Evaluation of a Novel Mitochondrial Pan-Mucorales Marker for the Detection, Identification, Quantification, and Growth Stage Determination of Mucormycetes

 ,
, 
 ,
,  , and
, and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Strain Collection and Cultivation
2.2. rnl Marker Design
2.3. rnl Real-Time PCR (Pan-Mucorales Marker)
2.4. rnl Real-Time PCR-HRM (Species-ID)
2.5. DNA Extraction and rnl/tef qPCR (Quantitative PCR)
2.6. rnl-tef qPCR (Growth Stage Determination)
2.7. rnl Real-Time PCR-HRM Evaluation of Pure Cultures
2.8. rnl Real-Time PCR-HRM Evaluation of Assay Performance in Samples Simulating the Human DNA Background of Clinical Specimens
2.9. rnl Real-Time PCR-HRM Evaluation of Assay Performance in Clinical Sample Mimics
2.10. Seminested rnl Real-Time PCR-HRM for Species ID in Clinical Sample Mimics
2.11. Paraffin-Embedded Tissue (FFPE) of Patients with Proven Fungal Infection and Suspected Mucormycosis
3. Results
3.1. rnl Marker
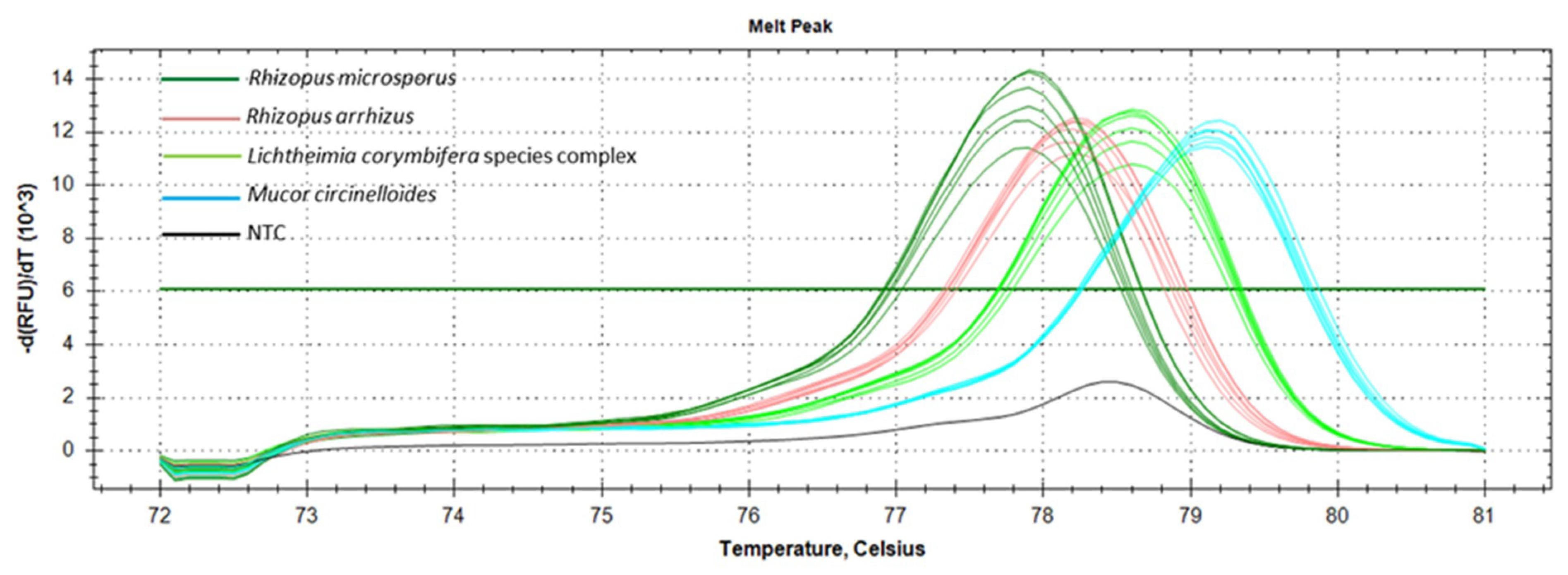
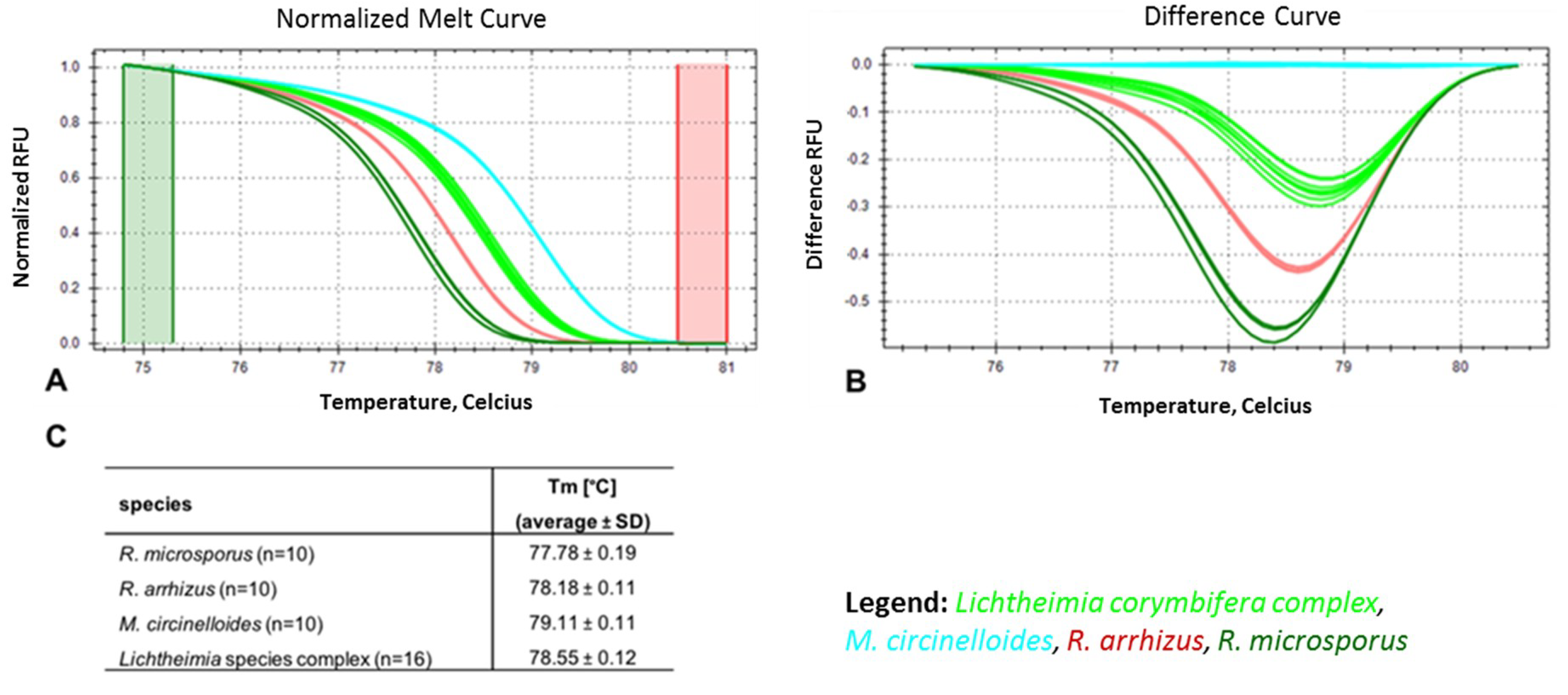
3.2. Marker Performance in Pure Cultures
3.3. Rnl Marker Evaluation with Human DNA Background
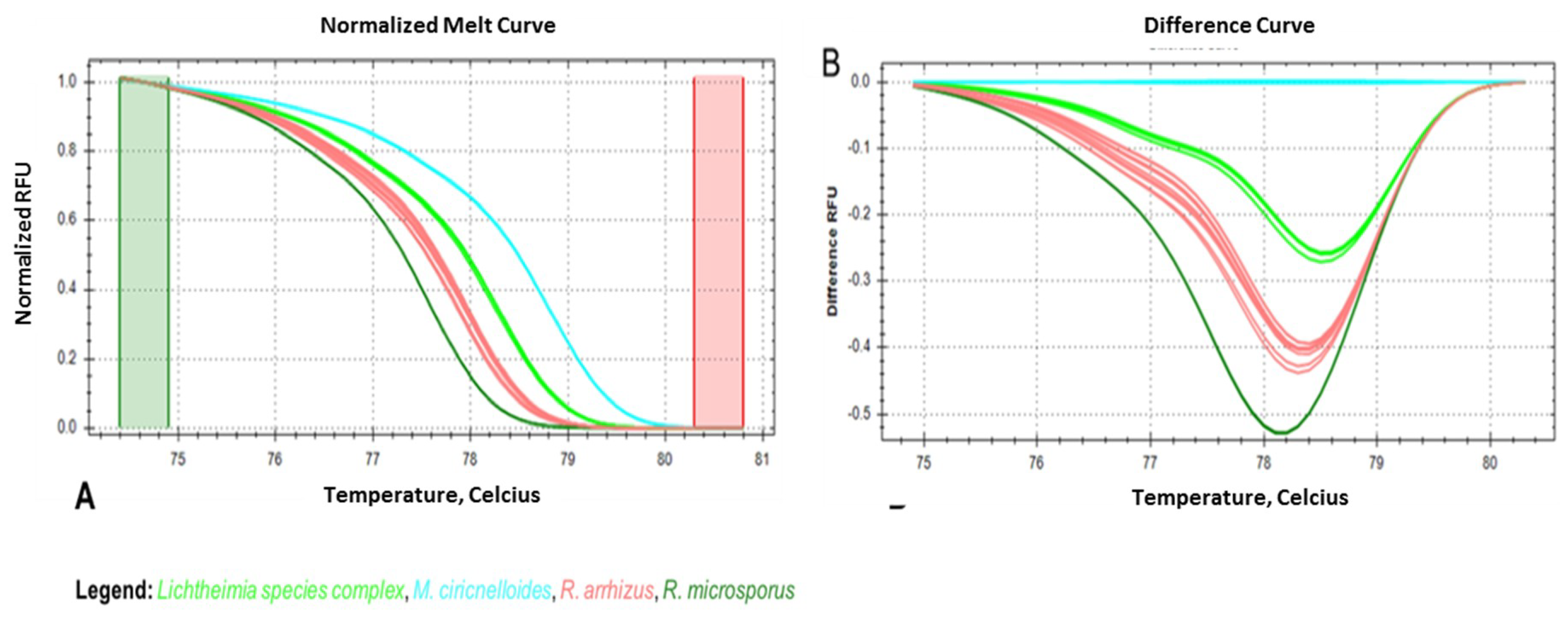
3.4. Seminested Real-Time PCR-HRM for Conidia-Spiked, EDTA-Treated Human Blood Samples
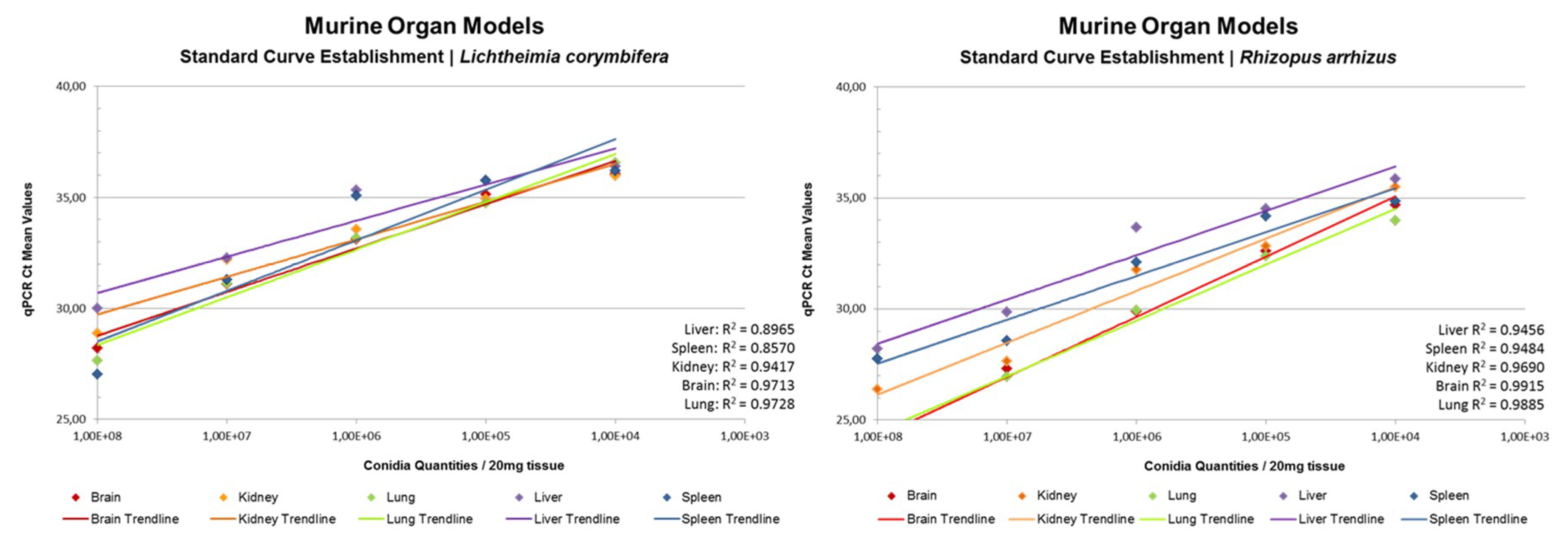
3.5. rnl Real-Time qPCR for the Detection of Fungal Burden in Murine Organs
3.6. Growth Stage Determination Using rnl/tef qPCR
3.7. rnl as a Pan-Mucorales Marker in FFPE Tissue Samples
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hibbett, D.S.; Binder, M.; Bischoff, J.F.; Blackwell, M.; Cannon, P.F.; Eriksson, O.E.; Huhndorf, S.; James, T.; Kirk, P.M.; Lücking, R.; et al. A higher-level phylogenetic classification of the fungi. Mycol. Res. 2007, 111, 509–547. [Google Scholar] [CrossRef] [PubMed]
- Gleissner, B.; Schilling, A.; Anagnostopolous, I.; Siehl, I.; Thiel, E. Improved outcome of zygomycosis in patients with hematological diseases? Leuk. Lymphoma 2004, 45, 1351–1360. [Google Scholar] [CrossRef] [PubMed]
- Kontoyiannis, D.P.; Lewis, R.E.; Lortholary, O.; Spellberg, B.; Petrikkos, G.; Roilides, E.; Ibrahim, A.; Walsh, T.J. Future directions in mucormycosis research. Clin. Infect. Dis. 2012, 54, S79–S85. [Google Scholar] [CrossRef] [PubMed]
- Boucher, H.W.; Groll, A.H.; Chiou, C.C.; Walsh, T.J. Newer systemic antifungal agents. Drugs 2004, 64, 1997–2020. [Google Scholar] [CrossRef] [PubMed]
- Millon, L.; Scherer, E.; Rocchi, S.; Bellanger, A.-P. Molecular strategies to diagnose mucormycosis. J. Fungi 2019, 5, 24. [Google Scholar] [CrossRef] [PubMed]
- Dannaoui, E. Molecular tools for identification of zygomycetes and the diagnosis of zygomycosis. Clin. Microbiol. Infect. 2009, 15, 66–70. [Google Scholar] [CrossRef] [PubMed]
- Richardson, M.D.; Cuenca-Estrella, M.; Maertens, J. An introduction to current standards of care in invasive fungal disease. J. Antimicrob. Chemother. 2019, 74, ii2. [Google Scholar] [CrossRef] [PubMed]
- Paiva, J.-A.; Pereira, J.M. Biomarkers of fungal lung infection. Curr. Opin. Infect. Dis. 2019, 32, 136–142. [Google Scholar] [CrossRef]
- Normand, A.-C.; Cassagne, C.; Gautier, M.; Becker, P.; Ranque, S.; Hendrickx, M.; Piarroux, R. Decision criteria for maldi-tof ms-based identification of filamentous fungi using commercial and in-house reference databases. BMC Microbiol. 2017, 17, 25. [Google Scholar] [CrossRef]
- Mery, A.; Sendid, B.; François, N.; Cornu, M.; Poissy, J.; Guerardel, Y.; Poulain, D. Application of mass spectrometry technology to early diagnosis of invasive fungal infections. J. Clin. Microbiol. 2016, 54, 2786–2797. [Google Scholar] [CrossRef]
- Hata, D.J.; Buckwalter, S.P.; Pritt, B.S.; Roberts, G.D.; Wengenack, N.L. Real-time pcr method for detection of zygomycetes. J. Clin. Microbiol. 2008, 46, 2353–2358. [Google Scholar] [CrossRef] [PubMed]
- Baldin, C.; Soliman, S.S.M.; Jeon, H.H.; Alkhazraji, S.; Gebremariam, T.; Gu, Y.; Bruno, V.M.; Cornely, O.A.; Leather, H.L.; Sugrue, M.W.; et al. Pcr-based approach targeting Mucorales-specific gene family for diagnosis of mucormycosis. J. Clin. Microbiol. 2018, 56, e00746-18. [Google Scholar] [CrossRef] [PubMed]
- Gebremariam, T.; Liu, M.; Luo, G.; Bruno, V.; Phan, Q.T.; Waring, A.J.; Edwards, J.E., Jr.; Filler, S.G.; Yeaman, M.R.; Ibrahim, A.S. Coth3 mediates fungal invasion of host cells during mucormycosis. J. Clin. Investig. 2014, 124, 237–250. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, P.; Bretagne, S.; Gantier, J.-C.; Garcia-Hermoso, D.; Lortholary, O.; Dromer, F.; Dannaoui, E. Molecular identification of zygomycetes from culture and experimentally infected tissues. J. Clin. Microbiol. 2006, 44, 340–349. [Google Scholar] [CrossRef] [PubMed]
- Merheb, M.; Matar, R.; Hodeify, R.; Siddiqui, S.S.; Vazhappilly, C.G.; Marton, J.; Azharuddin, S.; AL Zouabi, H. Mitochondrial DNA, a powerful tool to decipher ancient human civilization from domestication to music, and to uncover historical murder cases. Cells 2019, 8, 433. [Google Scholar] [CrossRef] [PubMed]
- Shokolenko, I.; LeDoux, S.; Wilson, G.; Alexeyev, M. Mitochondrial DNA damage, repair, degradation and experimental approaches to studying these phenomena. In DNA Repair-on the Pathways to Fixing DNA Damage and Errors; IntechOpen: London, UK, 2011. [Google Scholar]
- Lackner, M.; Caramalho, R.; Lass-Flörl, C. Laboratory diagnosis of mucormycosis: Current status and future perspectives. Future Microbiol. 2014, 9, 683–695. [Google Scholar] [CrossRef] [PubMed]
- Caramalho, R.; Maurer, E.; Binder, U.; Araújo, R.; Dolatabadi, S.; Lass-Flörl, C.; Lackner, M. Etest cannot be recommended for in vitro susceptibility testing of Mucorales. Antimicrob. Agents Chemother. 2015, 59, 3663–3665. [Google Scholar] [CrossRef] [PubMed]
- Wöstemeyer, J. Strain-dependent variation in ribosomal DNA arrangement in absidia glauca. Eur. J. Biochem. 1985, 146, 443–448. [Google Scholar] [CrossRef] [PubMed]
- White, T.J.; Bruns, T.; Lee, S.; Taylor, J. Pcr Protocols Amplification and Direct Sequencing of Fungal Ribosomal Rna Genes for Phylogenetics; Elsevier: Amsterdam, The Netherland, 1990. [Google Scholar]
- Möller, E.M.; Bahnweg, G.; Sandermann, H.; Geiger, H.H. A simple and efficient protocol for isolation of high molecular weight DNA from filamentous fungi, fruit bodies, and infected plant tissues. Nucleic Acids Res. 1992, 20, 6115–6116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caramalho, R. Mucorales Identification and Detection of Azole Resistance. Ph.D. Thesis, Medical University Innsbruck, Innrain, Austria, 2017. [Google Scholar]
- Prakash, H.; Chakrabarti, A. Global epidemiology of mucormycosis. J. Fungi 2019, 5, 26. [Google Scholar] [CrossRef] [PubMed]
- Seif, E.; Leigh, J.; Liu, Y.; Roewer, I.; Forget, L.; Lang, B.F. Comparative mitochondrial genomics in zygomycetes: Bacteria-like rnase p rnas, mobile elements and a close source of the group i intron invasion in angiosperms. Nucleic Acids Res. 2005, 33, 734–744. [Google Scholar] [CrossRef] [PubMed]
- Leung, S.-Y.; Huang, Y.; Lau, S.K.P.; Woo, P.C.Y. Complete mitochondrial genome sequence of lichtheimia ramosa (syn. Lichtheimia hongkongensis). Genome Announc. 2014, 2, e00644-14. [Google Scholar] [CrossRef] [PubMed]
- Serris, A.; Danion, F.; Lanternier, F. Disease entities in mucormycosis. J. Fungi 2019, 5, 23. [Google Scholar] [CrossRef] [PubMed]
- Klimko, N.; Khostelidi, S.; Shadrivova, O.; Volkova, A.; Popova, M.; Uspenskaya, O.; Shneyder, T.; Bogomolova, T.; Ignatyeva, S.; Zubarovskaya, L. Contrasts between mucormycosis and aspergillosis in oncohematological patients. Med. Mycol. 2019, 57, S138–S144. [Google Scholar] [CrossRef] [PubMed]
- Halliday, C.L.; Chen, S.C.A.; Kidd, S.E.; van Hal, S.; Chapman, B.; Heath, C.H.; Lee, A.; Kennedy, K.J.; Daveson, K.; Sorrell, T.C.; et al. Antifungal susceptibilities of non-aspergillus filamentous fungi causing invasive infection in australia: Support for current antifungal guideline recommendations. Int. J. Antimicrob. Agents 2016, 48, 453–458. [Google Scholar] [CrossRef] [PubMed]
- Dimopoulos, G.; Frantzeskaki, F.; Poulakou, G.; Armaganidis, A. Invasive aspergillosis in the intensive care unit. Ann. N. Y. Acad. Sci. 2012, 1272, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Cornely, O.A.; Arikan-Akdagli, S.; Dannaoui, E.; Groll, A.H.; Lagrou, K.; Chakrabarti, A.; Lanternier, F.; Pagano, L.; Skiada, A.; Akova, M.; et al. Escmid† and ecmm‡ joint clinical guidelines for the diagnosis and management of mucormycosis 2013. Clin. Microbiol. Infect. 2014, 20, 5–26. [Google Scholar] [CrossRef] [PubMed]
- Lackner, M.; Lass-Flörl, C. Commercial molecular tests for fungal diagnosis from a practical point of view. In Human Fungal Pathogen Identification; Springer: Berlin, Germany, 2017; pp. 85–105. [Google Scholar]
- Tacke, D.; Koehler, P.; Markiefka, B.; Cornely, O.A. Our 2014 approach to mucormycosis. Mycoses 2014, 57, 519–524. [Google Scholar] [CrossRef]
- Irinyi, L.; Lackner, M.; de Hoog, G.S.; Meyer, W. DNA barcoding of fungi causing infections in humans and animals. Fungal Biol. 2016, 120, 125–136. [Google Scholar] [CrossRef]
- Bernal-Martínez, L.; Buitrago, M.J.; Castelli, M.V.; Rodriguez-Tudela, J.L.; Cuenca-Estrella, M. Development of a single tube multiplex real-time pcr to detect the most clinically relevant mucormycetes species. Clin. Microbiol. Infect. 2013, 19, E1–E7. [Google Scholar] [CrossRef]
- Dannaoui, E.; Schwarz, P.; Slany, M.; Loeffler, J.; Jorde, A.T.; Cuenca-Estrella, M.; Hauser, P.M.; Shrief, R.; Huerre, M.; Freiberger, T.; et al. Molecular detection and identification of zygomycetes species from paraffin-embedded tissues in a murine model of disseminated zygomycosis: A collaborative european society of clinical microbiology and infectious diseases (escmid) fungal infection study group (efisg) evaluation. J. Clin. Microbiol. 2010, 48, 2043–2046. [Google Scholar] [PubMed]
- Hrncirova, K.; Lengerova, M.; Kocmanova, I.; Racil, Z.; Volfova, P.; Palousova, D.; Moulis, M.; Weinbergerova, B.; Winterova, J.; Toskova, M.; et al. Rapid detection and identification of mucormycetes from culture and tissue samples by use of high-resolution melt analysis. J. Clin. Microbiol. 2010, 48, 3392–3394. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, C.R.; Huang, L.; Bouchara, J.-P.; Barton, R.; Li, H.C.; Chang, T.C. Identification of medically important molds by an oligonucleotide array. J. Clin. Microbiol. 2005, 43, 3760–3768. [Google Scholar] [CrossRef] [PubMed]
- Lengerova, M.; Racil, Z.; Hrncirova, K.; Kocmanova, I.; Volfova, P.; Ricna, D.; Bejdak, P.; Moulis, M.; Pavlovsky, Z.; Weinbergerova, B.; et al. Rapid detection and identification of mucormycetes in bronchoalveolar lavage samples from immunocompromised patients with pulmonary infiltrates by use of high-resolution melt analysis. J. Clin. Microbiol. 2014, 52, 2824–2828. [Google Scholar] [CrossRef] [PubMed]
- Millon, L.; Larosa, F.; Lepiller, Q.; Legrand, F.; Rocchi, S.; Daguindau, E.; Scherer, E.; Bellanger, A.-P.; Leroy, J.; Grenouillet, F. Quantitative polymerase chain reaction detection of circulating DNA in serum for early diagnosis of mucormycosis in immunocompromised patients. Clin. Infect. Dis. 2013, 56, e95–e101. [Google Scholar] [CrossRef] [PubMed]
- Nagao, K.; Ota, T.; Tanikawa, A.; Takae, Y.; Mori, T.; Udagawa, S.-I.; Nishikawa, T. Genetic identification and detection of human pathogenic rhizopus species, a major mucormycosis agent, by multiplex pcr based on internal transcribed spacer region of rrna gene. J. Dermatol. Sci. 2005, 39, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, R.H.; Kristiansson, E.; Ryberg, M.; Hallenberg, N.; Larsson, K.-H. Intraspecific its variability in the kingdom fungi as expressed in the international sequence databases and its implications for molecular species identification. Evol. Bioinform. Online 2008, 4, 193–201. [Google Scholar] [CrossRef]
- Woo, P.C.Y.; Leung, S.-Y.; To, K.K.W.; Chan, J.F.W.; Ngan, A.H.Y.; Cheng, V.C.C.; Lau, S.K.P.; Yuen, K.-Y. Internal transcribed spacer region sequence heterogeneity in rhizopus microsporus: Implications for molecular diagnosis in clinical microbiology laboratories. J. Clin. Microbiol. 2010, 48, 208–214. [Google Scholar] [CrossRef]
- Santamaria, M.; Vicario, S.; Pappadà, G.; Scioscia, G.; Scazzocchio, C.; Saccone, C. Towards barcode markers in fungi: An intron map of ascomycota mitochondria. BMC Bioinform. 2009, 10, S15. [Google Scholar] [CrossRef]
- SEIFERT, K.A. Progress towards DNA barcoding of fungi. Mol. Ecol. Resour. 2009, 9, 83–89. [Google Scholar] [CrossRef]
- Garcia-Hermoso, D.; Hoinard, D.; Gantier, J.-C.; Grenouillet, F.; Dromer, F.; Dannaoui, E. Molecular and phenotypic evaluation of lichtheimia corymbifera (formerly absidia corymbifera) complex isolates associated with human mucormycosis: Rehabilitation of L. Ramosa. J. Clin. Microbiol. 2009, 47, 3862–3870. [Google Scholar] [CrossRef] [PubMed]
- Hjelmsø, M.H.; Hansen, L.H.; Bælum, J.; Feld, L.; Holben, W.E.; Jacobsen, C.S. High-resolution melt analysis for rapid comparison of bacterial community compositions. Appl. Environ. Microbiol. 2014, 80, 3568–3575. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.; Zorn, J.; Brielmeier, M. High-resolution melting curve analysis for identification of pasteurellaceae species in experimental animal facilities. PLoS ONE 2015, 10, e0142560. [Google Scholar] [CrossRef] [PubMed]
- Chua, K.H.; Lim, S.C.; Ng, C.C.; Lee, P.C.; Lim, Y.A.L.; Lau, T.P.; Chai, H.C. Development of high resolution melting analysis for the diagnosis of human malaria. Sci. Rep. 2015, 5, 15671. [Google Scholar] [CrossRef] [PubMed]
- Arancia, S.; Sandini, S.; De Bernardis, F.; Fortini, D. Rapid, simple, and low-cost identification of candida species using high-resolution melting analysis. Diagn. Microbiol. Infect. Dis. 2011, 69, 283–285. [Google Scholar] [CrossRef] [PubMed]
- Didehdar, M.; Khansarinejad, B.; Amirrajab, N.; Shokohi, T. Development of a high-resolution melting analysis assay for rapid and high-throughput identification of clinically important dermatophyte species. Mycoses 2016, 59, 442–449. [Google Scholar] [CrossRef] [PubMed]
- Duyvejonck, H.; Cools, P.; Decruyenaere, J.; Roelens, K.; Noens, L.; Vermeulen, S.; Claeys, G.; Decat, E.; Van Mechelen, E.; Vaneechoutte, M. Validation of high resolution melting analysis (hrm) of the amplified its2 region for the detection and identification of yeasts from clinical samples: Comparison with culture and maldi-tof based identification. PLoS ONE 2015, 10, e0132149. [Google Scholar]
- Gago, S.; Zaragoza, Ó.; Cuesta, I.; Rodríguez-Tudela, J.L.; Cuenca-Estrella, M.; Buitrago, M.J. High-resolution melting analysis for identification of the cryptococcus neoformans-cryptococcus gattii complex. J. Clin. Microbiol. 2011, 49, 3663–3666. [Google Scholar] [CrossRef] [PubMed]
- Nemcova, E.; Cernochova, M.; Ruzicka, F.; Malisova, B.; Freiberger, T.; Nemec, P. Rapid identification of medically important candida isolates using high resolution melting analysis. PLoS ONE 2015, 10, e0116940. [Google Scholar] [CrossRef]
- Guo, N.; Wang, B.; Ren, W.; Liu, M.; Chu, M.; Meng, D.; Yao, L.; Xue, W. Application of pcr and high-resolution melting for rapid identification of yeasts routinely isolated in a clinical microbiology laboratory. Ann. Clin. Lab. Sci. 2015, 45, 680–685. [Google Scholar]
- Goldschmidt, P.; Degorge, S.; Che Sarria, P.; Benallaoua, D.; Semoun, O.; Borderie, V.; Laroche, L.; Chaumeil, C. New strategy for rapid diagnosis and characterization of fungal infections: The example of corneal scrapings. PLoS ONE 2012, 7, e37660. [Google Scholar] [CrossRef] [PubMed]
- Bialek, R.; Konrad, F.; Kern, J.; Aepinus, C.; Cecenas, L.; Gonzalez, G.M.; Just-Nübling, G.; Willinger, B.; Presterl, E.; Lass-Flörl, C.; et al. Pcr based identification and discrimination of agents of mucormycosis and aspergillosis in paraffin wax embedded tissue. J. Clin. Pathol. 2005, 58, 1180–1184. [Google Scholar] [CrossRef] [PubMed]
- Millon, L.; Herbrecht, R.; Grenouillet, F.; Morio, F.; Alanio, A.; Letscher-Bru, V.; Cassaing, S.; Chouaki, T.; Kauffmann-Lacroix, C.; Poirier, P.; et al. Early diagnosis and monitoring of mucormycosis by detection of circulating DNA in serum: Retrospective analysis of 44 cases collected through the french surveillance network of invasive fungal infections (ressif). Clin. Microbiol. Infect. 2016, 22, 810-e1. [Google Scholar] [CrossRef] [PubMed]
- Bellanger, A.-P.; Berceanu, A.; Rocchi, S.; Valot, B.; Fontan, J.; Chauchet, A.; Belin, N.; Scherer, E.; Deconinck, E.; Navellou, J.-C.; et al. Development of a quantitative pcr detecting cunninghamella bertholletiae to help in diagnosing this rare and aggressive mucormycosis. Bone Marrow Transplant. 2018, 53, 1180–1183. [Google Scholar] [CrossRef] [PubMed]
- Springer, J.; Lackner, M.; Ensinger, C.; Risslegger, B.; Morton, C.O.; Nachbaur, D.; Lass-Flörl, C.; Einsele, H.; Heinz, W.J.; Loeffler, J. Clinical evaluation of a Mucorales-specific real-time pcr assay in tissue and serum samples. J. Med. Microbiol. 2016, 65, 1414–1421. [Google Scholar] [CrossRef]
- Jornayvaz, F.R.; Shulman, G.I. Regulation of mitochondrial biogenesis. Essays Biochem. 2010, 47, 69–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perfect, J.R. Fungal diagnosis: How do we do it and can we do better? Curr. Med. Res. Opin. 2013, 29, 3–11. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Histology | Routine Diagnostic PCR | Novel PCR | |||||
|---|---|---|---|---|---|---|---|
| Patient ID | Sample Type | Tissue Type | Histology/Pathology Diagnosis | Microscopy (Grocott Staining) | Pan-Fungal PCR | Culture or Pan-Fungal PCR ID | Pan-Mucorales |
| TR | histology | lung | necrotizing mucormycosis with tissue invasion | (+); mucormycosis | (-) | no ID | (-) |
| HJ | histology | lung | inflammatory reaction with macrophages of connective tissue, with embedded fungal structures, most probably mucormycetes | (+); mucormycosis | (-) | no ID | (-) |
| KA | histology | lymph node, lung | necrotizing pneumonia caused by a fungus | (-); single unidentifiable hyphae | (-) | no ID | (-) |
| VM | histology | skin ulcer | abscess building panniculitis due to sepsis with fungi, suspected mucormycosis | (+); mucormycosis | not performed | not available | (+) |
| PG | histology | lung | lung tissue with bronchiolitis obliterans with organizing pneumonia (BOOP) and fungal infection (mucormycosis) | (+); fungal elements suspected mucormycosis | not performed | Rhizopus arrhizus (culture) | (+) |
| WC | histology | jaw | necrotic, inflammatory connective tissue with evidence of fungi, correlating to a mucormycosis | (+); mucormycosis | (+) | Candida sp., Cryptococcus sp. | (+) |
| HI | histology | maxillary sinus | extensive fungal infection of sinus maxillaris | Grocott stain not performed | not performed | not available | (+) |
| ML | histology | lung | mucormycosis of lingula | (+); mucormycosis | (-) | mucormycete (culture) | (+) |
| LR | histology | lung | fungal structures | Grocott stain not performed | not performed | not available | (-) |
| EJ | histology | skin ulcer | suspected mucormycosis | (+); mucormycosis | (+) | Candida spp. | (-) |
| NN | histology | stomach | the material of ulcer and fungal structures, invasive mucormycosis | (+); no septate hyphae, mucormycosis | (+) | Rhizomucor pusillus | (+) |
| BH | histology | the soft tissue of groin | both samples fat tissue necrosis of the soft tissue | Grocott stain not performed | (+) | Rhizopus microsporus | (+) |
| BH | histology | (+) | |||||
| PN | autopsy | pleura aspirate | invasive mucormycosis with organizing pneumonia; epicardial myocarditis with fungal elements; two “Kissing” ulcers in the stomach with a fungal infection, suspected mucormycosis | (+); no septate hyphae; mucormycosis | (+) | Rhizopus arrhizus | (+) |
| GS | autopsy | lung | irreversible lung collapse due to mucormycosis | Grocott stain not performed | (+) | Lichtheimia spp. | (+) |
| WE | autopsy | lung | invasive fungal infection, suspected aspergillosis or mucormycosis | (+); no septate hyphae, mucormycosis | (+) | Rhizomucor pusillus | (+) |
| TM | autopsy | lung | fungal pneumonia, suspected aspergillosis | Grocott stain not performed | insufficient DNA quality | not available | (+) |
| BB | autopsy | lung | invasive sepsis due to generalized invasive fungal infection | (+); no septate hyphae, mucormycosis | insufficient DNA quality | Rhizopus microsporus (culture) | (-) |
| LRo | autopsy | lung | multiple infected sides, suspected mucormycosis | (+); no septate hyphae, mucormycosis | insufficient DNA quality | not available | (+) |
| SI | autopsy | lung | periphery pulmonary embolism and pulmonary infraction including fungal elements, suspected mucormycosis | (+); no septate hyphae, mucormycosis | (-) | Lichtheimia corymbifera | (+) |
| VL | autopsy | tissue of the cheek | fungal burden, mucormycosis, evidence of mixed yeast and mucormycete infection within pulmonary tissue | (+); no septate hyphae, mucormycosis | (+) | Lichtheimia corymbifera | (+) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caramalho, R.; Madl, L.; Rosam, K.; Rambach, G.; Speth, C.; Pallua, J.; Larentis, T.; Araujo, R.; Alastruey-Izquierdo, A.; Lass-Flörl, C.; et al. Evaluation of a Novel Mitochondrial Pan-Mucorales Marker for the Detection, Identification, Quantification, and Growth Stage Determination of Mucormycetes. J. Fungi 2019, 5, 98. https://doi.org/10.3390/jof5040098
Caramalho R, Madl L, Rosam K, Rambach G, Speth C, Pallua J, Larentis T, Araujo R, Alastruey-Izquierdo A, Lass-Flörl C, et al. Evaluation of a Novel Mitochondrial Pan-Mucorales Marker for the Detection, Identification, Quantification, and Growth Stage Determination of Mucormycetes. Journal of Fungi. 2019; 5(4):98. https://doi.org/10.3390/jof5040098
Chicago/Turabian StyleCaramalho, Rita, Lisa Madl, Katharina Rosam, Günter Rambach, Cornelia Speth, Johannes Pallua, Thomas Larentis, Ricardo Araujo, Ana Alastruey-Izquierdo, Cornelia Lass-Flörl, and et al. 2019. "Evaluation of a Novel Mitochondrial Pan-Mucorales Marker for the Detection, Identification, Quantification, and Growth Stage Determination of Mucormycetes" Journal of Fungi 5, no. 4: 98. https://doi.org/10.3390/jof5040098
APA StyleCaramalho, R., Madl, L., Rosam, K., Rambach, G., Speth, C., Pallua, J., Larentis, T., Araujo, R., Alastruey-Izquierdo, A., Lass-Flörl, C., & Lackner, M. (2019). Evaluation of a Novel Mitochondrial Pan-Mucorales Marker for the Detection, Identification, Quantification, and Growth Stage Determination of Mucormycetes. Journal of Fungi, 5(4), 98. https://doi.org/10.3390/jof5040098