Diversity within Aspergillus niger Clade and Description of a New Species: Aspergillus vinaceus sp. nov.

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Origin of CaM Sequences
2.2. Diversity and Phylogenetic Relationships of CaM Haplotypes
2.3. Sequencing Beta-Tubulin (BenA) and RNA Polymerase II (RPB2) Genes
2.4. Genealogical Concordance Phylogenetic Species Recognition—GCPSR
2.5. Random Amplified Length Polymorphism (RAPD)
2.6. Morphological Analysis
2.7. Secondary Metabolites Characterisation of PS3 Strains
2.8. Secondary Metabolites Characterisation of Sclerotia in PS3 Strains
3. Results and Discussion
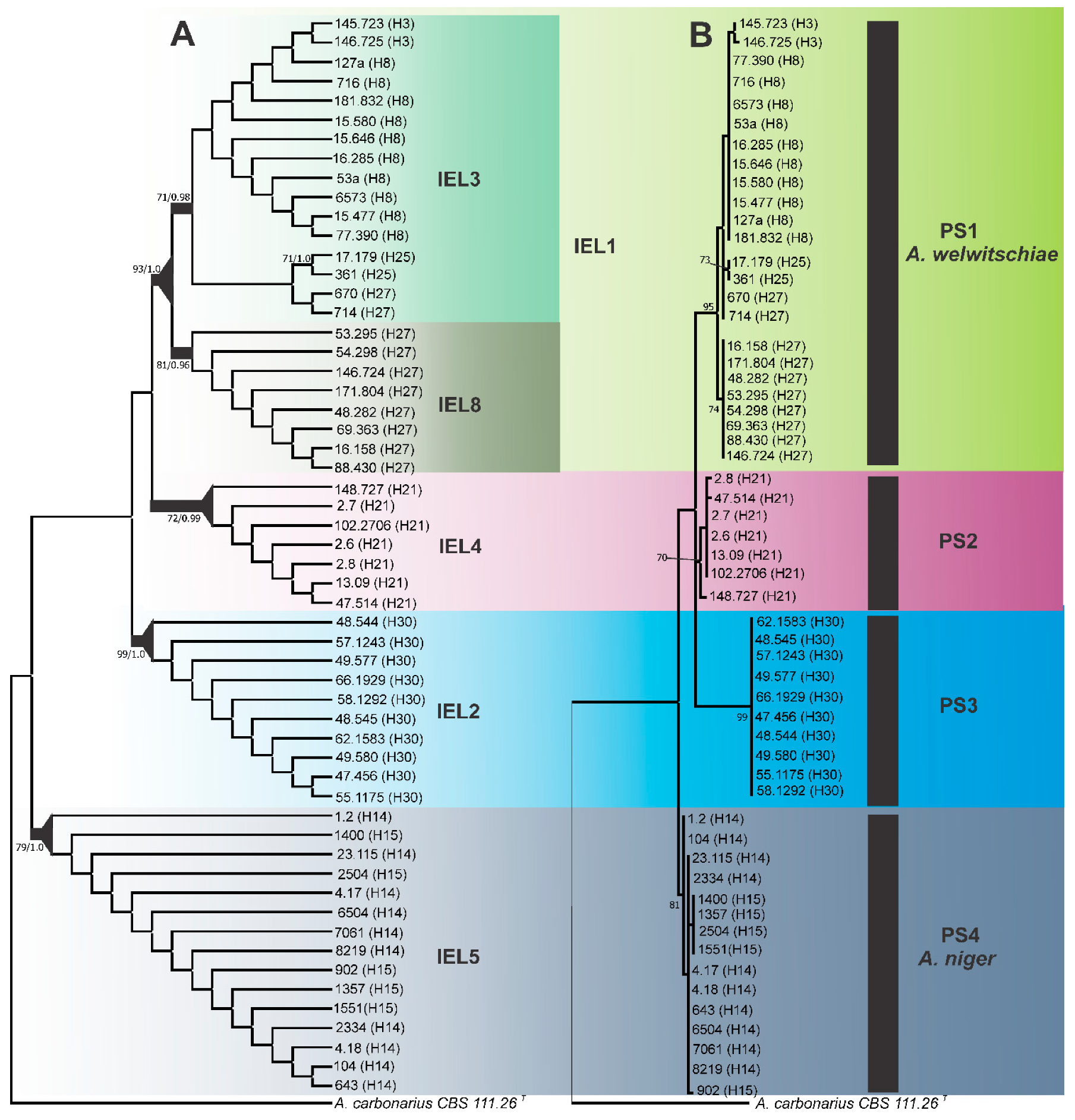
3.1. Diversity and Phylogenetic Relationships of CaM Haplotypes
3.2. Genealogical Concordance Phylogenetic Species Recognition (GCPSR)
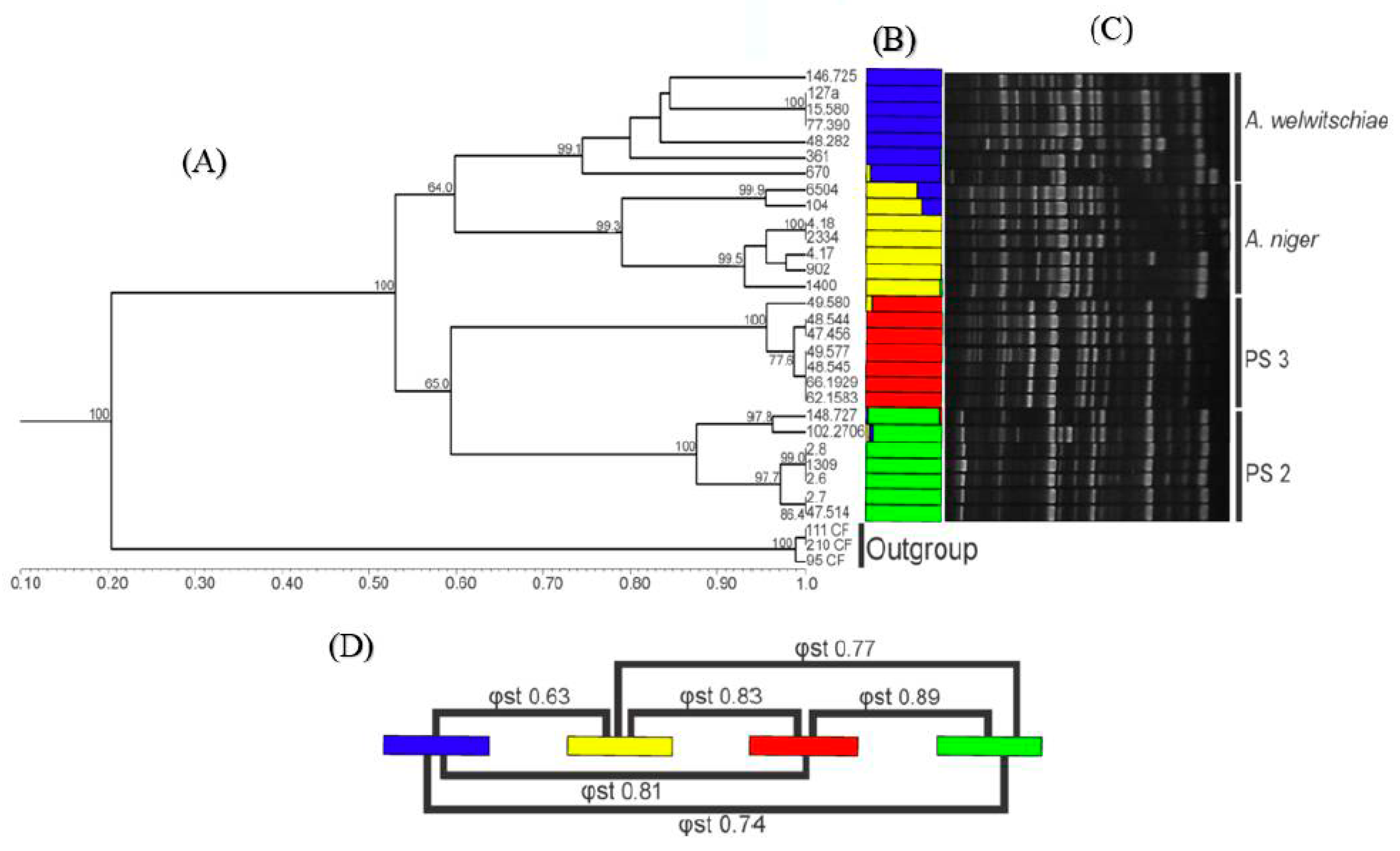
3.3. RAPD Analysis
3.4. Taxonomy
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hong, S.-B.; Lee, M.; Kim, D.-H.; Varga, J.; Frisvad, J.C.; Perrone, G.; Gomi, K.; Yamada, O.; Machida, M.; Houbraken, J.; et al. Aspergillus luchuensis, an industrially important black Aspergillus in east Asia. PLoS ONE 2013, 8, e63769. [Google Scholar] [CrossRef] [Green Version]
- Howard, S.J.; Harrison, E.; Bowyer, P.; Varga, J.; Denning, D.W. Cryptic species and azole resistance in the Aspergillus niger complex. Antimicrob. Agents Chemother. 2011, 55, 4802–4809. [Google Scholar] [CrossRef] [Green Version]
- Varga, J.; Kocsube, S.; Szigeti, G.; Baranyi, N.; Vagvolgyi, C.; Jaksic Despot, D.; Magyar, D.; Meijer, M.; Samson, R.A.; Segvic Klaric, M. Occurrence of black Aspergilli in indoor environments of six countries. Arh. Hig. Rada Toksikol. 2014, 65, 219–223. [Google Scholar] [CrossRef] [Green Version]
- Park, H.S.; Jun, S.C.; Han, K.H.; Hong, S.B.; Yu, J.H. Diversity, application, and synthetic biology of industrially important Aspergillus fungi. Adv. Appl. Microbiol. 2017, 100, 161–202. [Google Scholar]
- Raja, H.A.; Baker, T.R.; Little, J.G.; Oberlies, N.H. DNA barcoding for identification of consumer-relevant mushrooms: A partial solution for product certification? Food Chem. 2017, 214, 383–392. [Google Scholar] [CrossRef] [Green Version]
- Samson, R.A.; Visagie, C.M.; Houbraken, J.; Hong, S.B.; Hubka, V.; Klaassen, C.H.W.; Perrone, G.; Seifert, K.A.; Susca, A.; Tanney, J.B.; et al. Phylogeny, identification and nomenclature of the genus Aspergillus. Stud. Mycol. 2014, 78, 141–173. [Google Scholar] [CrossRef] [Green Version]
- Fungaro, M.H.P.; Ferranti, L.S.; Massi, F.P.; Da Silva, J.J.; Sartori, D.; Taniwaki, M.H.; Frisvad, J.C.; Iamanaka, B.T. Aspergillus labruscus sp. nov., a new species of Aspergillus section Nigri discovered in Brazil. Sci. Rep. 2017, 7, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Ferracin, L.M.; Frisvad, J.C.; Taniwaki, M.H.; Iamanaka, B.T.; Sartori, D.; Schapovaloff, M.E.; Fungaro, M.H.P. Genetic relationships among strains of the Aspergillus niger aggregate. Braz. Arch. Biol. Technol. 2009, 52, 241–248. [Google Scholar] [CrossRef]
- Ferranti, L.S.; Correa, B.; Fungaro, M.H.P.; Iamanaka, B.T.; Massi, F.P.; Phippen, C.B.W.; Frisvad, J.C.; Taniwaki, M.H. Occurrence and fumonisin B2 producing potential of Aspergillus section Nigri in Brazil nuts. Mycotoxin Res. 2017, 33, 49–58. [Google Scholar] [CrossRef]
- De Souza Ferranti, L.; Fungaro, M.H.P.; Massi, F.P.; da Silva, J.J.; Penha, R.E.S.; Frisvad, J.C.; Taniwaki, M.H.; Iamanaka, B.T. Diversity of Aspergillus section Nigri on the surface of Vitis labrusca and its hybrid grapes. Int. J. Food Microbiol. 2018, 268, 53–60. [Google Scholar] [CrossRef]
- Magnani, M.; Fernandes, T.; Prete, C.E.C.; Homechim, M.; Ono, E.Y.S.; Vilas-Boas, L.A.; Sartori, D.; Furlaneto, M.C.; Fungaro, M.H.P. Identificação molecular de Aspergillus spp. Isolados de grãos de café. Sci. Agric. 2005, 62, 45–49. [Google Scholar] [CrossRef]
- Massi, F.P.; Sartori, D.; de Souza Ferranti, L.; Iamanaka, B.T.; Taniwaki, M.H.; Vieira, M.L.C.; Fungaro, M.H.P. Prospecting for the incidence of genes involved in ochratoxin and fumonisin biosynthesis in Brazilian strains of Aspergillus niger and Aspergillus welwitschiae. Int. J. Food Microbiol. 2016, 221, 19–28. [Google Scholar] [CrossRef]
- Sartori, D.; Furlaneto, M.C.; Martins, M.K.; Ferreira de Paula, M.R.; Pizzirani-Kleiner, A.A.; Taniwaki, M.H.; Fungaro, M.H.P. PCR method for the detection of potential ochratoxin-producing Aspergillus species in coffee beans. Res. Microbiol. 2006, 157, 350–354. [Google Scholar] [CrossRef]
- Silva, J.J.; Puel, O.; Lorber, S.; Ferranti, L.S.; Ortiz, L.F.; Taniwaki, M.H.; Iamanaka, B.T.; Fungaro, M.H.P. Occurrence and diversity of Aspergillus in commercial yerba mate elaborated for the Brazilian beverage ‘chimarrão’. Food Res. Int. 2019, 121, 940–946. [Google Scholar] [CrossRef]
- Taniwaki, M.H.; Pitt, J.I.; Teixeira, A.A.; Iamanaka, B.T. The source of ochratoxin A in Brazilian coffee and its formation in relation to processing methods. Int. J. Food Microbiol. 2003, 82, 173–179. [Google Scholar] [CrossRef]
- Hong, S.B.; Cho, H.S.; Shin, H.D.; Frisvad, J.C.; Samson, R.A. Novel Neosartorya species isolated from soil in Korea. Int. J. Syst. Evol. Microbiol. 2006, 56, 477–486. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Taniwaki, M.H.; Pitt, J.I.; Iamanaka, B.T.; Sartori, D.; Copetti, M.V.; Balajee, A.; Fungaro, M.H.P.; Frisvad, J.C. Aspergillus bertholletius sp. nov. from Brazil nuts. PLoS ONE 2012, 7, e42480. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Rozas, J.; Ferrer-Mata, A.; Sanchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sanchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Kimura, M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef]
- Glass, N.L.; Donaldson, G.C. Development of primer sets designed for use with the PCR to amplify conserved genes from filamentous ascomycetes. Appl. Environ. Microbiol. 1995, 61, 1323–1330. [Google Scholar] [CrossRef] [Green Version]
- Dettman, J.R.; Jacobson, D.J.; Taylor, J.W. A multilocus genealogical approach to phylogenetic species recognition in the model eukaryote Neurospora. Evolution 2003, 57, 2703–2720. [Google Scholar] [CrossRef]
- Margush, T.; McMorris, F.R. Consensusn-trees. Bull. Math. Biol. 1981, 43, 239–244. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; Van Der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. Mrbayes 3.2: Efficient bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [Green Version]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. JModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [Green Version]
- Miller, M.A.; Pfeiffer, W.; Schwartz, T. Creating the CIPRES science gateway for inference of large phylogenetic trees. In Proceedings of the 2010 Gateway Computing Environments Workshop (GCE), New Orleans, LA, USA, 14 November 2010; pp. 1–8. [Google Scholar]
- Lanfear, R.; Frandsen, P.B.; Wright, A.M.; Senfeld, T.; Calcott, B. Partitionfinder 2: New methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol. Biol. Evol. 2017, 34, 772–773. [Google Scholar] [CrossRef] [Green Version]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Fungaro, M.H.P.; Vieira, M.L.C.; Pizzirani-Kleiner, A.A.; Azevedo, J.L. Diversity among soil and insect isolates of Metarhizium anisopliae var. anisopliae detected by RAPD. Lett. Appl. Microbiol. 1996, 22, 389–392. [Google Scholar] [CrossRef]
- Excoffier, L.; Lischer, H.E.L. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [PubMed]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [Green Version]
- Earl, D.; vonHoldt, B. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Frisvad, J.C.; Thrane, U. Standardized high-performance liquid chromatography of 182 mycotoxins and other fungal metabolites based on alkylphenone retention indices and UV-VIS spectra (diodearray detection). J. Chromatogr. A 1987, 404, 195–214. [Google Scholar] [CrossRef]
- Nielsen, K.F.; Månsson, M.; Rank, C.; Frisvad, J.C.; Larsen, T.O. Dereplication of microbial natural products by LC-DAD-TOFMS. J. Nat. Prod. 2011, 74, 2338–2348. [Google Scholar] [CrossRef]
- Carvajal-Campos, A.; Manizan, A.L.; Tadrist, S.; Akaki, D.K.; Koffi-Nevry, R.; Moore, G.G.; Fapohunda, S.O.; Bailly, S.; Montet, D.; Oswald, I.P.; et al. Aspergillus korhogoensis, a novel aflatoxin producing species from the Côte d’Ivoire. Toxins 2017, 9, 353. [Google Scholar] [CrossRef] [Green Version]
- Susca, A.; Moretti, A.; Stea, G.; Villani, A.; Haidukowski, M.; Logrieco, A.; Munkvold, G. Comparison of species composition and fumonisin production in Aspergillus section Nigri populations in maize kernels from USA and Italy. Int. J. Food Microbiol. 2014, 188, 75–82. [Google Scholar] [CrossRef]
- Taylor, J.W.; Jacobson, D.J.; Kroken, S.; Kasuga, T.; Geiser, D.M.; Hibbett, D.S.; Fisher, M.C. Phylogenetic species recognition and species concepts in fungi. Fungal Genet. Biol. 2000, 31, 21–32. [Google Scholar] [CrossRef] [Green Version]
- Avise, J.C.; Ball, J.R.M. Principles of genealogical concordance in species concepts and biological taxonomy. Oxf. Surv. Evol. Biol. 1990, 7, 45–67. [Google Scholar]
- Perrone, G.; Stea, G.; Epifani, F.; Varga, J.; Frisvad, J.C.; Samson, R.A. Aspergillus niger contains the cryptic phylogenetic species A. awamori. Fungal Biol. 2011, 115, 1138–1150. [Google Scholar] [CrossRef] [PubMed]
- D’hooge, E.; Becker, P.; Stubbe, D.; Normand, A.C.; Piarroux, R.; Hendrickx, M. Black aspergilli: A remaining challenge in fungal taxonomy? Med. Mycol. 2019, 57, 773–780. [Google Scholar] [CrossRef] [PubMed]
- Abe, A.; Asano, K.; Sone, T. A molecular phylogeny-based taxonomy of the genus Rhizopus. Biosci. Biotechnol. Biochem. 2010, 74, 1325–1331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirata, K.; Kusaba, M.; Chuma, I.; Osue, J.; Nakayashiki, H.; Mayama, S.; Tosa, Y. Speciation in Pyricularia inferred from multilocus phylogenetic analysis. Mycol. Res. 2007, 111, 799–808. [Google Scholar] [CrossRef] [Green Version]
- Laurence, M.H.; Summerell, B.A.; Burgess, L.W.; Liew, E.C.Y. Genealogical concordance phylogenetic species recognition in the Fusarium oxysporum species complex. Fungal Biol. 2014, 118, 374–384. [Google Scholar] [CrossRef]
- Liu, F.; Wang, M.; Damm, U.; Crous, P.W.; Cai, L. Species boundaries in plant pathogenic fungi: A Colletotrichum case study. BMC Evol. Biol. 2016, 16, 81. [Google Scholar] [CrossRef] [Green Version]
- O’Donnell, K.; Ward, T.J.; Geiser, D.M.; Kistler, H.C.; Aoki, T. Genealogical concordance between the mating type locus and seven other nuclear genes supports formal recognition of nine phylogenetically distinct species within the Fusarium graminearum clade. Fungal Genet. Biol. 2004, 41, 600–623. [Google Scholar] [CrossRef]
- Secor, G.A.; Rivera-Varas, V.; Christ, D.S.; Mathew, F.M.; Khan, M.F.R.; Varrelmann, M.; Bolton, M.D. Characterization of Fusarium secorum, a new species causing Fusarium yellowing decline ofsugar beet in north central USA. Fungal Biol. 2014, 118, 764–775. [Google Scholar] [CrossRef]
- Stewart, J.E.; Timmer, L.W.; Lawrence, C.B.; Pryor, B.M.; Peever, T.L. Discord between morphological and phylogenetic species boundaries: Incomplete lineage sorting and recombination results in fuzzy species boundaries in an asexual fungal pathogen. BMC Evol. Biol. 2014, 14, 38. [Google Scholar] [CrossRef]
- Visagie, C.M.; Seifert, K.A.; Houbraken, J.; Samson, R.A.; Jacobs, K. A phylogenetic revision of Penicillium sect. Exilicaulis, including nine new species from fynbos in South Africa. IMA Fungus 2016, 7, 75–117. [Google Scholar]
- Frisvad, J.C.; Hubka, V.; Ezekiel, C.N.; Hong, S.B.; Nováková, A.; Chen, A.J.; Arzanlou, M.; Larsen, T.O.; Sklenář, F.; Mahakarnchanakul, W.; et al. Taxonomy of Aspergillus section Flavi and their production of aflatoxins, ochratoxins and other mycotoxins. Stud. Mycol. 2019, 93, 1–63. [Google Scholar] [CrossRef]
- Rintoul, T.L.; Eggertson, Q.A.; Lévesque, C.A. Multigene phylogenetic analyses to delimit new species in fungal plant pathogens. Methods Mol. Biol. 2012, 835, 549–569. [Google Scholar]
- Matute, D.R.; McEwen, J.G.; Puccia, R.; Montes, B.A.; San-Blas, G.; Bagagli, E.; Rauscher, J.T.; Restrepo, A.; Morais, F.; Niño-Vega, G.; et al. Cryptic speciation and recombination in the fungus Paracoccidioides brasiliensis as revealed by gene genealogies. Mol. Biol. Evol. 2006, 23, 65–73. [Google Scholar] [CrossRef]
- Al-Sadi, A.M.; Al-Oweisi, F.A.; Edwards, S.G.; Al-Nadabi, H.; Al-Fahdi, A.M. Genetic analysis reveals diversity and genetic relationship among Trichoderma isolates from potting media, cultivated soil and uncultivated soil. BMC Microbiol. 2015, 15, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Zeng, D.F.; Chen, B. Genetic variability and bottleneck detection of four Tricholoma matsutake populations from northeastern and southwestern China. Environ. Microbiol. 2015, 17, 2870–2881. [Google Scholar] [CrossRef]
- Silva, J.J.; Viaro, H.P.; Ferranti, L.S.; Oliveira, A.L.M.; Ferreira, J.M.; Ruas, C.F.; Ono, E.Y.S.; Fungaro, M.H.P. Genetic structure of Fusarium verticillioides populations and occurrence of fumonisins in maize grown in Southern Brazil. Crop Prot. 2017, 99, 160–167. [Google Scholar] [CrossRef]
- Asadollahi, M.; Fekete, E.; Karaffa, L.; Flipphi, M.; Árnyasi, M.; Esmaeili, M.; Váczy, K.Z.; Sándor, E. Comparison of Botrytis cinerea populations isolated from two open-field cultivated host plants. Microbiol. Res. 2013, 168, 379–388. [Google Scholar] [CrossRef]
- Tsehaye, H.; Elameen, A.; Tronsmo, A.M.; Sundheim, L.; Tronsmo, A.; Assefa, D.; Brurberg, M.B. Genetic variation among Fusarium verticillioides isolates associated with Ethiopian maize kernels as revealed by AFLP analysis. Eur. J. Plant Pathol. 2016, 146, 807–816. [Google Scholar] [CrossRef]
- Liu, Q.; Xin, Y.H.; Zhou, Y.G.; Chen, W.X. Multilocus sequence analysis of homologous recombination and diversity in Arthrobacter sensu lato named species and glacier-inhabiting strains. Syst. Appl. Microbiol. 2018, 41, 23–29. [Google Scholar] [CrossRef]
- Araujo, R.; Amorim, A.; Gusmão, L. Genetic diversity of Aspergillus fumigatus in indoor hospital environments. Med. Mycol. 2010, 48, 832–838. [Google Scholar] [CrossRef] [Green Version]
- Zhan, J.; McDonald, B.A. Analytical and experimental methods for estimating population genetic structure of fungi. In The Fungal Community: Its Organization and Role in the Ecosystem; Dighton, J., White, J., Oudemans, P., Eds.; CRC Press: New York, NY, USA, 2005; pp. 241–264. [Google Scholar]
- Massi, F.P.; Vieira, M.L.C.; Sartori, D.; Penha, R.E.S.; de Freitas Munhoz, C.; Ferreira, J.M.; Iamanaka, B.T.; Taniwaki, M.H.; Frisvad, J.C.; Fungaro, M.H.P. Brazil nuts are subject to infection with B and G aflatoxin-producing fungus, Aspergillus pseudonomius. Int. J. Food Microbiol. 2014, 186, 14–21. [Google Scholar] [CrossRef]
- Frisvad, J.C.; Petersen, L.M.; Lyhne, E.K.; Larsen, T.O. Formation of sclerotia and production of indoloterpenes by Aspergillus niger and other species in section Nigri. PLoS ONE 2014, 9, e94857. [Google Scholar] [CrossRef] [Green Version]
- Frisvad, J.C.; Larsen, T.O.; De Vries, R.; Meijer, M.; Houbraken, J.; Cabañes, F.J.; Ehrlich, K.; Samson, R.A. Secondary metabolite profiling, growth profiles and other tools for species recognition and important Aspergillus mycotoxins. Stud. Mycol. 2007, 59, 31–37. [Google Scholar] [CrossRef]
- Frisvad, J.C.; Larsen, T.O.; Thrane, U.; Meijer, M.; Varga, J.; Samson, R.A.; Nielsen, K.F. Fumonisin and ochratoxin production in industrial Aspergillus niger strains. PLoS ONE 2011, 6, e23496. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 35 Cycles | ||||||
|---|---|---|---|---|---|---|
| 1 min | 45 s | 45 s | 1 min * | 5 min | ||
| Primers | Initial Denaturation | Denaturation | Annealing | Extension | Final Extension | |
| Thermal program | CaM (Hong et al. [15]) | 95 °C | 94 °C | 55 °C | 72 °C | 72 °C |
| BenA (Glass and Donaldson [17]) | 95 °C | 94 °C | 60 °C | 72 °C | 72 °C | |
| RPB2 (this study) | 95 °C | 94 °C | 58 °C | 72 °C | 72 °C | |
| PCR conditions | Taq DNA polymerase | 1 U | ||||
| PCR buffer | 1× | |||||
| Primer | 0.4 µM—each primer | |||||
| dNTP | 0.2 Mm | |||||
| MgCl2 | 1.5 Mm | |||||
| DNA template | 10 ng | |||||
| Growth Rate (mm/7 days) | ||||||
|---|---|---|---|---|---|---|
| Temperature | PS1 (A. welwitschiae) | PS2 | PS3 (A. vinaceus sp. nov.) | PS4 (A. niger) | ||
| Macromorphology | 25 °C | 63.3 ± 0.9 | 63 ± 0.8 | 65.2 ± 1.2 | 64.7 ± 0.5 | |
| CYA | 37 °C | 62.3 ± 0.8 | 61 ± 0.3 | 63.2 ± 0.4 | 62.3 ± 1.2 | |
| 42 °C | 34 ± 4.3 | 39 ± 0.2 | 23.6 ± 1.2 | 28 ± 0.8 | ||
| MEA | 25 °C | 54.2 ± 5.3 | 50 ± 0.4 | 38.2 ± 1.2 | 58.7 ± 4.7 | |
| YESA | 25 °C | 65.7 ± 2.3 | 68 ± 0.7 | 67.2 ± 0.8 | 67 ± 1.4 | |
| Sclerotia | Absent | Absent | White to cream | Absent | ||
| Conidia colour | Black | Black | Black | Black | ||
| Colour in mycelial area | Black | Black | White | Black | ||
| Micromorphology * | Vesicle shape | Globose to sub-globose | Globose to sub-globose | Globose to sub-globose | Globose to sub-globose | |
| Conidial ornamentation | Smooth to roughened | Smooth to finely roughened | Finely roughened to echinulate | Smooth to finely roughened | ||
| Vesicle size | 40.5 ± 5.2 | 45.8 ± 4.1 | 68.2 ± 8.8 | 41.7 ± 3.2 | ||
| Conidiophore size | 76.5 ± 12.7 | 86.6 ± 11.9 | 169 ± 23.1 | 82.2 ± 12.8 | ||
| Conidial size | 4.1 ± 0.6 | 4 ± 0.6 | 4.6 ± 0.6 | 3.8 ± 0.5 | ||
| Stipe width | 12.8 ± 1.6 | 14.6 ± 1.1 | 18.3 ± 2.1 | 12.4 ± 0.8 | ||
| Fungal Sample | Metabolites | Aspergillus niger Clade Species | ||
|---|---|---|---|---|
| A. vinaceus sp. nov. (PS3) | A. niger | A. welwitschiae | ||
| Whole fungal culture * | Acetyl-leucomelone | + | + | + |
| Asperazine | + | - | - | |
| Aurasperone B | + | + | + | |
| Aurasperone C | + | + | + | |
| Aurasperone F | + | + | + | |
| Fonsecin B | + | + | + | |
| Fumonisin B2 | - | + | + | |
| Fumonisin B4 | - | + | + | |
| Fumonisin B6 | - | + | + | |
| Funalenones | + | + | + | |
| HUTI | + | + | + | |
| Malformin A1 | + | + | + | |
| Malformin C | + | + | + | |
| Nigragillin | + | + | + | |
| Ochratoxin A | - | + | + | |
| Pyranonigrin A | + | + | + | |
| SURI | + | + | + | |
| Tensidol B | + | + | + | |
| Sclerotia extracts ** | 14-Epi-hydroxy-10,23-dihydro-24,25-dehydroaflavinine | + | - | - |
| 10,23-Dihydro-24,25-dehydroaflavinine | + | - | - | |
| 10,23-Dihydro-24,25-dehydro-21-oxo-aflavinine | + | - | - | |
| Aurasperone B | + | - | - | |
| Aurasperone C | + | - | - | |
| Aurasperone F | + | - | - | |
| Fonsecin | + | - | - | |
| Fonsecin B | + | - | - | |
| Funalenone | + | - | - | |
| Malformins A1, A2 and A4 | + | - | - | |
| Malformin C | + | - | - | |
| Nigragillin | + | - | - | |
| Pyrophen | + | - | - | |
| Tensidol A | + | - | - | |
| Tensidol B | + | - | - | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Silva, J.J.d.; Iamanaka, B.T.; Ferranti, L.S.; Massi, F.P.; Taniwaki, M.H.; Puel, O.; Lorber, S.; Frisvad, J.C.; Fungaro, M.H.P. Diversity within Aspergillus niger Clade and Description of a New Species: Aspergillus vinaceus sp. nov. J. Fungi 2020, 6, 371. https://doi.org/10.3390/jof6040371
Silva JJd, Iamanaka BT, Ferranti LS, Massi FP, Taniwaki MH, Puel O, Lorber S, Frisvad JC, Fungaro MHP. Diversity within Aspergillus niger Clade and Description of a New Species: Aspergillus vinaceus sp. nov. Journal of Fungi. 2020; 6(4):371. https://doi.org/10.3390/jof6040371
Chicago/Turabian StyleSilva, Josué J. da, Beatriz T. Iamanaka, Larissa S. Ferranti, Fernanda P. Massi, Marta H. Taniwaki, Olivier Puel, Sophie Lorber, Jens C. Frisvad, and Maria Helena P. Fungaro. 2020. "Diversity within Aspergillus niger Clade and Description of a New Species: Aspergillus vinaceus sp. nov." Journal of Fungi 6, no. 4: 371. https://doi.org/10.3390/jof6040371
APA StyleSilva, J. J. d., Iamanaka, B. T., Ferranti, L. S., Massi, F. P., Taniwaki, M. H., Puel, O., Lorber, S., Frisvad, J. C., & Fungaro, M. H. P. (2020). Diversity within Aspergillus niger Clade and Description of a New Species: Aspergillus vinaceus sp. nov. Journal of Fungi, 6(4), 371. https://doi.org/10.3390/jof6040371