
Pathogenic Variants in Cardiomyopathy Disorder Genes Underlie Pediatric Myocarditis—Further Impact of Heterozygous Immune Disorder Gene Variants?

 , , , , ,
, , , , ,  , and
, and
Abstract
:1. Introduction
2. Material and Methods
2.1. Study Population
2.2. Statistical Analysis for Clinical Data
2.3. Analysis of Endomyocardial Biopsies
2.4. Next-Generation Sequencing (NGS) and Variant Calling
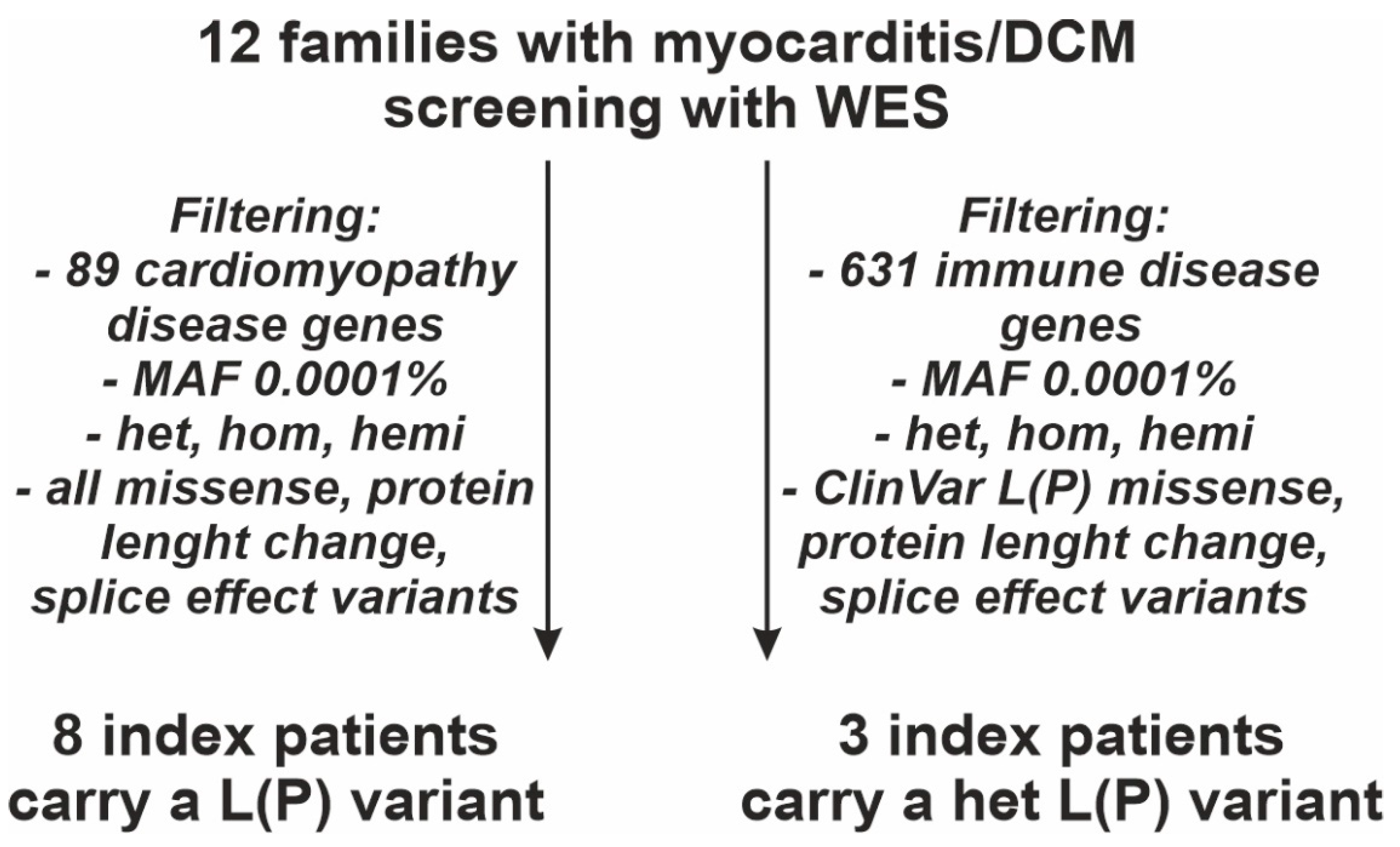
2.4.1. Gene List of 89 CMP Disease Genes
2.4.2. Gene List of 631 Immune Disease Genes
2.5. Genetic Analysis and Variant Classification
3. Results
3.1. Clinical Characterization
3.2. Identification of Genetic CMP Disease Variants
3.3. Identification of Immune Disease Gene Variants
4. Discussion
Study Limitations
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tschope, C.; Ammirati, E.; Bozkurt, B.; Caforio, A.L.P.; Cooper, L.T.; Felix, S.B.; Hare, J.M.; Heidecker, B.; Heymans, S.; Hubner, N.; et al. Myocarditis and inflammatory cardiomyopathy: Current evidence and future directions. Nat. Rev. Cardiol. 2021, 18, 169–193. [Google Scholar] [CrossRef] [PubMed]
- Canter, C.E.; Simpson, K.E. Diagnosis and treatment of myocarditis in children in the current era. Circulation 2014, 129, 115–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghelani, S.J.; Spaeder, M.C.; Pastor, W.; Spurney, C.F.; Klugman, D. Demographics, trends, and outcomes in pediatric acute myocarditis in the United States, 2006 to 2011. Circ. Cardiovasc. Qual. Outcomes 2012, 5, 622–627. [Google Scholar] [CrossRef] [Green Version]
- Messroghli, D.R.; Pickardt, T.; Fischer, M.; Opgen-Rhein, B.; Papakostas, K.; Bocker, D.; Jakob, A.; Khalil, M.; Mueller, G.C.; Schmidt, F.; et al. Toward evidence-based diagnosis of myocarditis in children and adolescents: Rationale, design, and first baseline data of MYKKE, a multicenter registry and study platform. Am. Heart J. 2017, 187, 133–144. [Google Scholar] [CrossRef]
- Schubert, S.; Opgen-Rhein, B.; Boehne, M.; Weigelt, A.; Wagner, R.; Muller, G.; Rentzsch, A.; Zu Knyphausen, E.; Fischer, M.; Papakostas, K.; et al. Severe heart failure and the need for mechanical circulatory support and heart transplantation in pediatric patients with myocarditis: Results from the prospective multicenter registry “MYKKE”. Pediatric Transplant. 2019, 23, e13548. [Google Scholar] [CrossRef]
- Seidel, F.; Opgen-Rhein, B.; Rentzsch, A.; Boehne, M.; Wannenmacher, B.; Boecker, D.; Reineker, K.; Grafmann, M.; Wiegand, G.; Hecht, T.; et al. Clinical characteristics and outcome of biopsy-proven myocarditis in children—Results of the German prospective multicentre registry “MYKKE”. Int. J. Cardiol. 2022, 357, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Ammirati, E.; Frigerio, M.; Adler, E.D.; Basso, C.; Birnie, D.H.; Brambatti, M.; Friedrich, M.G.; Klingel, K.; Lehtonen, J.; Moslehi, J.J.; et al. Management of Acute Myocarditis and Chronic Inflammatory Cardiomyopathy: An Expert Consensus Document. Circ. Heart Fail. 2020, 13, e007405. [Google Scholar] [CrossRef] [PubMed]
- Al-Wakeel-Marquard, N.; Degener, F.; Herbst, C.; Kuhnisch, J.; Dartsch, J.; Schmitt, B.; Kuehne, T.; Messroghli, D.; Berger, F.; Klaassen, S. RIKADA Study Reveals Risk Factors in Pediatric Primary Cardiomyopathy. J. Am. Heart Assoc. 2019, 8, e012531. [Google Scholar] [CrossRef] [Green Version]
- Lipshultz, S.E.; Sleeper, L.A.; Towbin, J.A.; Lowe, A.M.; Orav, E.J.; Cox, G.F.; Lurie, P.R.; McCoy, K.L.; McDonald, M.A.; Messere, J.E.; et al. The incidence of pediatric cardiomyopathy in two regions of the United States. N. Engl. J. Med. 2003, 348, 1647–1655. [Google Scholar] [CrossRef] [Green Version]
- Ammirati, E.; Buono, A.; Moroni, F.; Gigli, L.; Power, J.R.; Ciabatti, M.; Garascia, A.; Adler, E.D.; Pieroni, M. State-of-the-Art of Endomyocardial Biopsy on Acute Myocarditis and Chronic Inflammatory Cardiomyopathy. Curr. Cardiol. Rep. 2022, 24, 597–609. [Google Scholar] [CrossRef]
- Law, Y.M.; Lal, A.K.; Chen, S.; Cihakova, D.; Cooper, L.T., Jr.; Deshpande, S.; Godown, J.; Grosse-Wortmann, L.; Robinson, J.D.; Towbin, J.A.; et al. Diagnosis and Management of Myocarditis in Children: A Scientific Statement From the American Heart Association. Circulation 2021, 144, e123–e135. [Google Scholar] [CrossRef] [PubMed]
- Seidel, F.; Holtgrewe, M.; Al-Wakeel-Marquard, N.; Opgen-Rhein, B.; Dartsch, J.; Herbst, C.; Beule, D.; Pickardt, T.; Klingel, K.; Messroghli, D.; et al. Pathogenic Variants Associated With Dilated Cardiomyopathy Predict Outcome in Pediatric Myocarditis. Circ. Genom. Precis. Med. 2021, 14, e003250. [Google Scholar] [CrossRef] [PubMed]
- Caforio, A.L.; Pankuweit, S.; Arbustini, E.; Basso, C.; Gimeno-Blanes, J.; Felix, S.B.; Fu, M.; Helio, T.; Heymans, S.; Jahns, R.; et al. Current state of knowledge on aetiology, diagnosis, management, and therapy of myocarditis: A position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 2013, 34, 2636–2648, 2648a–2648d. [Google Scholar] [CrossRef] [PubMed]
- Kindermann, I.; Kindermann, M.; Kandolf, R.; Klingel, K.; Bultmann, B.; Muller, T.; Lindinger, A.; Bohm, M. Predictors of outcome in patients with suspected myocarditis. Circulation 2008, 118, 639–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maekawa, Y.; Ouzounian, M.; Opavsky, M.A.; Liu, P.P. Connecting the missing link between dilated cardiomyopathy and viral myocarditis: Virus, cytoskeleton, and innate immunity. Circulation 2007, 115, 5–8. [Google Scholar] [CrossRef] [Green Version]
- Trachtenberg, B.H.; Hare, J.M. Inflammatory Cardiomyopathic Syndromes. Circ. Res. 2017, 121, 803–818. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Cox, C.J.; Alvarez, K.M.; Cunningham, M.W. Cutting edge: Cardiac myosin activates innate immune responses through TLRs. J. Immunol. 2009, 183, 27–31. [Google Scholar] [CrossRef] [Green Version]
- Kontorovich, A.R.; Patel, N.; Moscati, A.; Richter, F.; Peter, I.; Purevjav, E.; Selejan, S.R.; Kindermann, I.; Towbin, J.A.; Bohm, M.; et al. Myopathic Cardiac Genotypes Increase Risk for Myocarditis. JACC Basic Transl. Sci. 2021, 6, 584–592. [Google Scholar] [CrossRef]
- Campuzano, O.; Fernandez-Falgueras, A.; Sarquella-Brugada, G.; Sanchez, O.; Cesar, S.; Mademont, I.; Allegue, C.; Mates, J.; Perez-Serra, A.; Coll, M.; et al. A Genetically Vulnerable Myocardium May Predispose to Myocarditis. J. Am. Coll. Cardiol. 2015, 66, 2913–2914. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.P.; Mason, J.W. Advances in the understanding of myocarditis. Circulation 2001, 104, 1076–1082. [Google Scholar] [CrossRef] [Green Version]
- Belkaya, S.; Kontorovich, A.R.; Byun, M.; Mulero-Navarro, S.; Bajolle, F.; Cobat, A.; Josowitz, R.; Itan, Y.; Quint, R.; Lorenzo, L.; et al. Autosomal Recessive Cardiomyopathy Presenting as Acute Myocarditis. J. Am. Coll. Cardiol. 2017, 69, 1653–1665. [Google Scholar] [CrossRef] [PubMed]
- Poller, W.; Haas, J.; Klingel, K.; Kuhnisch, J.; Gast, M.; Kaya, Z.; Escher, F.; Kayvanpour, E.; Degener, F.; Opgen-Rhein, B.; et al. Familial Recurrent Myocarditis Triggered by Exercise in Patients With a Truncating Variant of the Desmoplakin Gene. J. Am. Heart Assoc. 2020, 9, e015289. [Google Scholar] [CrossRef] [PubMed]
- Ader, F.; Surget, E.; Charron, P.; Redheuil, A.; Zouaghi, A.; Maltret, A.; Marijon, E.; Denjoy, I.; Hermida, A.; Fressart, V.; et al. Inherited Cardiomyopathies Revealed by Clinically Suspected Myocarditis: Highlights From Genetic Testing. Circ. Genom. Precis. Med. 2020, 13, e002744. [Google Scholar] [CrossRef] [PubMed]
- Reichl, K.; Kreykes, S.E.; Martin, C.M.; Shenoy, C. Desmoplakin Variant-Associated Arrhythmogenic Cardiomyopathy Presenting as Acute Myocarditis. Circ. Genom. Precis. Med. 2018, 11, e002373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Artico, J.; Merlo, M.; Delcaro, G.; Cannata, A.; Gentile, P.; De Angelis, G.; Paldino, A.; Bussani, R.; Ferro, M.D.; Sinagra, G. Lymphocytic Myocarditis: A Genetically Predisposed Disease? J. Am. Coll. Cardiol. 2020, 75, 3098–3100. [Google Scholar] [CrossRef]
- Carlquist, J.F.; Menlove, R.L.; Murray, M.B.; O’Connell, J.B.; Anderson, J.L. HLA class II (DR and DQ) antigen associations in idiopathic dilated cardiomyopathy. Validation study and meta-analysis of published HLA association studies. Circulation 1991, 83, 515–522. [Google Scholar] [CrossRef] [Green Version]
- Haerynck, F.; Holland, S.M.; Rosenzweig, S.D.; Casanova, J.L.; Schelstraete, P.; De Baets, F. Disseminated Mycobacterium avium infection in a patient with a novel mutation in the interleukin-12 receptor-beta1 chain. J. Pediatrics 2008, 153, 721–722. [Google Scholar] [CrossRef]
- Gorbea, C.; Makar, K.A.; Pauschinger, M.; Pratt, G.; Bersola, J.L.; Varela, J.; David, R.M.; Banks, L.; Huang, C.H.; Li, H.; et al. A role for Toll-like receptor 3 variants in host susceptibility to enteroviral myocarditis and dilated cardiomyopathy. J. Biol. Chem. 2010, 285, 23208–23223. [Google Scholar] [CrossRef] [Green Version]
- Degener, F.; Salameh, A.; Manuylova, T.; Pickardt, T.; Kostelka, M.; Daehnert, I.; Berger, F.; Messroghli, D.; Schubert, S.; Klingel, K. First paediatric cohort for the evaluation of inflammation in endomyocardial biopsies derived from congenital heart surgery. Int. J. Cardiol. 2020, 303, 36–40. [Google Scholar] [CrossRef]
- Aretz, H.T. Myocarditis: The Dallas criteria. Hum. Pathol. 1987, 18, 619–624. [Google Scholar] [CrossRef]
- Maisch, B. Cardio-Immunology of Myocarditis: Focus on Immune Mechanisms and Treatment Options. Front. Cardiovasc. Med. 2019, 6, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richardson, P.; McKenna, W.; Bristow, M.; Maisch, B.; Mautner, B.; O’Connell, J.; Olsen, E.; Thiene, G.; Goodwin, J.; Gyarfas, I.; et al. Report of the 1995 World Health Organization/International Society and Federation of Cardiology Task Force on the Definition and Classification of cardiomyopathies. Circulation 1996, 93, 841–842. [Google Scholar] [CrossRef] [PubMed]
- Kuhnisch, J.; Herbst, C.; Al-Wakeel-Marquard, N.; Dartsch, J.; Holtgrewe, M.; Baban, A.; Mearini, G.; Hardt, J.; Kolokotronis, K.; Gerull, B.; et al. Targeted panel sequencing in pediatric primary cardiomyopathy supports a critical role of TNNI3. Clin. Genet. 2019, 96, 549–559. [Google Scholar] [CrossRef] [Green Version]
- Holtgrewe, M.; Stolpe, O.; Nieminen, M.; Mundlos, S.; Knaus, A.; Kornak, U.; Seelow, D.; Segebrecht, L.; Spielmann, M.; Fischer-Zirnsak, B.; et al. VarFish: Comprehensive DNA variant analysis for diagnostics and research. Nucleic Acids Res. 2020, 48, W162–W169. [Google Scholar] [CrossRef]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [Green Version]
- Mazzarotto, F.; Tayal, U.; Buchan, R.J.; Midwinter, W.; Wilk, A.; Whiffin, N.; Govind, R.; Mazaika, E.; de Marvao, A.; Dawes, T.J.W.; et al. Reevaluating the Genetic Contribution of Monogenic Dilated Cardiomyopathy. Circulation 2020, 141, 387–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stray-Pedersen, A.; Sorte, H.S.; Samarakoon, P.; Gambin, T.; Chinn, I.K.; Coban Akdemir, Z.H.; Erichsen, H.C.; Forbes, L.R.; Gu, S.; Yuan, B.; et al. Primary immunodeficiency diseases: Genomic approaches delineate heterogeneous Mendelian disorders. J. Allergy Clin. Immunol. 2017, 139, 232–245. [Google Scholar] [CrossRef] [Green Version]
- Tangye, S.G.; Al-Herz, W.; Bousfiha, A.; Chatila, T.; Cunningham-Rundles, C.; Etzioni, A.; Franco, J.L.; Holland, S.M.; Klein, C.; Morio, T.; et al. Human Inborn Errors of Immunity: 2019 Update on the Classification from the International Union of Immunological Societies Expert Committee. J. Clin. Immunol. 2020, 40, 24–64. [Google Scholar] [CrossRef] [Green Version]
- Tangye, S.G.; Al-Herz, W.; Bousfiha, A.; Cunningham-Rundles, C.; Franco, J.L.; Holland, S.M.; Klein, C.; Morio, T.; Oksenhendler, E.; Picard, C.; et al. The Ever-Increasing Array of Novel Inborn Errors of Immunity: An Interim Update by the IUIS Committee. J. Clin. Immunol. 2021, 41, 666–679. [Google Scholar] [CrossRef]
- Hershberger, R.E.; Givertz, M.M.; Ho, C.Y.; Judge, D.P.; Kantor, P.F.; McBride, K.L.; Morales, A.; Taylor, M.R.G.; Vatta, M.; Ware, S.M. Genetic Evaluation of Cardiomyopathy-A Heart Failure Society of America Practice Guideline. J. Card. Fail. 2018, 24, 281–302. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, A.M.; Ware, J.S.; Herman, D.S.; Schafer, S.; Baksi, J.; Bick, A.G.; Buchan, R.J.; Walsh, R.; John, S.; Wilkinson, S.; et al. Integrated allelic, transcriptional, and phenomic dissection of the cardiac effects of titin truncations in health and disease. Sci. Transl. Med. 2015, 7, 270ra6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019, 47, D886–D894. [Google Scholar] [CrossRef] [PubMed]
- Gaertner, A.; Burr, L.; Klauke, B.; Brodehl, A.; Laser, K.-T.; Klingel, K.; Tiesmeier, J.; Schulz, U.; zu Knyphausen, E.; Gummert, J.; et al. Compound Heterozygous FKTN Variants in a Patient with Dilated Cardiomyopathy Led to an Aberrant a-Dystroglycan Pattern. Int. J. Mol. Sci. 2022, 23, 6685. [Google Scholar] [CrossRef]
- Rosenbaum, A.N.; Agre, K.E.; Pereira, N.L. Genetics of dilated cardiomyopathy: Practical implications for heart failure management. Nat. Rev. Cardiol. 2019, 17, 286–297. [Google Scholar] [CrossRef]
- Van Linthout, S.; Tschope, C. The Quest for Antiinflammatory and Immunomodulatory Strategies in Heart Failure. Clin. Pharmacol. 2019, 106, 1198–1208. [Google Scholar] [CrossRef] [Green Version]
- Schwabe, G.C.; Hoffmann, K.; Loges, N.T.; Birker, D.; Rossier, C.; de Santi, M.M.; Olbrich, H.; Fliegauf, M.; Failly, M.; Liebers, U.; et al. Primary ciliary dyskinesia associated with normal axoneme ultrastructure is caused by DNAH11 mutations. Hum. Mutat. 2008, 29, 289–298. [Google Scholar] [CrossRef]
- Chaudhry, B.; Henderson, D.J. Cilia, mitochondria, and cardiac development. J. Clin. Investig. 2019, 129, 2666–2668. [Google Scholar] [CrossRef] [Green Version]
- Wheway, G.; Mitchison, H.M.; Genomics England Research Consortium. Opportunities and Challenges for Molecular Understanding of Ciliopathies—The 100,000 Genomes Project. Front. Genet. 2019, 10, 127. [Google Scholar] [CrossRef] [Green Version]
- Douanne, T.; Stinchcombe, J.C.; Griffiths, G.M. Teasing out function from morphology: Similarities between primary cilia and immune synapses. J. Cell Biol. 2021, 220, e202102089. [Google Scholar] [CrossRef]
- Fassad, M.R.; Shoemark, A.; Legendre, M.; Hirst, R.A.; Koll, F.; le Borgne, P.; Louis, B.; Daudvohra, F.; Patel, M.P.; Thomas, L.; et al. Mutations in Outer Dynein Arm Heavy Chain DNAH9 Cause Motile Cilia Defects and Situs Inversus. Am. J. Hum. Genet. 2018, 103, 984–994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olcese, C.; Patel, M.P.; Shoemark, A.; Kiviluoto, S.; Legendre, M.; Williams, H.J.; Vaughan, C.K.; Hayward, J.; Goldenberg, A.; Emes, R.D.; et al. X-linked primary ciliary dyskinesia due to mutations in the cytoplasmic axonemal dynein assembly factor PIH1D3. Nat. Commun. 2017, 8, 14279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patir, A.; Fraser, A.M.; Barnett, M.W.; McTeir, L.; Rainger, J.; Davey, M.G.; Freeman, T.C. The transcriptional signature associated with human motile cilia. Sci. Rep. 2020, 10, 10814. [Google Scholar] [CrossRef] [PubMed]
- Pala, R.; Jamal, M.; Alshammari, Q.; Nauli, S.M. The Roles of Primary Cilia in Cardiovascular Diseases. Cells 2018, 7, 233. [Google Scholar] [CrossRef] [Green Version]
- Villalobos, E.; Criollo, A.; Schiattarella, G.G.; Altamirano, F.; French, K.M.; May, H.I.; Jiang, N.; Nguyen, N.U.N.; Romero, D.; Roa, J.C.; et al. Fibroblast Primary Cilia Are Required for Cardiac Fibrosis. Circulation 2019, 139, 2342–2357. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| n = 12 | |
|---|---|
| General information | |
| Male | 5 (42) |
| Age (years) | 1.6 (0.8–8.0) |
| BSA (m2) | 0.5 (0.3–0.9) |
| Symptoms | |
| NYHA | |
| I | 0 (0) |
| II | 1 (8) |
| III | 1 (8) |
| IV | 10 (84) |
| Angina pectoris | 1 (8) |
| Decompensation | 12 (100) |
| Gastrointestinal symptoms | 6 (50) |
| Infection (<6 weeks) | 7 (58) |
| Fever (<6 weeks) | 3 (25) |
| ECG | |
| ST-elevation | 0 (0) |
| T-inversion | 8 (67) |
| Arrhythmias * | 5 (42) |
| Laboratory | |
| NT-proBNP (pg/mL) (N = 8) | 23.025 (10.447–39.612) |
| Troponin elevated (N = 9) | 6 (67) |
| Echocardiography | |
| Z-score LVIDd | 6.6 (5.4–8.0) |
| LVEF (%) | 23 (21–30) |
| Endomyocardial Biopsy | |
| Myocardial virus detection | 6 (50) |
| Diagnosis EMB | |
| Acute myocarditis | 1 (8) |
| Chronic healing myocarditis | 8 (67) |
| Unspecific macrophages dominated inflammation | 1 (8) |
| Unspecific macrophages dominated inflammation & DCM | 2 (17) |
| Medical treatment | |
| Heart failure medication | 13 (100) |
| Inotropic medication | 13 (100) |
| Immunoglobulin | 5 (42) |
| Valganciclovir/Ganciclovir | 1 (8) |
| Azathioprine/Prednisolone | 0 (0) |
| Devices | |
| ICD | 1 (8) |
| Pacemaker | 0 (0) |
| VAD | 10 (83) |
| ECMO | 0 (0) |
| Weaned overall (N = 11) | 1 (8) |
| Complications | |
| Resuscitation | 3 (25) |
| HTx | 8 (67) |
| Death | 0 (0) |
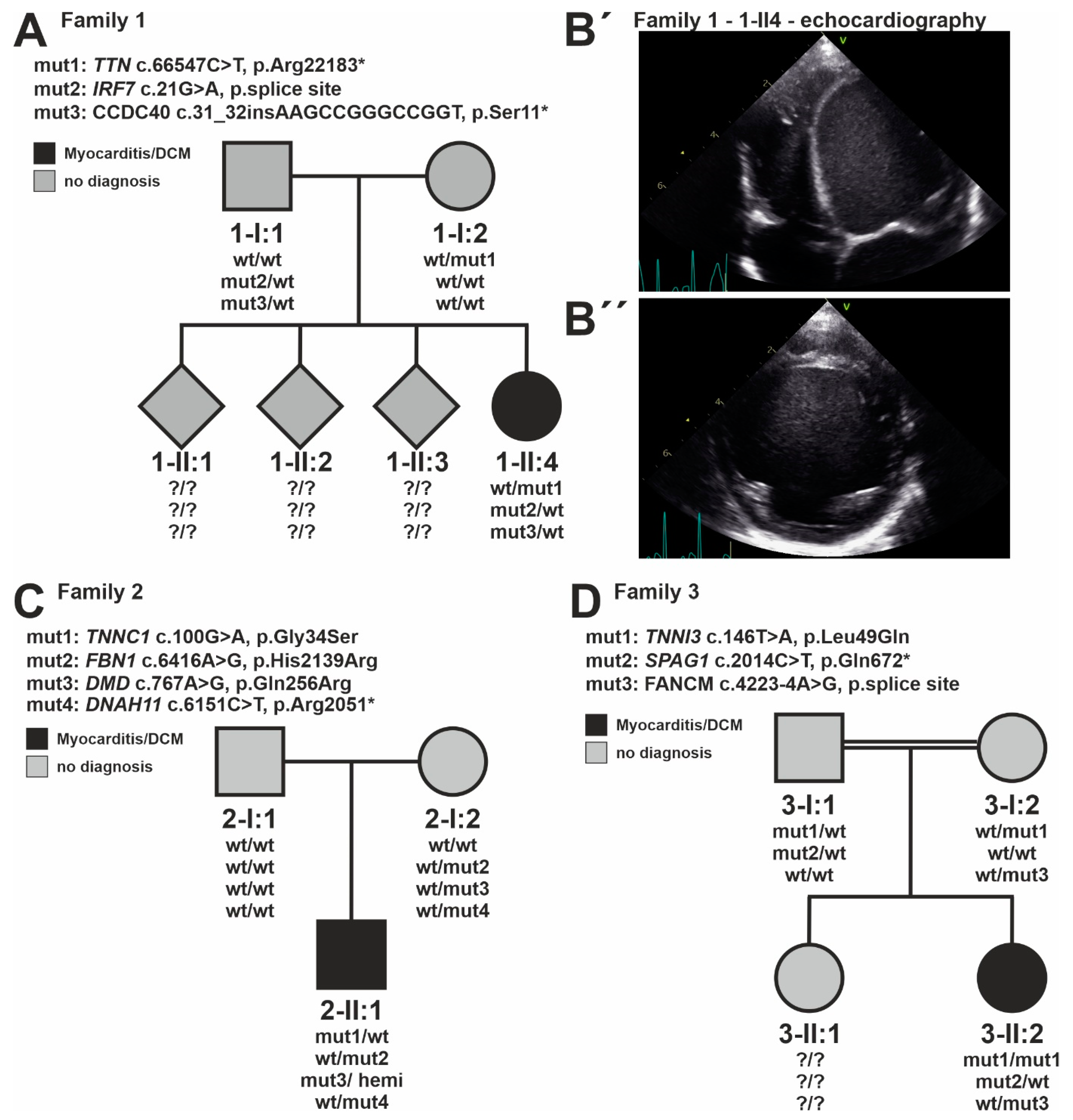
| Patient | Gene | Gene Class | Transcript | cDNA Position | Protein Position | Geno-Type | Consequence | ClinVar Annotation | Frequency GnomAD (Exomes) | CADD Value | ACMG Evaluation |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Family #1 | TTNAD | CMP | ENSG00000155657 | c.66547C > T | p.R22183* | het | stop gain | VCV000223326 (2LP, 4P) | 0.0000040 | 67.0 | LP |
| IRF7AR | IMMUNE | ENSG00000185507 | c.21G > A | p.= | het | splice region | no | 0 | 5.8 | VUS | |
| CCDC40AR | IMMUNE | ENSG00000141519 | c.31_32insAAGCCGGGCCGGT | p.S11* | het | stop gain | no | 0.0000308 | 21.9 | VUS | |
| Family #2 | TNNC1AD | CMP | ENSG00000114854 | c.100G > A | p.G34S | het | de novo, missense | no | 0 | 25.6 | LP |
| FBN1AD | CMP | ENSG00000166147 | c.6416A > G | p.H2139R | het | missense | VCV000200082 (4VUS) | 0.000008 | 23.1 | VUS | |
| DMDXLD,XLR | CMP | ENSG00000198947 | c.767A > G | p.Q256R | hemi | missense | no | 0.000066 | 23.6 | VUS | |
| DNAH11AR | IMMUNE | ENSG00000105877 | c.6151C > T | p.R2051* | het | stop gain | VCV000454692 (1P) | 0.000017 | 40.0 | LP (het) | |
| Family #3 | TNNI3 AD | CMP | ENSG00000129991 | c.146T > A | p.L49Q | hom | missense | no | 0 | 29.2 | LP |
| SPAG1 AR | IMMUNE | ENSG00000104450 | c.2014C > T | p.Q672* | het | stop gain | VCV000088683 (1LP, 5P) | 0.000082 | 38.0 | LP (het) | |
| FANCMAR | IMMUNE | ENSG00000187790 | c.4223-4A > G | p.? | het | splice region | VCV001104556 (1VUS, 1LB) | 0.000058 | 8.3 | VUS | |
| Family #4 | MYH7 AD | CMP | ENSG00000092054 | c.644C > T | p.T215I | het | de novo, missense | VCV000837897 (1VUS) | 0 | 24.8 | LP |
| Family #5 | MYLK2 AD | CMP | ENSG00000101306 | c.266G > T | p.G89V | het | missense | no | 0 | 23.4 | VUS |
| SGCG AR | CMP | ENSG00000102683 | c.631A > G | p.I211V | het | missense | no | 0 | 0.17 | VUS | |
| Family #6 | DNAH9 AR | IMMUNE | ENSG00000007174 | c.10479C > T | p.= | het | splice region | no | 0.000079 | 6.5 | VUS |
| Family #7 | TNNI3 AD | CMP | ENSG00000129991 | c.544G > C | p.E182Q | het | de novo, missense | no | 0 | 23.9 | LP |
| Family #8 | MYH7 AD | CMP | ENSG00000092054 | c.1633G > A | p.D545N | het | missense | VCV000264607 (2VUS, 1LP, 3P) | 0 | 26.1 | LP |
| MYH7 AD | CMP | ENSG00000092054 | c.2863G > A | p.D955N | het | missense | VCV000264608 (2VUS, 1LP, 3P) | 0 | 31.0 | VUS | |
| ATRX XLD,XLR | IMMUNE | ENSG00000085224 | c.6871A > G | p.I2291V | hemi | missense | VCV000210499 (1B, 3LB, 1VUS) | 0 | 16.4 | VUS | |
| PIH1D3 XLR | IMMUNE | ENSG00000080572 | c.333G > A | p.= | hemi | splice region | no | 0 | 6.9 | VUS | |
| Family #9 | TTNAD | CMP | ENSG00000155657 | c.24597C > A | p.Y8199* | het | stop gain | no | 0 | 44.0 | LP |
| FANCCAR | IMMUNE | ENSG00000158169 | c.349_360del | p.V117_H120del | het | inframe deletion | VCV000970500 (2VUS) | 0 | 19.2 | VUS | |
| Family #10 [44] | FKTNAR | CMP | ENSG00000106692 | c.895A > C | p.S299R | het | missense | VCV000264590 (2VUS) | 0.000008 | 29.1 | VUS |
| FKTNAR | CMP | ENSG00000106692 | c.1325A > G | p.N442S | het | missense | VCV001022112 (1VUS) | 0.000004 | 25.2 | VUS | |
| FANCCAR | IMMUNE | ENSG00000158169 | c.1642C > T | p.R548* | het | stop gain | VCV000012047 (1LP, 13P) | 0.000025 | 36.0 | LP (het) | |
| FLNAXLR;XLD | IMMUNE | ENSG00000196924 | c.49C > G | p.P17A | hemi | missense | VCV000393067 (1VUS) | 0.000014 | 15.9 | VUS | |
| RAC2 AD,AR | IMMUNE | ENSG00000128340 | c.*2 + 1del | p.= | het | splice region | no | 0 | 31 | VUS | |
| Family #11 | RYR2AD | CMP | ENSG00000198626 | c.3265G > A | p.E1089K | het | missense | VCV000180493 (1VUS, 1LP) | 0.000008 | 27.2 | LP |
| PLGAD,AR | IMMUNE | ENSG00000122194 | c.1675C > T | p.Q559* | het | stop gain | no | 0 | 36.0 | VUS | |
| Family #12 | SDHA AD,AR | CMP | ENSG00000073578 | c.1951G > A | p.E651K | het | missense | VCV000220782 (2VUS) | 0.000019 | 21.9 | VUS |
| DSC2 AD,AR | CMP | ENSG00000134755 | c.1309G > C | p.V437L | het | missense | VCV000925401 (1VUS) | 0 | 22.8 | VUS | |
| RBCK1AR | IMMUNE | ENSG00000125826 | c.583_584dup | p.A196Efs* 39 | het | stop gain | no | 0 | 32.0 | VUS |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seidel, F.; Laser, K.T.; Klingel, K.; Dartsch, J.; Theisen, S.; Pickardt, T.; Holtgrewe, M.; Gärtner, A.; Berger, F.; Beule, D.; et al. Pathogenic Variants in Cardiomyopathy Disorder Genes Underlie Pediatric Myocarditis—Further Impact of Heterozygous Immune Disorder Gene Variants? J. Cardiovasc. Dev. Dis. 2022, 9, 216. https://doi.org/10.3390/jcdd9070216
Seidel F, Laser KT, Klingel K, Dartsch J, Theisen S, Pickardt T, Holtgrewe M, Gärtner A, Berger F, Beule D, et al. Pathogenic Variants in Cardiomyopathy Disorder Genes Underlie Pediatric Myocarditis—Further Impact of Heterozygous Immune Disorder Gene Variants? Journal of Cardiovascular Development and Disease. 2022; 9(7):216. https://doi.org/10.3390/jcdd9070216
Chicago/Turabian StyleSeidel, Franziska, Kai Thorsten Laser, Karin Klingel, Josephine Dartsch, Simon Theisen, Thomas Pickardt, Manuel Holtgrewe, Anna Gärtner, Felix Berger, Dieter Beule, and et al. 2022. "Pathogenic Variants in Cardiomyopathy Disorder Genes Underlie Pediatric Myocarditis—Further Impact of Heterozygous Immune Disorder Gene Variants?" Journal of Cardiovascular Development and Disease 9, no. 7: 216. https://doi.org/10.3390/jcdd9070216
APA StyleSeidel, F., Laser, K. T., Klingel, K., Dartsch, J., Theisen, S., Pickardt, T., Holtgrewe, M., Gärtner, A., Berger, F., Beule, D., Milting, H., Schubert, S., Klaassen, S., & Kühnisch, J. (2022). Pathogenic Variants in Cardiomyopathy Disorder Genes Underlie Pediatric Myocarditis—Further Impact of Heterozygous Immune Disorder Gene Variants? Journal of Cardiovascular Development and Disease, 9(7), 216. https://doi.org/10.3390/jcdd9070216