; Upadhyay, M.; Korzeniewski, K.; Viswanathan, P.; Abuaysheh, S.; Mohanty, P.; Dandona, P. Orange juice neutralizes the proinflammatory effect of a high-fat, high-carbohydrate meal and prevents endotoxin increase and Toll-like receptor expression. Am. J. Clin. Nutr. 2010, 91, 940–949. [Google Scholar] [CrossRef] [Green Version]
- Wong, X.M.N.; Madrid, A.M.; Tralma, K.; Castillo, R.; Carrasco-Pozo, C.; Navarrete, P.; Beltran, C.; Pastene, E.; Gotteland, M. Polyphenol extracts interfere with bacterial lipopolysaccharide in vitro and decrease postprandial endotoxemia in human volunteers. J. Funct. Foods 2016, 26, 406–417. [Google Scholar] [CrossRef]
- Ghanim, H.; Batra, M.; Abuaysheh, S.; Green, K.; Makdissi, A.; Kuhadiya, N.D.; Chaudhuri, A.; Dandona, P. Antiinflammatory and ROS Suppressive Effects of the Addition of Fiber to a High-Fat High-Calorie Meal. J. Clin. Endocrinol. Metab. 2017, 102, 858–869. [Google Scholar] [CrossRef] [Green Version]
- O’Hearn, A. Can a carnivore diet provide all essential nutrients? Curr. Opin. Endocrinol. Diabetes Obes. 2020, 27, 312–316. [Google Scholar] [CrossRef]
- Churuangsuk, C.; Griffiths, D.; Lean, M.E.J.; Combet, E. Impacts of carbohydrate-restricted diets on micronutrient intakes and status: A systematic review. Obes. Rev. 2019, 20, 1132–1147. [Google Scholar] [CrossRef]
- Suzuki, J.; Bayna, E.; Li, H.L.; Molle, E.D.; Lew, W.Y. Lipopolysaccharide activates calcineurin in ventricular myocytes. J. Am. Coll. Cardiol. 2007, 49, 491–499. [Google Scholar] [CrossRef] [Green Version]
- Yucel, G.; Zhao, Z.; El-Battrawy, I.; Lan, H.; Lang, S.; Li, X.; Buljubasic, F.; Zimmermann, W.H.; Cyganek, L.; Utikal, J.; et al. Lipopolysaccharides induced inflammatory responses and electrophysiological dysfunctions in human-induced pluripotent stem cell derived cardiomyocytes. Sci. Rep. 2017, 7, 2935. [Google Scholar] [CrossRef] [Green Version]
- Cowan, D.B.; Noria, S.; Stamm, C.; Garcia, L.M.; Poutias, D.N.; del Nido, P.J.; McGowan, F.X., Jr. Lipopolysaccharide internalization activates endotoxin-dependent signal transduction in cardiomyocytes. Circ. Res. 2001, 88, 491–498. [Google Scholar] [CrossRef] [Green Version]
- Pastori, D.; Carnevale, R.; Nocella, C.; Novo, M.; Santulli, M.; Cammisotto, V.; Menichelli, D.; Pignatelli, P.; Violi, F. Gut-Derived Serum Lipopolysaccharide is Associated With Enhanced Risk of Major Adverse Cardiovascular Events in Atrial Fibrillation: Effect of Adherence to Mediterranean Diet. J. Am. Heart Assoc. 2017, 6, e005784. [Google Scholar] [CrossRef] [PubMed]
- Carrera-Bastos, P.; Picazo, O.; Fontes-Villalba, M.; Pareja-Galeano, H.; Lindeberg, S.; Martinez-Selles, M.; Lucia, A.; Emanuele, E. Serum Zonulin and Endotoxin Levels in Exceptional Longevity versus Precocious Myocardial Infarction. Aging Dis. 2018, 9, 317–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flesch, M.; Kilter, H.; Cremers, B.; Laufs, U.; Sudkamp, M.; Ortmann, M.; Muller, F.U.; Bohm, M. Effects of endotoxin on human myocardial contractility involvement of nitric oxide and peroxynitrite. J. Am. Coll. Cardiol. 1999, 33, 1062–1070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, A.A.; Megson, I.L.; Gray, G.A. Inducible nitric oxide synthase-derived superoxide contributes to hypereactivity in small mesenteric arteries from a rat model of chronic heart failure. Br. J. Pharmacol. 2000, 131, 29–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, Y.; Roman, L.J.; Masters, B.S.; Zweier, J.L. Inducible nitric-oxide synthase generates superoxide from the reductase domain. J. Biol. Chem. 1998, 273, 22635–22639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, Y.; Zweier, J.L. Superoxide and peroxynitrite generation from inducible nitric oxide synthase in macrophages. Proc. Natl. Acad. Sci. USA 1997, 94, 6954–6958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savage, G.P.; Dutta, P.C.; Rodriguez-Estrada, M.T. Cholesterol oxides: Their occurrence and methods to prevent their generation in foods. Asia Pac. J. Clin. Nutr. 2002, 11, 72–78. [Google Scholar] [CrossRef]
- Staprans, I.; Pan, X.M.; Rapp, J.H.; Feingold, K.R. Oxidized cholesterol in the diet is a source of oxidized lipoproteins in human serum. J. Lipid Res. 2003, 44, 705–715. [Google Scholar] [CrossRef] [Green Version]
- Hur, S.J.; Park, G.B.; Joo, S.T. Formation of cholesterol oxidation products (COPs) in animal products. Food Control 2007, 18, 939–947. [Google Scholar] [CrossRef]
- Pie, J.E.; Spahis, K.; Seillan, C. Cholesterol oxidation in meat products during cooking and frozen storage. J. Agric. Food Chem. 1991, 39, 250–254. [Google Scholar] [CrossRef]
- Carr, A.C.; McCall, M.R.; Frei, B. Oxidation of LDL by myeloperoxidase and reactive nitrogen species: Reaction pathways and antioxidant protection. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1716–1723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mabile, L.; Meilhac, O.; Escargueil-Blanc, I.; Troly, M.; Pieraggi, M.T.; Salvayre, R.; Negre-Salvayre, A. Mitochondrial function is involved in LDL oxidation mediated by human cultured endothelial cells. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 1575–1582. [Google Scholar] [CrossRef] [PubMed]
- Levy, Y.; Maor, I.; Presser, D.; Aviram, M. Consumption of eggs with meals increases the susceptibility of human plasma and low-density lipoprotein to lipid peroxidation. Ann. Nutr. Metab. 1996, 40, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Schwab, U.S.; Ausman, L.M.; Vogel, S.; Li, Z.; Lammi-Keefe, C.J.; Goldin, B.R.; Ordovas, J.M.; Schaefer, E.J.; Lichtenstein, A.H. Dietary cholesterol increases the susceptibility of low density lipoprotein to oxidative modification. Atherosclerosis 2000, 149, 83–90. [Google Scholar] [CrossRef]
- Chiu, H.C.; Jeng, J.R.; Shieh, S.M. Increased oxidizability of plasma low density lipoprotein from patients with coronary artery disease. Biochim. Biophys. Acta 1994, 1225, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Diehl, K.J.; Stauffer, B.L.; Greiner, J.J.; Weil, B.R.; DeSouza, C.A. Nitric oxide-mediated endothlium-dependent vasodilation is impaired with borderline high-LDL cholesterol. Clin. Transl. Sci. 2012, 5, 21–26. [Google Scholar] [CrossRef] [Green Version]
- Blair, A.; Shaul, P.W.; Yuhanna, I.S.; Conrad, P.A.; Smart, E.J. Oxidized low density lipoprotein displaces endothelial nitric-oxide synthase (eNOS) from plasmalemmal caveolae and impairs eNOS activation. J. Biol. Chem. 1999, 274, 32512–32519. [Google Scholar] [CrossRef] [Green Version]
- Staprans, I.; Pan, X.M.; Rapp, J.H.; Feingold, K.R. Oxidized cholesterol in the diet accelerates the development of aortic atherosclerosis in cholesterol-fed rabbits. Arterioscler. Thromb. Vasc. Biol. 1998, 18, 977–983. [Google Scholar] [CrossRef] [Green Version]
- Staprans, I.; Pan, X.M.; Rapp, J.H.; Grunfeld, C.; Feingold, K.R. Oxidized cholesterol in the diet accelerates the development of atherosclerosis in LDL receptor- and apolipoprotein E-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 708–714. [Google Scholar] [CrossRef] [Green Version]
- Miller, Y.I. Toll-like receptors and atherosclerosis: Oxidized LDL as an endogenous Toll-like receptor ligand. Future Cardiol. 2005, 1, 785–792. [Google Scholar] [CrossRef]
- Khorrami, A.; Ziaee, M.; Rameshrad, M.; Nakhlband, A.; Maleki-Dizaji, N.; Garjani, A. Oxidized cholesterol exacerbates toll-like receptor 4 expression and activity in the hearts of rats with myocardial infarction. J. Cardiovasc. Thorac. Res. 2020, 12, 43–50. [Google Scholar] [CrossRef] [Green Version]
- Meisinger, C.; Baumert, J.; Khuseyinova, N.; Loewel, H.; Koenig, W. Plasma oxidized low-density lipoprotein, a strong predictor for acute coronary heart disease events in apparently healthy, middle-aged men from the general population. Circulation 2005, 112, 651–657. [Google Scholar] [CrossRef] [Green Version]
- Peiretti, P. Palmitic Acid: Effect of Diet Supplementation and Occurrence in Animal Origin Food. In Palmitic Acid: Occurrence, Biochemistry and Health Effects; Porto, L.F., Ed.; Nova Science Publishers: Hauppauge, NY, USA, 2014; p. 45. [Google Scholar]
- O’Neil, C.E.; Keast, D.R.; Fulgoni, V.L.; Nicklas, T.A. Food sources of energy and nutrients among adults in the US: NHANES 2003-2006. Nutrients 2012, 4, 2097–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Xia, G.; Zhang, Y.; Liu, J.; Liu, X.; Li, W.; Lv, Y.; Wei, S.; Liu, J.; Quan, J. Palmitate induces VSMC apoptosis via toll like receptor (TLR)4/ROS/p53 pathway. Atherosclerosis 2017, 263, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Qian, Y.; Fang, Q.; Zhong, P.; Li, W.; Wang, L.; Fu, W.; Zhang, Y.; Xu, Z.; Li, X.; et al. Saturated palmitic acid induces myocardial inflammatory injuries through direct binding to TLR4 accessory protein MD2. Nat. Commun. 2017, 8, 13997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maloney, E.; Sweet, I.R.; Hockenbery, D.M.; Pham, M.; Rizzo, N.O.; Tateya, S.; Handa, P.; Schwartz, M.W.; Kim, F. Activation of NF-kappaB by palmitate in endothelial cells: A key role for NADPH oxidase-derived superoxide in response to TLR4 activation. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1370–1375. [Google Scholar] [CrossRef] [Green Version]
- Nolan, C.J.; Larter, C.Z. Lipotoxicity: Why do saturated fatty acids cause and monounsaturates protect against it? J. Gastroenterol. Hepatol. 2009, 24, 703–706. [Google Scholar] [CrossRef]
- Hall, K.D.; Guo, J.; Courville, A.B.; Boring, J.; Brychta, R.; Chen, K.Y.; Darcey, V.; Forde, C.G.; Gharib, A.M.; Gallagher, I.; et al. Effect of a plant-based, low-fat diet versus an animal-based, ketogenic diet on ad libitum energy intake. Nat. Med. 2021, 27, 344–353. [Google Scholar] [CrossRef]
- You, Y.; Guo, Y.; Jia, P.; Zhuang, B.; Cheng, Y.; Deng, H.; Wang, X.; Zhang, C.; Luo, S.; Huang, B. Ketogenic diet aggravates cardiac remodeling in adult spontaneously hypertensive rats. Nutr. Metab. 2020, 17, 91. [Google Scholar] [CrossRef]
- Liu, J.; Wang, P.; Douglas, S.L.; Tate, J.M.; Sham, S.; Lloyd, S.G. Impact of high-fat, low-carbohydrate diet on myocardial substrate oxidation, insulin sensitivity, and cardiac function after ischemia-reperfusion. Am. J. Physiol. Heart Circ. Physiol. 2016, 311, H1–H10. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Wang, P.; Zou, L.; Qu, J.; Litovsky, S.; Umeda, P.; Zhou, L.; Chatham, J.; Marsh, S.A.; Dell’Italia, L.J.; et al. High-fat, low-carbohydrate diet promotes arrhythmic death and increases myocardial ischemia-reperfusion injury in rats. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H598–H608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, J.; Chen, H.; Wang, Y.J.; Qiu, J.X.; Meng, Q.Q.; Zou, R.J.; Li, L.; Huang, J.G.; Zhao, Z.K.; Huang, Y.L.; et al. Ketogenic Diet Suppressed T-Regulatory Cells and Promoted Cardiac Fibrosis via Reducing Mitochondria-Associated Membranes and Inhibiting Mitochondrial Function. Oxid. Med. Cell. Longev. 2021, 2021, 5512322. [Google Scholar] [CrossRef] [PubMed]
- Abdurrachim, D.; Teo, X.Q.; Woo, C.C.; Ong, S.Y.; Salleh, N.F.; Lalic, J.; Tan, R.S.; Lee, P.T.H. Cardiac metabolic modulation upon low-carbohydrate low-protein ketogenic diet in diabetic rats studied in vivo using hyperpolarized (13) C pyruvate, butyrate and acetoacetate probes. Diabetes Obes. Metab. 2019, 21, 949–960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, S.; Tao, H.; Cao, W.; Cao, L.; Lin, Y.; Zhao, S.M.; Xu, W.; Cao, J.; Zhao, J.Y. Ketogenic diets inhibit mitochondrial biogenesis and induce cardiac fibrosis. Signal. Transduct. Target. Ther. 2021, 6, 54. [Google Scholar] [CrossRef]
- Wang, P.; Tate, J.M.; Lloyd, S.G. Low carbohydrate diet decreases myocardial insulin signaling and increases susceptibility to myocardial ischemia. Life Sci. 2008, 83, 836–844. [Google Scholar] [CrossRef] [Green Version]
- Duda, M.K.; O’Shea, K.M.; Lei, B.; Barrows, B.R.; Azimzadeh, A.M.; McElfresh, T.E.; Hoit, B.D.; Kop, W.J.; Stanley, W.C. Low-carbohydrate/high-fat diet attenuates pressure overload-induced ventricular remodeling and dysfunction. J. Card. Fail. 2008, 14, 327–335. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, M.; Odanovic, N.; Nakada, Y.; Dohi, S.; Zhai, P.; Ivessa, A.; Yang, Z.; Abdellatif, M.; Sadoshima, J. Dietary carbohydrates restriction inhibits the development of cardiac hypertrophy and heart failure. Cardiovasc. Res. 2021, 117, 2365–2376. [Google Scholar] [CrossRef]
- Lancaster, G.I.; Langley, K.G.; Berglund, N.A.; Kammoun, H.L.; Reibe, S.; Estevez, E.; Weir, J.; Mellett, N.A.; Pernes, G.; Conway, J.R.W.; et al. Evidence that TLR4 Is Not a Receptor for Saturated Fatty Acids but Mediates Lipid-Induced Inflammation by Reprogramming Macrophage Metabolism. Cell. Metab. 2018, 27, 1096–1110.e1095. [Google Scholar] [CrossRef] [Green Version]
- Erridge, C.; Samani, N.J. Saturated fatty acids do not directly stimulate Toll-like receptor signaling. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1944–1949. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Qi, M.Y.; Liu, S.; Song, X.T.; Zhang, J.N.; Zhai, Y.F.; Lu, M.H.; Han, H.B.; Lian, Z.X.; Yao, Y.C. TLR4 overexpression enhances saturated fatty acid-induced inflammatory cytokine gene expression in sheep. Eur. J. Inflamm. 2018, 16, 2976. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.Y.; Zhao, L.; Youn, H.S.; Weatherill, A.R.; Tapping, R.; Feng, L.; Lee, W.H.; Fitzgerald, K.A.; Hwang, D.H. Saturated fatty acid activates but polyunsaturated fatty acid inhibits Toll-like receptor 2 dimerized with Toll-like receptor 6 or 1. J. Biol. Chem. 2004, 279, 16971–16979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mo, Z.; Huang, S.; Burnett, D.J.; Rutledge, J.C.; Hwang, D.H. Endotoxin May Not Be the Major Cause of Postprandial Inflammation in Adults Who Consume a Single High-Fat or Moderately High-Fat Meal. J. Nutr. 2020, 150, 1303–1312. [Google Scholar] [CrossRef] [PubMed]
- Roden, M. How free fatty acids inhibit glucose utilization in human skeletal muscle. News Physiol. Sci. 2004, 19, 92–96. [Google Scholar] [CrossRef] [Green Version]
- Valsdottir, T.D.; Henriksen, C.; Odden, N.; Nellemann, B.; Jeppesen, P.B.; Hisdal, J.; Westerberg, A.C.; Jensen, J. Effect of a Low-Carbohydrate High-Fat Diet and a Single Bout of Exercise on Glucose Tolerance, Lipid Profile and Endothelial Function in Normal Weight Young Healthy Females. Front. Physiol. 2019, 10, 1499. [Google Scholar] [CrossRef]
- Hernandez, T.L.; Sutherland, J.P.; Wolfe, P.; Allian-Sauer, M.; Capell, W.H.; Talley, N.D.; Wyatt, H.R.; Foster, G.D.; Hill, J.O.; Eckel, R.H. Lack of suppression of circulating free fatty acids and hypercholesterolemia during weight loss on a high-fat, low-carbohydrate diet. Am. J. Clin. Nutr. 2010, 91, 578–585. [Google Scholar] [CrossRef] [Green Version]
- Zderic, T.W.; Davidson, C.J.; Schenk, S.; Byerley, L.O.; Coyle, E.F. High-fat diet elevates resting intramuscular triglyceride concentration and whole body lipolysis during exercise. Am. J. Physiol. Endocrinol. Metab. 2004, 286, E217–E225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rachek, L.I. Free fatty acids and skeletal muscle insulin resistance. Prog. Mol. Biol. Transl. Sci. 2014, 121, 267–292. [Google Scholar] [CrossRef] [PubMed]
- Lovejoy, J.C.; Windhauser, M.M.; Rood, J.C.; de la Bretonne, J.A. Effect of a controlled high-fat versus low-fat diet on insulin sensitivity and leptin levels in African-American and Caucasian women. Metabolism 1998, 47, 1520–1524. [Google Scholar] [CrossRef]
- Numao, S.; Kawano, H.; Endo, N.; Yamada, Y.; Konishi, M.; Takahashi, M.; Sakamoto, S. Short-term low carbohydrate/high-fat diet intake increases postprandial plasma glucose and glucagon-like peptide-1 levels during an oral glucose tolerance test in healthy men. Eur. J. Clin. Nutr. 2012, 66, 926–931. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Yang, X.; Zhang, J.; Jiang, T.; Zhang, Z.; Wang, Z.; Gong, M.; Zhao, L.; Zhang, C. Ketogenic Diets Induced Glucose Intolerance and Lipid Accumulation in Mice with Alterations in Gut Microbiota and Metabolites. mBio 2021, 12, e03601–e03620. [Google Scholar] [CrossRef]
- Grandl, G.; Straub, L.; Rudigier, C.; Arnold, M.; Wueest, S.; Konrad, D.; Wolfrum, C. Short-term feeding of a ketogenic diet induces more severe hepatic insulin resistance than an obesogenic high-fat diet. J. Physiol. 2018, 596, 4597–4609. [Google Scholar] [CrossRef] [PubMed]
- Ellenbroek, J.H.; van Dijck, L.; Tons, H.A.; Rabelink, T.J.; Carlotti, F.; Ballieux, B.E.; de Koning, E.J. Long-term ketogenic diet causes glucose intolerance and reduced beta- and alpha-cell mass but no weight loss in mice. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E552–E558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinzig, K.P.; Honors, M.A.; Hargrave, S.L. Insulin sensitivity and glucose tolerance are altered by maintenance on a ketogenic diet. Endocrinology 2010, 151, 3105–3114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nowotny, B.; Zahiragic, L.; Krog, D.; Nowotny, P.J.; Herder, C.; Carstensen, M.; Yoshimura, T.; Szendroedi, J.; Phielix, E.; Schadewaldt, P.; et al. Mechanisms underlying the onset of oral lipid-induced skeletal muscle insulin resistance in humans. Diabetes 2013, 62, 2240–2248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, H.; Lum, H.; Alvarez, A.; Garduno-Garcia, J.J.; Daniel, B.J.; Musi, N. A low dose lipid infusion is sufficient to induce insulin resistance and a pro-inflammatory response in human subjects. PLoS ONE 2018, 13, e0195810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charidemou, E.; Ashmore, T.; Li, X.; McNally, B.D.; West, J.A.; Liggi, S.; Harvey, M.; Orford, E.; Griffin, J.L. A randomized 3-way crossover study indicates that high-protein feeding induces de novo lipogenesis in healthy humans. JCI Insight 2019, 4, e12481. [Google Scholar] [CrossRef] [Green Version]
- Rider, O.J.; Holloway, C.J.; Emmanuel, Y.; Bloch, E.; Clarke, K.; Neubauer, S. Increasing plasma free fatty acids in healthy subjects induces aortic distensibility changes seen in obesity. Circ. Cardiovasc. Imaging 2012, 5, 367–375. [Google Scholar] [CrossRef] [Green Version]
- Forstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837, 837a–837d. [Google Scholar] [CrossRef] [Green Version]
- Qian, J.; Fulton, D. Post-translational regulation of endothelial nitric oxide synthase in vascular endothelium. Front. Physiol. 2013, 4, 347. [Google Scholar] [CrossRef] [Green Version]
- Wartenberg, M.; Schallenberg, M.; Hescheler, J.; Sauer, H. Reactive oxygen species-mediated regulation of eNOS and iNOS expression in multicellular prostate tumor spheroids. Int. J. Cancer 2003, 104, 274–282. [Google Scholar] [CrossRef]
- Steinberg, H.O.; Paradisi, G.; Hook, G.; Crowder, K.; Cronin, J.; Baron, A.D. Free fatty acid elevation impairs insulin-mediated vasodilation and nitric oxide production. Diabetes 2000, 49, 1231–1238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, F.; Tysseling, K.A.; Rice, J.; Pham, M.; Haji, L.; Gallis, B.M.; Baas, A.S.; Paramsothy, P.; Giachelli, C.M.; Corson, M.A.; et al. Free fatty acid impairment of nitric oxide production in endothelial cells is mediated by IKKbeta. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 989–994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Youn, J.Y.; Cai, H. Mechanisms and consequences of endothelial nitric oxide synthase dysfunction in hypertension. J. Hypertens. 2015, 33, 1128–1136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawashima, S.; Yokoyama, M. Dysfunction of endothelial nitric oxide synthase and atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 998–1005. [Google Scholar] [CrossRef] [PubMed]
- Montagnani, M.; Chen, H.; Barr, V.A.; Quon, M.J. Insulin-stimulated activation of eNOS is independent of Ca2+ but requires phosphorylation by Akt at Ser(1179). J. Biol. Chem. 2001, 276, 30392–30398. [Google Scholar] [CrossRef] [Green Version]
- Mohan, S.; Konopinski, R.; Yan, B.; Centonze, V.E.; Natarajan, M. High glucose-induced IKK-Hsp-90 interaction contributes to endothelial dysfunction. Am. J. Physiol. Cell. Physiol. 2009, 296, C182–C192. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Wang, X.; Jia, P.; You, Y.; Cheng, Y.; Deng, H.; Luo, S.; Huang, B. Ketogenic diet aggravates hypertension via NF-kappaB-mediated endothelial dysfunction in spontaneously hypertensive rats. Life Sci. 2020, 258, 118124. [Google Scholar] [CrossRef]
- Yan, G.; You, B.; Chen, S.P.; Liao, J.K.; Sun, J. Tumor necrosis factor-alpha downregulates endothelial nitric oxide synthase mRNA stability via translation elongation factor 1-alpha 1. Circ. Res. 2008, 103, 591–597. [Google Scholar] [CrossRef] [Green Version]
- Chavez, J.A.; Summers, S.A. Lipid oversupply, selective insulin resistance, and lipotoxicity: Molecular mechanisms. Biochim. Biophys. Acta 2010, 1801, 252–265. [Google Scholar] [CrossRef] [Green Version]
- Chavez, J.A.; Summers, S.A. Characterizing the effects of saturated fatty acids on insulin signaling and ceramide and diacylglycerol accumulation in 3T3-L1 adipocytes and C2C12 myotubes. Arch. Biochem. Biophys. 2003, 419, 101–109. [Google Scholar] [CrossRef]
- Gao, D.; Pararasa, C.; Dunston, C.R.; Bailey, C.J.; Griffiths, H.R. Palmitate promotes monocyte atherogenicity via de novo ceramide synthesis. Free Radic. Biol. Med. 2012, 53, 796–806. [Google Scholar] [CrossRef] [PubMed]
- Holland, W.L.; Bikman, B.T.; Wang, L.P.; Yuguang, G.; Sargent, K.M.; Bulchand, S.; Knotts, T.A.; Shui, G.; Clegg, D.J.; Wenk, M.R.; et al. Lipid-induced insulin resistance mediated by the proinflammatory receptor TLR4 requires saturated fatty acid-induced ceramide biosynthesis in mice. J. Clin. Invest. 2011, 121, 1858–1870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Memon, R.A.; Holleran, W.M.; Moser, A.H.; Seki, T.; Uchida, Y.; Fuller, J.; Shigenaga, J.K.; Grunfeld, C.; Feingold, K.R. Endotoxin and cytokines increase hepatic sphingolipid biosynthesis and produce lipoproteins enriched in ceramides and sphingomyelin. Arterioscler. Thromb. Vasc. Biol. 1998, 18, 1257–1265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Memon, R.A.; Holleran, W.M.; Uchida, Y.; Moser, A.H.; Grunfeld, C.; Feingold, K.R. Regulation of sphingolipid and glycosphingolipid metabolism in extrahepatic tissues by endotoxin. J. Lipid Res. 2001, 42, 452–459. [Google Scholar] [CrossRef] [PubMed]
- Hanada, K. Serine palmitoyltransferase, a key enzyme of sphingolipid metabolism. BBA-Mol. Cell. Biol. Lipids 2003, 1632, 16–30. [Google Scholar] [CrossRef]
- Zhang, D.X.; Zou, A.P.; Li, P.L. Ceramide-induced activation of NADPH oxidase and endothelial dysfunction in small coronary arteries. Am. J. Physiol. Heart Circ. Physiol. 2003, 284, H605–H612. [Google Scholar] [CrossRef] [Green Version]
- Beckman, J.S.; Koppenol, W.H. Nitric oxide, superoxide, and peroxynitrite: The good, the bad, and ugly. Am. J. Physiol. 1996, 271, C1424–C1437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Junk, P.; Huwiler, A.; Burkhardt, C.; Wallerath, T.; Pfeilschifter, J.; Forstermann, U. Dual effect of ceramide on human endothelial cells: Induction of oxidative stress and transcriptional upregulation of endothelial nitric oxide synthase. Circulation 2002, 106, 2250–2256. [Google Scholar] [CrossRef] [Green Version]
- Piepoli, M.F.; The Sixth Joint Task Force of the European Society of Cardiology and Other Societies on Cardiovascular Disease Prevention in Clinical Practice (Constituted by Representatives of 10 Societies and by Invited Experts). 2016 European Guidelines on cardiovascular disease prevention in clinical practice. Int. J. Behav. Med. 2017, 24, 321–419. [Google Scholar] [CrossRef]
- Singh, K.D.; Karnik, S.S. Angiotensin Type 1 Receptor Blockers in Heart Failure. Curr. Drug. Targets 2020, 21, 125–131. [Google Scholar] [CrossRef]
- Civieri, G.; Iop, L.; Tona, F. Antibodies against Angiotensin II Type 1 and Endothelin 1 Type A Receptors in Cardiovascular Pathologies. Int. J. Mol. Sci. 2022, 23, 927. [Google Scholar] [CrossRef] [PubMed]
- Biancardi, V.C.; Bomfim, G.F.; Reis, W.L.; Al-Gassimi, S.; Nunes, K.P. The interplay between Angiotensin II, TLR4 and hypertension. Pharmacol. Res. 2017, 120, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Laghlam, D.; Jozwiak, M.; Nguyen, L.S. Renin-Angiotensin-Aldosterone System and Immunomodulation: A State-of-the-Art Review. Cells 2021, 10, 1767. [Google Scholar] [CrossRef] [PubMed]
- Paul, M.; Wagner, J.; Dzau, V.J. Gene expression of the renin-angiotensin system in human tissues. Quantitative analysis by the polymerase chain reaction. J. Clin. Invest. 1993, 91, 2058–2064. [Google Scholar] [CrossRef]
- Nguyen Dinh Cat, A.; Montezano, A.C.; Burger, D.; Touyz, R.M. Angiotensin II, NADPH oxidase, and redox signaling in the vasculature. Antioxid. Redox Signal. 2013, 19, 1110–1120. [Google Scholar] [CrossRef] [Green Version]
- Su, K.H.; Tsai, J.Y.; Kou, Y.R.; Chiang, A.N.; Hsiao, S.H.; Wu, Y.L.; Hou, H.H.; Pan, C.C.; Shyue, S.K.; Lee, T.S. Valsartan regulates the interaction of angiotensin II type 1 receptor and endothelial nitric oxide synthase via Src/PI3K/Akt signalling. Cardiovasc. Res. 2009, 82, 468–475. [Google Scholar] [CrossRef] [Green Version]
- Ding, J.; Yu, M.; Jiang, J.; Luo, Y.; Zhang, Q.; Wang, S.; Yang, F.; Wang, A.; Wang, L.; Zhuang, M.; et al. Angiotensin II Decreases Endothelial Nitric Oxide Synthase Phosphorylation via AT(1)R Nox/ROS/PP2A Pathway. Front. Physiol. 2020, 11, 566410. [Google Scholar] [CrossRef]
- Wynne, B.M.; Chiao, C.W.; Webb, R.C. Vascular Smooth Muscle Cell Signaling Mechanisms for Contraction to Angiotensin II and Endothelin-1. J. Am. Soc. Hypertens. 2009, 3, 84–95. [Google Scholar] [CrossRef] [Green Version]
- Ushio-Fukai, M.; Zafari, A.M.; Fukui, T.; Ishizaka, N.; Griendling, K.K. p22phox is a critical component of the superoxide-generating NADH/NADPH oxidase system and regulates angiotensin II-induced hypertrophy in vascular smooth muscle cells. J. Biol. Chem. 1996, 271, 23317–23321. [Google Scholar] [CrossRef] [Green Version]
- Dubey, R.K.; Jackson, E.K.; Luscher, T.F. Nitric oxide inhibits angiotensin II-induced migration of rat aortic smooth muscle cell. Role of cyclic-nucleotides and angiotensin1 receptors. J. Clin. Invest. 1995, 96, 141–149. [Google Scholar] [CrossRef] [Green Version]
- Ainscough, J.F.; Drinkhill, M.J.; Sedo, A.; Turner, N.A.; Brooke, D.A.; Balmforth, A.J.; Ball, S.G. Angiotensin II type-1 receptor activation in the adult heart causes blood pressure-independent hypertrophy and cardiac dysfunction. Cardiovasc. Res. 2009, 81, 592–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paradis, P.; Dali-Youcef, N.; Paradis, F.W.; Thibault, G.; Nemer, M. Overexpression of angiotensin II type I receptor in cardiomyocytes induces cardiac hypertrophy and remodeling. Proc. Natl. Acad. Sci. USA 2000, 97, 931–936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mansoor, N.; Vinknes, K.J.; Veierod, M.B.; Retterstol, K. Effects of low-carbohydrate diets v. low-fat diets on body weight and cardiovascular risk factors: A meta-analysis of randomised controlled trials. Br. J. Nutr. 2016, 115, 466–479. [Google Scholar] [CrossRef] [PubMed]
- Daugherty, A.; Rateri, D.L.; Lu, H.; Inagami, T.; Cassis, L.A. Hypercholesterolemia stimulates angiotensin peptide synthesis and contributes to atherosclerosis through the AT1A receptor. Circulation 2004, 110, 3849–3857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nickenig, G.; Sachinidis, A.; Michaelsen, F.; Bohm, M.; Seewald, S.; Vetter, H. Upregulation of vascular angiotensin II receptor gene expression by low-density lipoprotein in vascular smooth muscle cells. Circulation 1997, 95, 473–478. [Google Scholar] [CrossRef]
- Nickenig, G.; Jung, O.; Strehlow, K.; Zolk, O.; Linz, W.; Scholkens, B.A.; Bohm, M. Hypercholesterolemia is associated with enhanced angiotensin AT1-receptor expression. Am. J. Physiol. 1997, 272, H2701–H2707. [Google Scholar] [CrossRef]
- Li, D.; Saldeen, T.; Romeo, F.; Mehta, J.L. Oxidized LDL upregulates angiotensin II type 1 receptor expression in cultured human coronary artery endothelial cells: The potential role of transcription factor NF-kappaB. Circulation 2000, 102, 1970–1976. [Google Scholar] [CrossRef] [Green Version]
- Schuler, R.; Osterhoff, M.A.; Frahnow, T.; Seltmann, A.C.; Busjahn, A.; Kabisch, S.; Xu, L.; Mosig, A.S.; Spranger, J.; Mohlig, M.; et al. High-Saturated-Fat Diet Increases Circulating Angiotensin-Converting Enzyme, Which Is Enhanced by the rs4343 Polymorphism Defining Persons at Risk of Nutrient-Dependent Increases of Blood Pressure. J. Am. Heart Assoc. 2017, 6, e004465. [Google Scholar] [CrossRef]
- Sun, J.; Luo, J.; Ruan, Y.; Xiu, L.; Fang, B.; Zhang, H.; Wang, M.; Chen, H. Free Fatty Acids Activate Renin-Angiotensin System in 3T3-L1 Adipocytes through Nuclear Factor-kappa B Pathway. J. Diabetes Res. 2016, 2016, 1587594. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, S.; Tagawa, T.; Yamakawa, K.; Shimabukuro, M.; Ueda, S. Inhibition of the renin-angiotensin system prevents free fatty acid-induced acute endothelial dysfunction in humans. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 2376–2380. [Google Scholar] [CrossRef] [Green Version]
- Office of Dietary Supplements. Choline: Fact Sheet for Health Professionals. Available online: https://ods.od.nih.gov/factsheets/Choline-HealthProfessional/#en11 (accessed on 3 March 2023).
- Office of Dietary Supplements. Carnitine: Fact Sheet for Health Professionals. Available online: https://ods.od.nih.gov/factsheets/Carnitine-HealthProfessional/#en10 (accessed on 3 March 2023).
- Rebouche, C.J. Carnitine function and requirements during the life cycle. FASEB J. 1992, 6, 3379–3386. [Google Scholar] [CrossRef] [PubMed]
- Michel, V.; Yuan, Z.; Ramsubir, S.; Bakovic, M. Choline transport for phospholipid synthesis. Exp. Biol. Med. 2006, 231, 490–504. [Google Scholar] [CrossRef] [PubMed]
- Sam, C.; Bordoni, B. Physiology, Acetylcholine. In StatPearls; StatPearls Publishing LLC.: Treasure Island, FL, USA, 2023. [Google Scholar]
- Koeth, R.A.; Wang, Z.; Levison, B.S.; Buffa, J.A.; Org, E.; Sheehy, B.T.; Britt, E.B.; Fu, X.; Wu, Y.; Li, L.; et al. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat. Med. 2013, 19, 576–585. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Qiu, J.; Lian, J.; Yang, X.; Zhou, J. Gut Metabolite Trimethylamine-N-Oxide in Atherosclerosis: From Mechanism to Therapy. Front. Cardiovasc. Med. 2021, 8, 723886. [Google Scholar] [CrossRef] [PubMed]
- Seldin, M.M.; Meng, Y.; Qi, H.; Zhu, W.; Wang, Z.; Hazen, S.L.; Lusis, A.J.; Shih, D.M. Trimethylamine N-Oxide Promotes Vascular Inflammation Through Signaling of Mitogen-Activated Protein Kinase and Nuclear Factor-kappaB. J. Am. Heart Assoc. 2016, 5, e002767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, K.; Yuan, Y.; Yu, H.; Dai, X.; Wang, S.; Sun, Z.; Wang, F.; Fei, H.; Lin, Q.; Jiang, H.; et al. The gut microbial metabolite trimethylamine N-oxide aggravates GVHD by inducing M1 macrophage polarization in mice. Blood 2020, 136, 501–515. [Google Scholar] [CrossRef]
- Hakhamaneshi, M.S.; Abdolahi, A.; Vahabzadeh, Z.; Abdi, M.; Andalibi, P. Toll-Like Receptor 4: A Macrophage Cell Surface Receptor Is Activated By Trimethylamine-N-Oxide. Cell. J. 2021, 23, 516–522. [Google Scholar] [CrossRef]
- Zhang, X.; Li, Y.; Yang, P.; Liu, X.; Lu, L.; Chen, Y.; Zhong, X.; Li, Z.; Liu, H.; Ou, C.; et al. Trimethylamine-N-Oxide Promotes Vascular Calcification Through Activation of NLRP3 (Nucleotide-Binding Domain, Leucine-Rich-Containing Family, Pyrin Domain-Containing-3) Inflammasome and NF-kappaB (Nuclear Factor kappaB) Signals. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 751–765. [Google Scholar] [CrossRef]
- Li, X.; Fan, Z.; Cui, J.; Li, D.; Lu, J.; Cui, X.; Xie, L.; Wu, Y.; Lin, Q.; Li, Y. Trimethylamine N-Oxide in Heart Failure: A Meta-Analysis of Prognostic Value. Front. Cardiovasc. Med. 2022, 9, 817396. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, Y.; Ke, B.; Du, J. TMAO: How gut microbiota contributes to heart failure. Transl. Res. 2021, 228, 109–125. [Google Scholar] [CrossRef]
- Savi, M.; Bocchi, L.; Bresciani, L.; Falco, A.; Quaini, F.; Mena, P.; Brighenti, F.; Crozier, A.; Stilli, D.; Del Rio, D. Trimethylamine-N-Oxide (TMAO)-Induced Impairment of Cardiomyocyte Function and the Protective Role of Urolithin B-Glucuronide. Molecules 2018, 23, 549. [Google Scholar] [CrossRef] [Green Version]
- Makrecka-Kuka, M.; Volska, K.; Antone, U.; Vilskersts, R.; Grinberga, S.; Bandere, D.; Liepinsh, E.; Dambrova, M. Trimethylamine N-oxide impairs pyruvate and fatty acid oxidation in cardiac mitochondria. Toxicol. Lett. 2017, 267, 32–38. [Google Scholar] [CrossRef]
- Yang, J.J.; Shu, X.O.; Herrington, D.M.; Moore, S.C.; Meyer, K.A.; Ose, J.; Menni, C.; Palmer, N.D.; Eliassen, H.; Harada, S.; et al. Circulating trimethylamine N-oxide in association with diet and cardiometabolic biomarkers: An international pooled analysis. Am. J. Clin. Nutr. 2021, 113, 1145–1156. [Google Scholar] [CrossRef]
- Wang, Z.; Bergeron, N.; Levison, B.S.; Li, X.S.; Chiu, S.; Jia, X.; Koeth, R.A.; Li, L.; Wu, Y.; Tang, W.H.W.; et al. Impact of chronic dietary red meat, white meat, or non-meat protein on trimethylamine N-oxide metabolism and renal excretion in healthy men and women. Eur. Heart J. 2019, 40, 583–594. [Google Scholar] [CrossRef]
- Lombardo, M.; Aulisa, G.; Marcon, D.; Rizzo, G. The Influence of Animal- or Plant-Based Diets on Blood and Urine Trimethylamine-N-Oxide (TMAO) Levels in Humans. Curr. Nutr. Rep. 2022, 11, 56–68. [Google Scholar] [CrossRef] [PubMed]
- Argyridou, S.; Davies, M.J.; Biddle, G.J.H.; Bernieh, D.; Suzuki, T.; Dawkins, N.P.; Rowlands, A.V.; Khunti, K.; Smith, A.C.; Yates, T. Evaluation of an 8-Week Vegan Diet on Plasma Trimethylamine-N-Oxide and Postchallenge Glucose in Adults with Dysglycemia or Obesity. J. Nutr. 2021, 151, 1844–1853. [Google Scholar] [CrossRef] [PubMed]
- Park, J.E.; Miller, M.; Rhyne, J.; Wang, Z.; Hazen, S.L. Differential effect of short-term popular diets on TMAO and other cardio-metabolic risk markers. Nutr. Metab. Cardiovasc. Dis. 2019, 29, 513–517. [Google Scholar] [CrossRef]
- Cho, C.E.; Taesuwan, S.; Malysheva, O.V.; Bender, E.; Tulchinsky, N.F.; Yan, J.; Sutter, J.L.; Caudill, M.A. Trimethylamine-N-oxide (TMAO) response to animal source foods varies among healthy young men and is influenced by their gut microbiota composition: A randomized controlled trial. Mol. Nutr. Food Res. 2017, 61, 1600324. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Xiong, K.; Cai, J.; Ma, A. Fish Consumption and Coronary Heart Disease: A Meta-Analysis. Nutrients 2020, 12, 2278. [Google Scholar] [CrossRef]
- Zhao, L.G.; Sun, J.W.; Yang, Y.; Ma, X.; Wang, Y.Y.; Xiang, Y.B. Fish consumption and all-cause mortality: A meta-analysis of cohort studies. Eur. J. Clin. Nutr. 2016, 70, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Jayedi, A.; Shab-Bidar, S.; Eimeri, S.; Djafarian, K. Fish consumption and risk of all-cause and cardiovascular mortality: A dose-response meta-analysis of prospective observational studies. Public. Health Nutr. 2018, 21, 1297–1306. [Google Scholar] [CrossRef] [Green Version]
- Tonstad, S.; Butler, T.; Yan, R.; Fraser, G.E. Type of vegetarian diet, body weight, and prevalence of type 2 diabetes. Diabetes Care 2009, 32, 791–796. [Google Scholar] [CrossRef] [Green Version]
- Orlich, M.J.; Fraser, G.E. Vegetarian diets in the Adventist Health Study 2: A review of initial published findings. Am. J. Clin. Nutr. 2014, 100 (Suppl. S1), 353S–358S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orlich, M.J.; Singh, P.N.; Sabate, J.; Jaceldo-Siegl, K.; Fan, J.; Knutsen, S.; Beeson, W.L.; Fraser, G.E. Vegetarian dietary patterns and mortality in Adventist Health Study 2. JAMA Intern. Med. 2013, 173, 1230–1238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Outzen, M.; Tjonneland, A.; Larsen, E.H.; Hansen, M.; Andersen, K.K.; Christensen, J.; Overvad, K.; Olsen, A. Effect of increased intake of fish and mussels on exposure to toxic trace elements in a healthy, middle-aged population. Food Addit. Contam. Part A Chem. Anal. Control. Expo. Risk Assess. 2015, 32, 1858–1866. [Google Scholar] [CrossRef]
- Lopez, S.; Bermudez, B.; Ortega, A.; Varela, L.M.; Pacheco, Y.M.; Villar, J.; Abia, R.; Muriana, F.J. Effects of meals rich in either monounsaturated or saturated fat on lipid concentrations and on insulin secretion and action in subjects with high fasting triglyceride concentrations. Am. J. Clin. Nutr. 2011, 93, 494–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miles, J.M.; Wooldridge, D.; Grellner, W.J.; Windsor, S.; Isley, W.L.; Klein, S.; Harris, W.S. Nocturnal and postprandial free fatty acid kinetics in normal and type 2 diabetic subjects: Effects of insulin sensitization therapy. Diabetes 2003, 52, 675–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karpe, F.; Olivecrona, T.; Walldius, G.; Hamsten, A. Lipoprotein lipase in plasma after an oral fat load: Relation to free fatty acids. J. Lipid Res. 1992, 33, 975–984. [Google Scholar] [CrossRef] [PubMed]
- Keirns, B.H.; Sciarrillo, C.M.; Koemel, N.A.; Emerson, S.R. Fasting, non-fasting and postprandial triglycerides for screening cardiometabolic risk. J. Nutr. Sci. 2021, 10, e75. [Google Scholar] [CrossRef]
- Strong, J.P.; McGill, H.C., Jr. The pediatric aspects of atherosclerosis. J. Atheroscler. Res. 1969, 9, 251–265. [Google Scholar] [CrossRef]
- McMahan, C.A.; Gidding, S.S.; Malcom, G.T.; Tracy, R.E.; Strong, J.P.; McGill, H.C., Jr.; Pathobiological Determinants of Atherosclerosis in Youth Research Group. Pathobiological determinants of atherosclerosis in youth risk scores are associated with early and advanced atherosclerosis. Pediatrics 2006, 118, 1447–1455. [Google Scholar] [CrossRef] [PubMed]
- Newman, W.P., 3rd; Freedman, D.S.; Voors, A.W.; Gard, P.D.; Srinivasan, S.R.; Cresanta, J.L.; Williamson, G.D.; Webber, L.S.; Berenson, G.S. Relation of serum lipoprotein levels and systolic blood pressure to early atherosclerosis. The Bogalusa Heart Study. N. Engl. J. Med. 1986, 314, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Berenson, G.S.; Wattigney, W.A.; Tracy, R.E.; Newman, W.P., 3rd; Srinivasan, S.R.; Webber, L.S.; Dalferes, E.R., Jr.; Strong, J.P. Atherosclerosis of the aorta and coronary arteries and cardiovascular risk factors in persons aged 6 to 30 years and studied at necropsy (The Bogalusa Heart Study). Am. J. Cardiol. 1992, 70, 851–858. [Google Scholar] [CrossRef] [PubMed]
- Zieske, A.W.; Tracy, R.P.; McMahan, C.A.; Herderick, E.E.; Homma, S.; Malcom, G.T.; McGill, H.C., Jr.; Strong, J.P.; Pathobiological Determinants of Atherosclerosis in Youth Research Group. Elevated serum C-reactive protein levels and advanced atherosclerosis in youth. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1237–1243. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.K.; Sawhney, R.C.; Rai, L.; Chavan, V.D.; Dani, S.; Arora, R.C.; Selvamurthy, W.; Chopra, H.K.; Nanda, N.C. Regression of coronary atherosclerosis through healthy lifestyle in coronary artery disease patients--Mount Abu Open Heart Trial. Indian Heart J. 2011, 63, 461–469. [Google Scholar]
- Gould, K.L.; Ornish, D.; Scherwitz, L.; Brown, S.; Edens, R.P.; Hess, M.J.; Mullani, N.; Bolomey, L.; Dobbs, F.; Armstrong, W.T.; et al. Changes in myocardial perfusion abnormalities by positron emission tomography after long-term, intense risk factor modification. JAMA 1995, 274, 894–901. [Google Scholar] [CrossRef]
- Melina, V.; Craig, W.; Levin, S. Position of the Academy of Nutrition and Dietetics: Vegetarian Diets. J. Acad. Nutr. Diet. 2016, 116, 1970–1980. [Google Scholar] [CrossRef]
- Tsao, C.W.; Aday, A.W.; Almarzooq, Z.I.; Alonso, A.; Beaton, A.Z.; Bittencourt, M.S.; Boehme, A.K.; Buxton, A.E.; Carson, A.P.; Commodore-Mensah, Y.; et al. Heart Disease and Stroke Statistics-2022 Update: A Report From Cell.the American Heart Association. Circulation 2022, 145, e153–e639. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Najjar, R.S. The Impacts of Animal-Based Diets in Cardiovascular Disease Development: A Cellular and Physiological Overview. J. Cardiovasc. Dev. Dis. 2023, 10, 282. https://doi.org/10.3390/jcdd10070282
Najjar RS. The Impacts of Animal-Based Diets in Cardiovascular Disease Development: A Cellular and Physiological Overview. Journal of Cardiovascular Development and Disease. 2023; 10(7):282. https://doi.org/10.3390/jcdd10070282
Chicago/Turabian StyleNajjar, Rami Salim. 2023. "The Impacts of Animal-Based Diets in Cardiovascular Disease Development: A Cellular and Physiological Overview" Journal of Cardiovascular Development and Disease 10, no. 7: 282. https://doi.org/10.3390/jcdd10070282
APA StyleNajjar, R. S. (2023). The Impacts of Animal-Based Diets in Cardiovascular Disease Development: A Cellular and Physiological Overview. Journal of Cardiovascular Development and Disease, 10(7), 282. https://doi.org/10.3390/jcdd10070282