


Does Supramolecular Gelation Require an External Trigger?

 , , ,
, , ,  , and
, and 
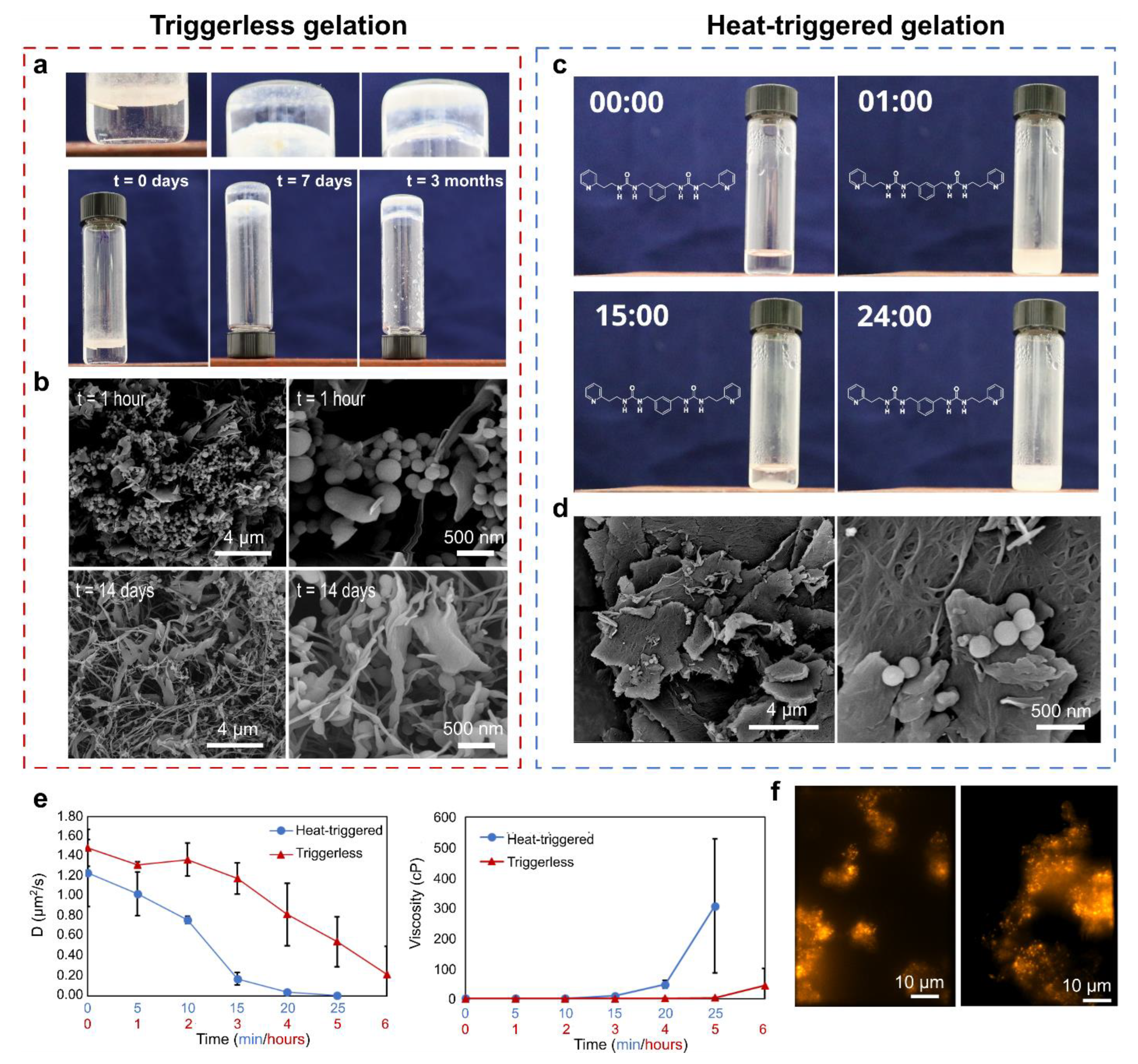{kind=link}
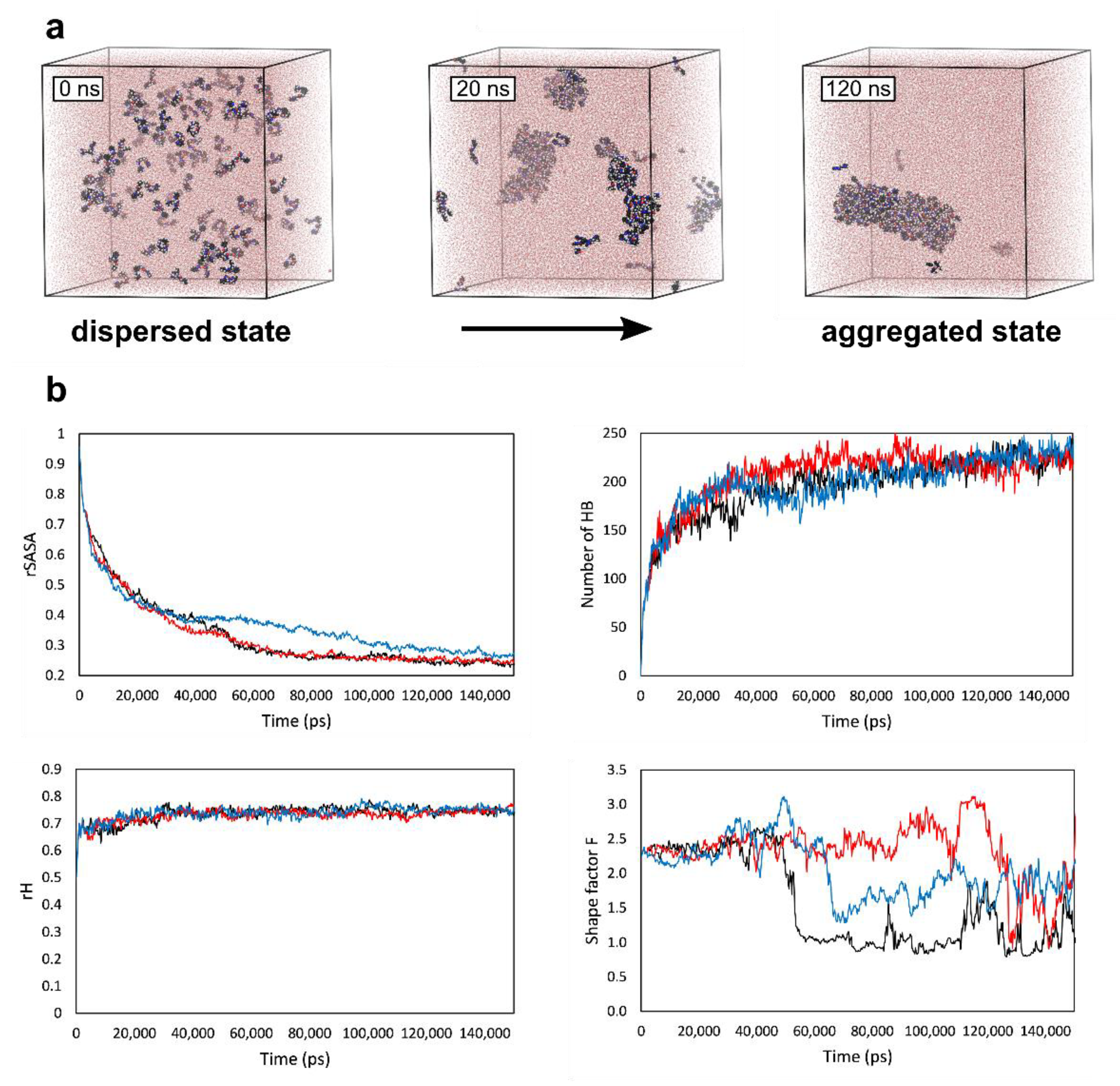{kind=link}
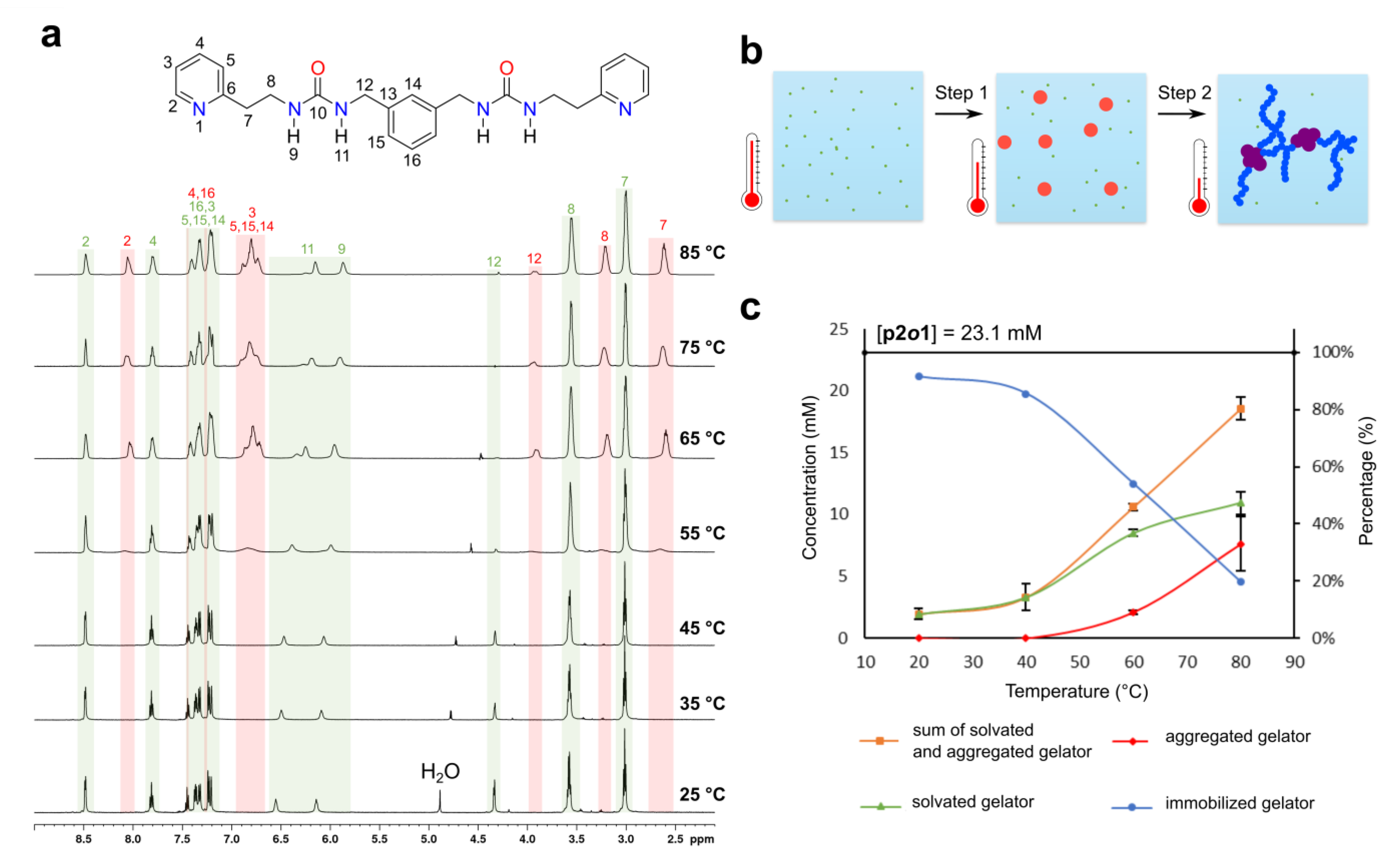{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
- (i)
- For each narrow signal, a corresponding broader signal appeared upfield with a similar 1H-13C HSQC cross-peak pattern (Supplementary Figure S14). Note that for the urea hydrogen atoms H-11 and H-9, the original narrow and broader signal (partially) overlapped with one another.
- (ii)
- The 1H-1H NOESY spectrum of the gel at 60 °C showed several cross-peaks for the well-resolved signals, which can all be accounted for by intramolecular proximity. On the other hand, the broader set of signals showed cross-peaks between the aromatic (7.0–6.7 ppm) and aliphatic (3.95, 3.24 and 2.62 ppm) region, which points toward intermolecular interactions and/or conformational changes (Supplementary Figure S16).
- (iii)
- Upon decreasing the concentration of p2o1 in the sample, the broader signals were absent, highlighting the importance of intermolecular interactions (Supplementary Figure S17).
3. Conclusions
4. Materials and Methods
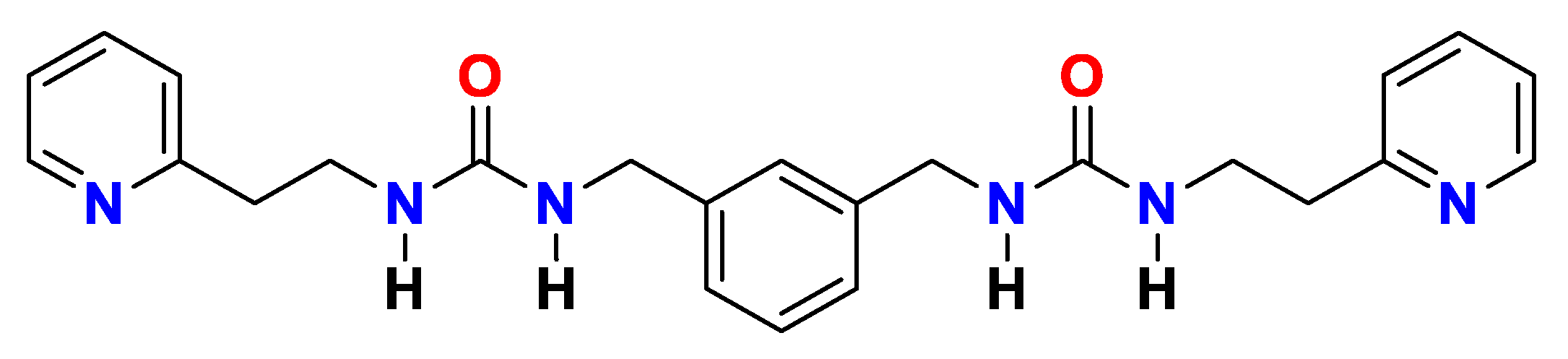
4.1. Synthesis, Characterization and Hydrogelation of p2o1
4.2. NMR Experiments on Gelled Sample of p2o1
4.3. Molecular Dynamics Simulations
4.4. Scanning Electron Microscopy Imaging
4.5. Single Particle Tracking Experiments
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Draper, E.R.; Adams, D.J. Low-molecular-weight gels: The state of the art. Chem 2017, 3, 390–410. [Google Scholar] [CrossRef] [Green Version]
- Du, X.W.; Zhou, J.; Shi, J.F.; Xu, B. Supramolecular hydrogelators and hydrogels: From soft matter to molecular biomaterials. Chem. Rev. 2015, 115, 13165–13307. [Google Scholar] [CrossRef] [PubMed]
- Sangeetha, N.M.; Maitra, U. Supramolecular gels: Functions and uses. Chem. Soc. Rev. 2005, 34, 821–836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okesola, B.O.; Vieira, V.M.P.; Cornwell, D.J.; Whitelaw, N.K.; Smith, D.K. 1,3:2,4-Dibenzylidene-d-sorbitol (DBS) and its derivatives—Efficient, versatile and industrially-relevant low-molecular-weight gelators with over 100 years of history and a bright future. Soft Matter 2015, 11, 4768–4787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sang, Y.; Liu, M. Nanoarchitectonics through supramolecular gelation: Formation and switching of diverse nanostructures. Mol. Syst. Des. Eng. 2019, 4, 11–28. [Google Scholar] [CrossRef]
- Wojciechowski, J.P.; Martin, A.D.; Du, E.Y.; Garvey, C.J.; Nordon, R.E.; Thordarson, P. Non-reversible heat-induced gelation of a biocompatible Fmoc-hexapeptide in water. Nanoscale 2020, 12, 8262–8267. [Google Scholar] [CrossRef]
- Patra, T.; Pal, A.; Dey, J. A Smart supramolecular hydrogel of Nα-(4-n-alkyloxybenzoyl)-l-histidine exhibiting pH-modulated properties. Langmuir 2010, 26, 7761–7767. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.-W.; Stricker, L.; Kirse, T.M.; Hayduk, M.; Ravoo, B.J. Light-responsive arylazopyrazole gelators: From organic to aqueous media and from supramolecular to dynamic covalent chemistry. Chem. Eur. J. 2019, 25, 6131–6140. [Google Scholar] [CrossRef] [Green Version]
- Himabindu, M.; Palanisamy, A. Ultrasound- and temperature-induced gelation of gluconosemicarbazide gelator in dmso and water mixtures. Gels 2017, 3, 12. [Google Scholar] [CrossRef] [Green Version]
- Jones, C.D.; Steed, J.W. Gels with sense: Supramolecular materials that respond to heat, light and sound. Chem. Soc. Rev. 2016, 45, 6546–6596. [Google Scholar] [CrossRef]
- Yu, X.D.; Chen, L.M.; Zhang, M.M.; Yi, T. Low-molecular-mass gels responding to ultrasound and mechanical stress: Towards self-healing materials. Chem. Soc. Rev. 2014, 43, 5346–5371. [Google Scholar] [CrossRef] [PubMed]
- Dudukovic, N.A.; Zukoski, C.F. Evidence for equilibrium gels of valence-limited particles. Soft Matter 2014, 10, 7849–7856. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, K.; Patterson, A.K.; Clarke, P.A.; Smith, D.K. Catalytic gels for a prebiotically relevant asymmetric aldol reaction in water: From organocatalyst design to hydrogel discovery and back again. J. Am. Chem. Soc. 2020, 142, 4379–4389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozbas, B.; Kretsinger, J.; Rajagopal, K.; Schneider, J.P.; Pochan, D.J. Salt-triggered peptide folding and consequent self-assembly into hydrogels with tunable modulus. Macromolecules 2004, 37, 7331–7337. [Google Scholar] [CrossRef]
- Shao, T.; Falcone, N.; Kraatz, H.-B. Supramolecular peptide gels: Influencing properties by metal ion coordination and their wide-ranging applications. ACS Omega 2020, 5, 1312–1317. [Google Scholar] [CrossRef] [PubMed]
- Andrews, J.L.; Pearson, E.; Yufit, D.S.; Steed, J.W.; Edkins, K. Supramolecular gelation as the first stage in Ostwald’s rule. Cryst. Growth Des. 2018, 18, 7690–7700. [Google Scholar] [CrossRef] [Green Version]
- Ramos Sasselli, I.; Halling, P.J.; Ulijn, R.V.; Tuttle, T. Supramolecular fibers in gels can be at thermodynamic equilibrium: A simple packing model reveals preferential fibril formation versus crystallization. ACS Nano 2016, 10, 2661–2668. [Google Scholar] [CrossRef] [Green Version]
- Christoff-Tempesta, T.; Lew, A.J.; Ortony, J.H. Beyond covalent crosslinks: Applications of supramolecular gels. Gels 2018, 4, 40. [Google Scholar] [CrossRef] [Green Version]
- Mehwish, N.; Dou, X.; Zhao, Y.; Feng, C.-L. Supramolecular fluorescent hydrogelators as bio-imaging probes. Mater. Horiz. 2019, 6, 14–44. [Google Scholar] [CrossRef]
- Lim, J.Y.C.; Lin, Q.; Xue, K.; Loh, X.J. Recent advances in supramolecular hydrogels for biomedical applications. Mater. Today Adv. 2019, 3, 100021. [Google Scholar] [CrossRef]
- Mehwish, N.; Dou, X.; Zhao, C.; Feng, C.; Fu, Q. Chirality transfer in supramolecular co-assembled fibrous material enabling the visual recognition of sucrose. Adv. Fiber Mater. 2020, 2, 204–211. [Google Scholar] [CrossRef] [Green Version]
- Skilling, K.J.; Citossi, F.; Bradshaw, T.D.; Ashford, M.; Kellam, B.; Marlow, M. Insights into low molecular mass organic gelators: A focus on drug delivery and tissue engineering applications. Soft Matter 2014, 10, 237–256. [Google Scholar] [CrossRef] [PubMed]
- Webber, M.J.; Appel, E.A.; Meijer, E.W.; Langer, R. Supramolecular biomaterials. Nat. Mater. 2016, 15, 13–26. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Park, J.; Park, J.-W.; Ahn, H.-J.; Jaworski, J.; Jung, J.H. Supramolecular gels with high strength by tuning of calix[4]arene-derived networks. Nat. Commun. 2015, 6, 6650. [Google Scholar] [CrossRef]
- Chivers, P.R.A.; Smith, D.K. Shaping and structuring supramolecular gels. Nat. Rev. Mater. 2019, 4, 463–478. [Google Scholar] [CrossRef] [Green Version]
- Boekhoven, J.; Poolman, J.M.; Maity, C.; Li, F.; van der Mee, L.; Minkenberg, C.B.; Mendes, E.; van Esch, J.H.; Eelkema, R. Catalytic control over supramolecular gel formation. Nat. Chem. 2013, 5, 433–437. [Google Scholar] [CrossRef]
- Piras, C.C.; Slavik, P.; Smith, D.K. Self-assembling supramolecular hybrid hydrogel beads. Angew. Chem. Int. Ed. 2020, 59, 853–859. [Google Scholar] [CrossRef] [Green Version]
- Draper, E.R.; Eden, E.G.B.; McDonald, T.O.; Adams, D.J. Spatially resolved multicomponent gels. Nat. Chem. 2015, 7, 848–852. [Google Scholar] [CrossRef]
- Korevaar, P.A.; Newcomb, C.J.; Meijer, E.W.; Stupp, S.I. Pathway Selection in Peptide Amphiphile Assembly. J. Am. Chem. Soc. 2014, 136, 8540–8543. [Google Scholar] [CrossRef]
- de Jong, J.J.; Lucas, L.N.; Kellogg, R.M.; van Esch, J.H.; Feringa, B.L. Reversible optical transcription of supramolecular chirality into molecular chirality. Science 2004, 304, 278–281. [Google Scholar] [CrossRef]
- Wang, X.; Ding, Z.; Ma, Y.; Zhang, Y.; Shang, H.; Jiang, S. Multi-stimuli responsive supramolecular gels based on a D–π–A structural cyanostilbene derivative with aggregation induced emission properties. Soft Matter 2019, 15, 1658–1665. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Cong, Y.; He, L.; Wang, Y.; Wang, J.; Zhang, F.-M.; Bu, W. Multiple stimuli-responsive supramolecular gels constructed from metal–organic cycles. Polym. Chem. 2016, 7, 6288–6292. [Google Scholar] [CrossRef]
- Cheng, W.; Zhao, D.; Qiu, Y.; Hu, H.; Wang, H.; Wang, Q.; Liao, Y.; Peng, H.; Xie, X. Robust multi-responsive supramolecular hydrogel based on a mono-component host–guest gelator. Soft Matter 2018, 14, 5213–5221. [Google Scholar] [CrossRef]
- Basak, S.; Nanda, J.; Banerjee, A. Multi-stimuli responsive self-healing metallo-hydrogels: Tuning of the gel recovery property. Chem. Commun. 2014, 50, 2356–2359. [Google Scholar] [CrossRef]
- Arango-Restrepo, A.; Rubi, J.M.; Barragán, D. Understanding gelation as a nonequilibrium self-assembly process. J. Phys. Chem. B 2018, 122, 4937–4945. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.K.; Steed, J.W. Supramolecular gel phase crystallization: Orthogonal self-assembly under non-equilibrium conditions. Chem. Soc. Rev. 2014, 43, 2080–2088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rutgeerts, L.A.J.; Soultan, A.H.; Subramani, R.; Toprakhisar, B.; Ramon, H.; Paderes, M.C.; De Borggraeve, W.M.; Patterson, J. Robust scalable synthesis of a bis-urea derivative forming thixotropic and cytocompatible supramolecular hydrogels. Chem. Commun. 2019, 55, 7323–7326. [Google Scholar] [CrossRef]
- Sawada, H.; Yamanaka, M. Synthesis of a bis-urea dimer and its effects on the physical properties of an amphiphilic tris-urea supramolecular hydrogel. Chem. Asian J. 2018, 13, 929–933. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, M. Urea derivatives as low-molecular-weight gelators. J. Incl. Phenom. Macrocycl. Chem. 2013, 77, 33–48. [Google Scholar] [CrossRef]
- George, M.; Tan, G.; John, V.T.; Weiss, R.G. Urea and thiourea derivatives as low molecular-mass organogelators. Chem. Eur. J. 2005, 11, 3243–3254. [Google Scholar] [CrossRef]
- van Esch, J.; Kellogg, R.M.; Feringa, B.L. Di-urea compounds as gelators for organic solvents. Tetrahedron Lett. 1997, 38, 281–284. [Google Scholar] [CrossRef] [Green Version]
- van Esch, J.; Schoonbeek, F.; de Loos, M.; Kooijman, H.; Spek, A.L.; Kellogg, R.M.; Feringa, B.L. Cyclic bis-urea compounds as gelators for organic solvents. Chem. Eur. J. 1999, 5, 937–950. [Google Scholar] [CrossRef]
- Van Lommel, R.; Rutgeerts, L.A.J.; De Borggraeve, W.M.; De Proft, F.; Alonso, M. Rationalising supramolecular hydrogelation of bis-urea-based gelators through a multiscale approach. ChemPlusChem 2020, 85, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Van Lommel, R. Towards a Better Understanding and Development Strategy for Supramolecular Gels. Ph.D. Thesis, KU Leuven and Vrije Universiteit Brussels, Leuven, Belgium, 13 May 2022. [Google Scholar]
- Mears, L.L.E.; Draper, E.R.; Castilla, A.M.; Su, H.; Zhuola; Dietrich, B.; Nolan, M.C.; Smith, G.N.; Doutch, J.; Rogers, S.; et al. Drying affects the fiber network in low molecular weight hydrogels. Biomacromolecules 2017, 18, 3531–3540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louis, B.; Camacho, R.; Bresolí-Obach, R.; Abakumov, S.; Vandaele, J.; Kudo, T.; Masuhara, H.; Scheblykin, I.G.; Hofkens, J.; Rocha, S. Fast-tracking of single emitters in large volumes with nanometer precision. Opt. Express 2020, 28, 28656–28671. [Google Scholar] [CrossRef]
- Korson, L.; Drost-Hansen, W.; Millero, F.J. Viscosity of water at various temperatures. J. Phys. Chem. 1969, 73, 34–39. [Google Scholar] [CrossRef]
- Van Lommel, R.; Zhao, J.; De Borggraeve, W.M.; De Proft, F.; Alonso, M. Molecular dynamics based descriptors for predicting supramolecular gelation. Chem. Sci. 2020, 11, 4226–4238. [Google Scholar] [CrossRef] [Green Version]
- Hirst, A.R.; Coates, I.A.; Boucheteau, T.R.; Miravet, J.F.; Escuder, B.; Castelletto, V.; Hamley, I.W.; Smith, D.K. Low-molecular-weight gelators: Elucidating the principles of gelation based on gelator solubility and a cooperative self-assembly model. J. Am. Chem. Soc. 2008, 130, 9113–9121. [Google Scholar] [CrossRef]
- Wallace, M.; Iggo, J.A.; Adams, D.J. Using solution state NMR spectroscopy to probe NMR invisible gelators. Soft Matter 2015, 11, 7739–7747. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.; Zhang, Q.; Deng, Y.; Li, X.; Luo, Z.; Zheng, B.; Dong, S. Assembly pattern of supramolecular hydrogel induced by lower critical solution temperature behavior of low-molecular-weight gelator. J. Am. Chem. Soc. 2020, 142, 448–455. [Google Scholar] [CrossRef]
- Liu, J.; He, P.; Yan, J.; Fang, X.; Peng, J.; Liu, K.; Fang, Y. An Organometallic super-gelator with multiple-stimulus responsive properties. Adv. Mater. 2008, 20, 2508–2511. [Google Scholar] [CrossRef]
- Escuder, B.; Llusar, M.; Miravet, J.F. Insight on the NMR Study of supramolecular gels and its application to monitor molecular recognition on self-assembled fibers. J. Org. Chem. 2006, 71, 7747–7752. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Xu, D.; Chi, X.; Chen, J.; Dong, S.; Ding, X.; Yu, Y.; Huang, F. A Multiresponsive, shape-persistent, and elastic supramolecular polymer network gel constructed by orthogonal self-assembly. Adv. Mater. 2012, 24, 362–369. [Google Scholar] [CrossRef] [PubMed]
- Tyburn, J.M.; Coutant, J. TopSpin ERETIC2 Electronic to Access In-Vivo Concentratation User Manual, 1st ed.; Corporation, B., Ed.; Bruker Corporation: Billerica, MD, USA, 2016; p. 33. [Google Scholar]
- Wider, G.; Dreier, L. Measuring protein concentrations by NMR spectroscopy. J. Am. Chem. Soc. 2006, 128, 2571–2576. [Google Scholar] [CrossRef]
- Smulders, M.M.J.; Nieuwenhuizen, M.M.L.; de Greef, T.F.A.; van der Schoot, P.; Schenning, A.P.H.J.; Meijer, E.W. How to Distinguish isodesmic from cooperative supramolecular polymerisation. Chem. Eur. J. 2010, 16, 362–367. [Google Scholar] [CrossRef]
- Chakraborty, P.; Tang, Y.; Yamamoto, T.; Yao, Y.; Guterman, T.; Zilberzwige-Tal, S.; Adadi, N.; Ji, W.; Dvir, T.; Ramamoorthy, A.; et al. Unusual two-step assembly of a minimalistic dipeptide-based functional hypergelator. Adv. Mater. 2020, 32, 1906043. [Google Scholar] [CrossRef] [PubMed]
- Fichman, G.; Guterman, T.; Damron, J.; Adler-Abramovich, L.; Schmidt, J.; Kesselman, E.; Shimon, L.J.; Ramamoorthy, A.; Talmon, Y.; Gazit, E. Spontaneous structural transition and crystal formation in minimal supramolecular polymer model. Sci. Adv. 2016, 2, e1500827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Foloppe, N.; MacKerell, J.A.D. All-atom empirical force field for nucleic acids: I. Parameter optimization based on small molecule and condensed phase macromolecular target data. J. Comput. Chem. 2000, 21, 86–104. [Google Scholar] [CrossRef]
- Zoete, V.; Cuendet, M.A.; Grosdidier, A.; Michielin, O. SwissParam: A fast force field generation tool for small organic molecules. J. Comput. Chem. 2011, 32, 2359–2368. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Edelstein, A.D.; Tsuchida, M.A.; Amodaj, N.; Pinkard, H.; Vale, R.D.; Stuurman, N. Advanced methods of microscope control using μManager software. J. Biomol. Meth. 2014, 1, e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, T.L.; Shaka, A.J. Water suppression that works. excitation sculpting using arbitrary wave-forms and pulsed-field gradients. J. Magn. Reson., Series A 1995, 112, 275–279. [Google Scholar] [CrossRef]
- Lagerquist, L.; Rahkila, J.; Eklund, P. Utilization of 31P PULCON for quantitative hydroxyl group determination in lignin by NMR Spectroscopy. ACS Sust. Chem. Eng. 2019, 7, 9002–9006. [Google Scholar] [CrossRef] [Green Version]
- Martinez, L.; Andrade, R.; Birgin, E.G.; Martinez, J.M. PACKMOL: A package for building initial configurations for molecular dynamics simulations. J. Comput. Chem. 2009, 30, 2157–2164. [Google Scholar] [CrossRef]
- MacKerell, A.D.; Bashford, D.; Bellott, M.; Dunbrack, R.L.; Evanseck, J.D.; Field, M.J.; Fischer, S.; Gao, J.; Guo, H.; Ha, S.; et al. All-Atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Van Lommel, R.; Van Hooste, J.; Vandaele, J.; Steurs, G.; Van der Donck, T.; De Proft, F.; Rocha, S.; Sakellariou, D.; Alonso, M.; De Borggraeve, W.M. Does Supramolecular Gelation Require an External Trigger? Gels 2022, 8, 813. https://doi.org/10.3390/gels8120813
Van Lommel R, Van Hooste J, Vandaele J, Steurs G, Van der Donck T, De Proft F, Rocha S, Sakellariou D, Alonso M, De Borggraeve WM. Does Supramolecular Gelation Require an External Trigger? Gels. 2022; 8(12):813. https://doi.org/10.3390/gels8120813
Chicago/Turabian StyleVan Lommel, Ruben, Julie Van Hooste, Johannes Vandaele, Gert Steurs, Tom Van der Donck, Frank De Proft, Susana Rocha, Dimitrios Sakellariou, Mercedes Alonso, and Wim M. De Borggraeve. 2022. "Does Supramolecular Gelation Require an External Trigger?" Gels 8, no. 12: 813. https://doi.org/10.3390/gels8120813
APA StyleVan Lommel, R., Van Hooste, J., Vandaele, J., Steurs, G., Van der Donck, T., De Proft, F., Rocha, S., Sakellariou, D., Alonso, M., & De Borggraeve, W. M. (2022). Does Supramolecular Gelation Require an External Trigger? Gels, 8(12), 813. https://doi.org/10.3390/gels8120813