
Non-Coding Variants in Cancer: Mechanistic Insights and Clinical Potential for Personalized Medicine


Abstract
:1. Introduction
2. Genetic Variability in the Cancer Genome
2.1. Structural Classification of Mutations in Cancer
2.2. Functional Classification of Mutations in Cancer
3. Non-Transcribed Regulatory Variants
3.1. Genetic Variability in Promoters
3.2. Genetic Variability in Enhancers
3.3. Genetic Variability in Silencer Elements
3.4. Genetic Variability in Insulator Elements
4. Transcribed Non-Coding Variants
4.1. Non-Coding Variants Affecting miRNA Targeting and Biogenesis
4.2. Non-Coding Variants Affecting lncRNA Function
5. Methodologies to Functionally Characterize Non-Coding Variants
5.1. Scanning for Regulatory Sequences Based on Open-Chromatin State
5.2. Scanning for Regulatory Variants in Trans Regulatory Elements
5.3. Scanning for Regulatory Variants Based on Chromatin Interactions
5.4. Scanning for Regulatory Variants Based on RNA-Chromatin Interactions
5.5. Methodologies to Functionally Dissect Transcribed Non-Coding Variants
5.6. Validating Regulatory Variants with CRISPR-Based Approaches
6. Utilizing Non-Coding Variants in Clinomics
7. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Della Chiara, G.; Gervasoni, F.; Fakiola, M.; Godano, C.; D’Oria, C.; Azzolin, L.; Bonnal, R.J.P.; Moreni, G.; Drufuca, L.; Rossetti, G.; et al. Epigenomic landscape of human colorectal cancer unveils an aberrant core of pan-cancer enhancers orchestrated by YAP/TAZ. Nat. Commun. 2021, 12, 2340. [Google Scholar] [CrossRef]
- Huang, H.; Hu, J.; Maryam, A.; Huang, Q.; Zhang, Y.; Ramakrishnan, S.; Li, J.; Ma, H.; Ma, V.W.S.; Cheuk, W.; et al. Defining super-enhancer landscape in triple-negative breast cancer by multiomic profiling. Nat. Commun. 2021, 12, 2242. [Google Scholar] [CrossRef]
- Xiong, L.; Wu, F.; Wu, Q.; Xu, L.; Cheung, O.K.; Kang, W.; Mok, M.T.; Szeto, L.L.M.; Lun, C.Y.; Lung, R.W.; et al. Aberrant enhancer hypomethylation contributes to hepatic carcinogenesis through global transcriptional reprogramming. Nat. Commun. 2019, 10, 335. [Google Scholar] [CrossRef]
- Sur, I.; Taipale, J. The role of enhancers in cancer. Nat. Rev. Cancer 2016, 16, 483–493. [Google Scholar] [CrossRef]
- See, Y.X.; Wang, B.Z.; Fullwood, M.J. Chromatin Interactions and Regulatory Elements in Cancer: From Bench to Bedside. Trends Genet. 2019, 35, 145–158. [Google Scholar] [CrossRef]
- Savarese, F.; Grosschedl, R. Blurring cis and trans in gene regulation. Cell 2006, 126, 248–250. [Google Scholar] [CrossRef] [Green Version]
- Andersson, R.; Sandelin, A. Determinants of enhancer and promoter activities of regulatory elements. Nat. Rev. Genet 2020, 21, 71–87. [Google Scholar] [CrossRef]
- Schoenfelder, S.; Fraser, P. Long-range enhancer-promoter contacts in gene expression control. Nat. Rev. Genet. 2019, 20, 437–455. [Google Scholar] [CrossRef]
- Schoenfelder, S.; Furlan-Magaril, M.; Mifsud, B.; Tavares-Cadete, F.; Sugar, R.; Javierre, B.M.; Nagano, T.; Katsman, Y.; Sakthidevi, M.; Wingett, S.W.; et al. The pluripotent regulatory circuitry connecting promoters to their long-range interacting elements. Genome Res. 2015, 25, 582–597. [Google Scholar] [CrossRef] [Green Version]
- Furlong, E.E.M.; Levine, M. Developmental enhancers and chromosome topology. Science 2018, 361, 1341–1345. [Google Scholar] [CrossRef] [Green Version]
- Maass, P.G.; Barutcu, A.R.; Rinn, J.L. Interchromosomal interactions: A genomic love story of kissing chromosomes. J. Cell Biol. 2019, 218, 27–38. [Google Scholar] [CrossRef] [Green Version]
- Gasperini, M.; Tome, J.M.; Shendure, J. Towards a comprehensive catalogue of validated and target-linked human enhancers. Nat. Rev. Genet. 2020, 21, 292–310. [Google Scholar] [CrossRef]
- Shlyueva, D.; Stampfel, G.; Stark, A. Transcriptional enhancers: From properties to genome-wide predictions. Nat. Rev. Genet. 2014, 15, 272–286. [Google Scholar] [CrossRef] [PubMed]
- Roadmap Epigenomics, C.; Kundaje, A.; Meuleman, W.; Ernst, J.; Bilenky, M.; Yen, A.; Heravi-Moussavi, A.; Kheradpour, P.; Zhang, Z.; Wang, J.; et al. Integrative analysis of 111 reference human epigenomes. Nature 2015, 518, 317–330. [Google Scholar] [CrossRef] [Green Version]
- Soutourina, J. Transcription regulation by the Mediator complex. Nat. Rev. Mol. Cell Biol. 2018, 19, 262–274. [Google Scholar] [CrossRef]
- Szabo, Q.; Bantignies, F.; Cavalli, G. Principles of genome folding into topologically associating domains. Sci. Adv. 2019, 5, eaaw1668. [Google Scholar] [CrossRef] [Green Version]
- Vietri Rudan, M.; Barrington, C.; Henderson, S.; Ernst, C.; Odom, D.T.; Tanay, A.; Hadjur, S. Comparative Hi-C reveals that CTCF underlies evolution of chromosomal domain architecture. Cell Rep. 2015, 10, 1297–1309. [Google Scholar] [CrossRef] [Green Version]
- Nora, E.P.; Goloborodko, A.; Valton, A.L.; Gibcus, J.H.; Uebersohn, A.; Abdennur, N.; Dekker, J.; Mirny, L.A.; Bruneau, B.G. Targeted Degradation of CTCF Decouples Local Insulation of Chromosome Domains from Genomic Compartmentalization. Cell 2017, 169, 930.e922–944.e922. [Google Scholar] [CrossRef] [Green Version]
- Wutz, G.; Varnai, C.; Nagasaka, K.; Cisneros, D.A.; Stocsits, R.R.; Tang, W.; Schoenfelder, S.; Jessberger, G.; Muhar, M.; Hossain, M.J.; et al. Topologically associating domains and chromatin loops depend on cohesin and are regulated by CTCF, WAPL, and PDS5 proteins. EMBO J. 2017, 36, 3573–3599. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.S.; Huntley, M.H.; Durand, N.C.; Stamenova, E.K.; Bochkov, I.D.; Robinson, J.T.; Sanborn, A.L.; Machol, I.; Omer, A.D.; Lander, E.S.; et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 2014, 159, 1665–1680. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Yu, N.K.; Kaang, B.K. CTCF as a multifunctional protein in genome regulation and gene expression. Exp. Mol. Med. 2015, 47, e166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Notani, D.; Rosenfeld, M.G. Enhancers as non-coding RNA transcription units: Recent insights and future perspectives. Nat. Rev. Genet. 2016, 17, 207–223. [Google Scholar] [CrossRef]
- Kim, T.K.; Hemberg, M.; Gray, J.M. Enhancer RNAs: A class of long noncoding RNAs synthesized at enhancers. Cold Spring Harb. Perspect. Biol. 2015, 7, a018622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, Q.; Chen, S.; Tan, Y.; Li, Y.; Tang, F. Oncogenic super-enhancer formation in tumorigenesis and its molecular mechanisms. Exp. Mol. Med. 2020, 52, 713–723. [Google Scholar] [CrossRef]
- Pott, S.; Lieb, J.D. What are super-enhancers? Nat. Genet. 2015, 47, 8–12. [Google Scholar] [CrossRef] [PubMed]
- Sartorelli, V.; Lauberth, S.M. Enhancer RNAs are an important regulatory layer of the epigenome. Nat. Struct. Mol. Biol. 2020, 27, 521–528. [Google Scholar] [CrossRef]
- Zhang, Z.; Lee, J.H.; Ruan, H.; Ye, Y.; Krakowiak, J.; Hu, Q.; Xiang, Y.; Gong, J.; Zhou, B.; Wang, L.; et al. Transcriptional landscape and clinical utility of enhancer RNAs for eRNA-targeted therapy in cancer. Nat. Commun. 2019, 10, 4562. [Google Scholar] [CrossRef] [Green Version]
- Sakabe, N.J.; Savic, D.; Nobrega, M.A. Transcriptional enhancers in development and disease. Genome Biol. 2012, 13, 238. [Google Scholar] [CrossRef] [Green Version]
- Xia, J.H.; Wei, G.H. Enhancer Dysfunction in 3D Genome and Disease. Cells 2019, 8, 1281. [Google Scholar] [CrossRef] [Green Version]
- Chakravarty, D.; Solit, D.B. Clinical cancer genomic profiling. Nat. Rev. Genet. 2021. [Google Scholar] [CrossRef]
- Ramirez, M.; Rajaram, S.; Steininger, R.J.; Osipchuk, D.; Roth, M.A.; Morinishi, L.S.; Evans, L.; Ji, W.; Hsu, C.H.; Thurley, K.; et al. Diverse drug-resistance mechanisms can emerge from drug-tolerant cancer persister cells. Nat. Commun. 2016, 7, 10690. [Google Scholar] [CrossRef]
- Spielmann, M.; Lupianez, D.G.; Mundlos, S. Structural variation in the 3D genome. Nat. Rev. Genet. 2018, 19, 453–467. [Google Scholar] [CrossRef] [Green Version]
- Beckmann, J.S.; Estivill, X.; Antonarakis, S.E. Copy number variants and genetic traits: Closer to the resolution of phenotypic to genotypic variability. Nat. Rev. Genet. 2007, 8, 639–646. [Google Scholar] [CrossRef]
- Bu, D.; Yu, K.; Sun, S.; Xie, C.; Skogerbo, G.; Miao, R.; Xiao, H.; Liao, Q.; Luo, H.; Zhao, G.; et al. NONCODE v3.0: Integrative annotation of long noncoding RNAs. Nucleic Acids Res. 2012, 40, D210–D215. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas Research, N. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef]
- Carrot-Zhang, J.; Chambwe, N.; Damrauer, J.S.; Knijnenburg, T.A.; Robertson, A.G.; Yau, C.; Zhou, W.; Berger, A.C.; Huang, K.L.; Newberg, J.Y.; et al. Comprehensive Analysis of Genetic Ancestry and Its Molecular Correlates in Cancer. Cancer Cell 2020, 37, 639.e636–654.e636. [Google Scholar] [CrossRef]
- Hong, E.L.; Sloan, C.A.; Chan, E.T.; Davidson, J.M.; Malladi, V.S.; Strattan, J.S.; Hitz, B.C.; Gabdank, I.; Narayanan, A.K.; Ho, M.; et al. Principles of metadata organization at the ENCODE data coordination center. Database 2016, 2016. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Lee, D.; Dhiman, V.; Jiang, P.; Xu, J.; McGillivray, P.; Yang, H.; Liu, J.; Meyerson, W.; Clarke, D.; et al. An integrative ENCODE resource for cancer genomics. Nat. Commun. 2020, 11, 3696. [Google Scholar] [CrossRef]
- Liu, C.; Bai, B.; Skogerbo, G.; Cai, L.; Deng, W.; Zhang, Y.; Bu, D.; Zhao, Y.; Chen, R. NONCODE: An integrated knowledge database of non-coding RNAs. Nucleic Acids Res. 2005, 33, D112–D115. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas Research, N.; Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- Karki, R.; Pandya, D.; Elston, R.C.; Ferlini, C. Defining “mutation” and “polymorphism” in the era of personal genomics. BMC Med. Genom. 2015, 8, 37. [Google Scholar] [CrossRef] [Green Version]
- Maston, G.A.; Evans, S.K.; Green, M.R. Transcriptional regulatory elements in the human genome. Annu Rev. Genom. Hum. Genet. 2006, 7, 29–59. [Google Scholar] [CrossRef] [Green Version]
- Kalender Atak, Z.; Imrichova, H.; Svetlichnyy, D.; Hulselmans, G.; Christiaens, V.; Reumers, J.; Ceulemans, H.; Aerts, S. Identification of cis-regulatory mutations generating de novo edges in personalized cancer gene regulatory networks. Genome Med. 2017, 9, 80. [Google Scholar] [CrossRef] [Green Version]
- Erson-Bensan, A.E. RNA-biology ruling cancer progression? Focus on 3’UTRs and splicing. Cancer Metastasis Rev. 2020, 39, 887–901. [Google Scholar] [CrossRef] [PubMed]
- Slack, F.J.; Chinnaiyan, A.M. The Role of Non-coding RNAs in Oncology. Cell 2019, 179, 1033–1055. [Google Scholar] [CrossRef]
- Anna, A.; Monika, G. Splicing mutations in human genetic disorders: Examples, detection, and confirmation. J. Appl. Genet. 2018, 59, 253–268. [Google Scholar] [CrossRef] [Green Version]
- Bashyam, M.D.; Animireddy, S.; Bala, P.; Naz, A.; George, S.A. The Yin and Yang of cancer genes. Gene 2019, 704, 121–133. [Google Scholar] [CrossRef]
- Baylin, S.B.; Jones, P.A. Epigenetic Determinants of Cancer. Cold Spring Harb. Perspect. Biol. 2016, 8. [Google Scholar] [CrossRef] [Green Version]
- Futreal, P.A.; Coin, L.; Marshall, M.; Down, T.; Hubbard, T.; Wooster, R.; Rahman, N.; Stratton, M.R. A census of human cancer genes. Nat. Rev. Cancer 2004, 4, 177–183. [Google Scholar] [CrossRef]
- Tam, V.; Patel, N.; Turcotte, M.; Bosse, Y.; Pare, G.; Meyre, D. Benefits and limitations of genome-wide association studies. Nat. Rev. Genet. 2019, 20, 467–484. [Google Scholar] [CrossRef]
- Yong, S.Y.; Raben, T.G.; Lello, L.; Hsu, S.D.H. Genetic architecture of complex traits and disease risk predictors. Sci. Rep. 2020, 10, 12055. [Google Scholar] [CrossRef]
- Slatkin, M. Linkage disequilibrium--understanding the evolutionary past and mapping the medical future. Nat. Rev. Genet. 2008, 9, 477–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soong, D.; Stratford, J.; Avet-Loiseau, H.; Bahlis, N.; Davies, F.; Dispenzieri, A.; Sasser, A.K.; Schecter, J.M.; Qi, M.; Brown, C.; et al. CNV Radar: An improved method for somatic copy number alteration characterization in oncology. BMC Bioinform. 2020, 21, 98. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Roberts, N.D.; Wala, J.A.; Shapira, O.; Schumacher, S.E.; Kumar, K.; Khurana, E.; Waszak, S.; Korbel, J.O.; Haber, J.E.; et al. Patterns of somatic structural variation in human cancer genomes. Nature 2020, 578, 112–121. [Google Scholar] [CrossRef] [Green Version]
- Kumaran, M.; Cass, C.E.; Graham, K.; Mackey, J.R.; Hubaux, R.; Lam, W.; Yasui, Y.; Damaraju, S. Germline copy number variations are associated with breast cancer risk and prognosis. Sci. Rep. 2017, 7, 14621. [Google Scholar] [CrossRef]
- Granados-Soler, J.L.; Bornemann-Kolatzki, K.; Beck, J.; Brenig, B.; Schutz, E.; Betz, D.; Junginger, J.; Hewicker-Trautwein, M.; Murua Escobar, H.; Nolte, I. Analysis of Copy-Number Variations and Feline Mammary Carcinoma Survival. Sci. Rep. 2020, 10, 1003. [Google Scholar] [CrossRef] [PubMed]
- Camps, J.; Grade, M.; Nguyen, Q.T.; Hormann, P.; Becker, S.; Hummon, A.B.; Rodriguez, V.; Chandrasekharappa, S.; Chen, Y.; Difilippantonio, M.J.; et al. Chromosomal breakpoints in primary colon cancer cluster at sites of structural variants in the genome. Cancer Res. 2008, 68, 1284–1295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lahortiga, I.; De Keersmaecker, K.; Van Vlierberghe, P.; Graux, C.; Cauwelier, B.; Lambert, F.; Mentens, N.; Beverloo, H.B.; Pieters, R.; Speleman, F.; et al. Duplication of the MYB oncogene in T cell acute lymphoblastic leukemia. Nat. Genet. 2007, 39, 593–595. [Google Scholar] [CrossRef]
- Basecke, J.; Whelan, J.T.; Griesinger, F.; Bertrand, F.E. The MLL partial tandem duplication in acute myeloid leukaemia. Br. J. Haematol. 2006, 135, 438–449. [Google Scholar] [CrossRef]
- Kumaran, M.; Krishnan, P.; Cass, C.E.; Hubaux, R.; Lam, W.; Yasui, Y.; Damaraju, S. Breast cancer associated germline structural variants harboring small noncoding RNAs impact post-transcriptional gene regulation. Sci Rep. 2018, 8, 7529. [Google Scholar] [CrossRef]
- Li, Y.R.; Glessner, J.T.; Coe, B.P.; Li, J.; Mohebnasab, M.; Chang, X.; Connolly, J.; Kao, C.; Wei, Z.; Bradfield, J.; et al. Rare copy number variants in over 100,000 European ancestry subjects reveal multiple disease associations. Nat. Commun. 2020, 11, 255. [Google Scholar] [CrossRef] [PubMed]
- Hennessey, R.C.; Brown, K.M. Cancer regulatory variation. Curr Opin Genet. Dev. 2021, 66, 41–49. [Google Scholar] [CrossRef]
- Spielmann, M.; Klopocki, E. CNVs of noncoding cis-regulatory elements in human disease. Curr. Opin. Genet. Dev. 2013, 23, 249–256. [Google Scholar] [CrossRef]
- Bonetti, A.; Agostini, F.; Suzuki, A.M.; Hashimoto, K.; Pascarella, G.; Gimenez, J.; Roos, L.; Nash, A.J.; Ghilotti, M.; Cameron, C.J.F.; et al. RADICL-seq identifies general and cell type-specific principles of genome-wide RNA-chromatin interactions. Nat. Commun. 2020, 11, 1018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, R.; Deng, L.; Xia, Y.; Wei, X.; Cao, Y.; Guo, R.; Zhang, R.; Guo, J.; Liang, D.; Wu, L. A clear bias in parental origin of de novo pathogenic CNVs related to intellectual disability, developmental delay and multiple congenital anomalies. Sci. Rep. 2017, 7, 44446. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Ji, T.; Zhou, X.; Wang, J.; Wang, X.; Wang, J.; Zhu, D.; Zhang, X.; Sham, P.C.; Zhang, X.; et al. CNV analysis in Chinese children of mental retardation highlights a sex differentiation in parental contribution to de novo and inherited mutational burdens. Sci. Rep. 2016, 6, 25954. [Google Scholar] [CrossRef] [Green Version]
- Rheinbay, E.; Nielsen, M.M.; Abascal, F.; Wala, J.A.; Shapira, O.; Tiao, G.; Hornshoj, H.; Hess, J.M.; Juul, R.I.; Lin, Z.; et al. Analyses of non-coding somatic drivers in 2,658 cancer whole genomes. Nature 2020, 578, 102–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hua, J.T.; Ahmed, M.; Guo, H.; Zhang, Y.; Chen, S.; Soares, F.; Lu, J.; Zhou, S.; Wang, M.; Li, H.; et al. Risk SNP-Mediated Promoter-Enhancer Switching Drives Prostate Cancer through lncRNA PCAT19. Cell 2018, 174, 564.e518–575.e518. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Ma, R.; Liu, B.; Kong, J.; Lin, H.; Yu, X.; Wang, R.; Li, L.; Gao, M.; Zhou, B.; et al. SNP rs17079281 decreases lung cancer risk through creating an YY1-binding site to suppress DCBLD1 expression. Oncogene 2020, 39, 4092–4102. [Google Scholar] [CrossRef] [Green Version]
- Motawi, T.M.K.; Sadik, N.A.H.; Sabry, D.; Shahin, N.N.; Fahim, S.A. rs2267531, a promoter SNP within glypican-3 gene in the X chromosome, is associated with hepatocellular carcinoma in Egyptians. Sci. Rep. 2019, 9, 6868. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, N.; Zhang, L.; Song, Y.; Liu, J.; Yu, J.; Yang, M. Oncogene HSPH1 modulated by the rs2280059 genetic variant diminishes EGFR-TKIs efficiency in advanced lung adenocarcinoma. Carcinogenesis 2020, 41, 1195–1202. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Ahmed, M.; Zhang, F.; Yao, C.Q.; Li, S.; Liang, Y.; Hua, J.; Soares, F.; Sun, Y.; Langstein, J.; et al. Modulation of long noncoding RNAs by risk SNPs underlying genetic predispositions to prostate cancer. Nat. Genet. 2016, 48, 1142–1150. [Google Scholar] [CrossRef]
- Grampp, S.; Platt, J.L.; Lauer, V.; Salama, R.; Kranz, F.; Neumann, V.K.; Wach, S.; Stohr, C.; Hartmann, A.; Eckardt, K.U.; et al. Genetic variation at the 8q24.21 renal cancer susceptibility locus affects HIF binding to a MYC enhancer. Nat. Commun. 2016, 7, 13183. [Google Scholar] [CrossRef]
- Tuupanen, S.; Turunen, M.; Lehtonen, R.; Hallikas, O.; Vanharanta, S.; Kivioja, T.; Bjorklund, M.; Wei, G.; Yan, J.; Niittymaki, I.; et al. The common colorectal cancer predisposition SNP rs6983267 at chromosome 8q24 confers potential to enhanced Wnt signaling. Nat. Genet. 2009, 41, 885–890. [Google Scholar] [CrossRef] [Green Version]
- Walker, L.C.; BCFR; Marquart, L.; Pearson, J.F.; Wiggins, G.A.R.; O’Mara, T.A.; Parsons, M.T.; Barrowdale, D.; McGuffog, L.; Dennis, J.; et al. Evaluation of copy-number variants as modifiers of breast and ovarian cancer risk for BRCA1 pathogenic variant carriers. Eur. J. Hum. Genet. 2017, 25, 432–438. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Zou, Y.; Du, B.; Li, W.; Yu, G.; Li, L.; Zhou, L.; Gu, X.; Song, S.; Liu, Y.; et al. SNP-mediated lncRNA-ENTPD3-AS1 upregulation suppresses renal cell carcinoma via miR-155/HIF-1alpha signaling. Cell Death Dis. 2021, 12, 672. [Google Scholar] [CrossRef] [PubMed]
- Ostrovsky, O.; Grushchenko-Polaq, A.H.; Beider, K.; Mayorov, M.; Canaani, J.; Shimoni, A.; Vlodavsky, I.; Nagler, A. Identification of strong intron enhancer in the heparanase gene: Effect of functional rs4693608 variant on HPSE enhancer activity in hematological and solid malignancies. Oncogenesis 2018, 7, 51. [Google Scholar] [CrossRef] [Green Version]
- Painter, J.N.; Kaufmann, S.; O’Mara, T.A.; Hillman, K.M.; Sivakumaran, H.; Darabi, H.; Cheng, T.H.T.; Pearson, J.; Kazakoff, S.; Waddell, N.; et al. A Common Variant at the 14q32 Endometrial Cancer Risk Locus Activates AKT1 through YY1 Binding. Am. J. Hum. Genet. 2016, 98, 1159–1169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, M.; Zheng, R.; Ma, G.; Chu, H.; Lu, J.; Li, S.; Xin, J.; Tong, N.; Zhang, G.; Wang, W.; et al. Remote modulation of lncRNA GCLET by risk variant at 16p13 underlying genetic susceptibility to gastric cancer. Sci. Adv. 2020, 6, eaay5525. [Google Scholar] [CrossRef]
- Helmsauer, K.; Valieva, M.E.; Ali, S.; Chamorro Gonzalez, R.; Schopflin, R.; Roefzaad, C.; Bei, Y.; Dorado Garcia, H.; Rodriguez-Fos, E.; Puiggros, M.; et al. Enhancer hijacking determines extrachromosomal circular MYCN amplicon architecture in neuroblastoma. Nat. Commun. 2020, 11, 5823. [Google Scholar] [CrossRef]
- El Hajj, P.; Gilot, D.; Migault, M.; Theunis, A.; van Kempen, L.C.; Sales, F.; Fayyad-Kazan, H.; Badran, B.; Larsimont, D.; Awada, A.; et al. SNPs at miR-155 binding sites of TYRP1 explain discrepancy between mRNA and protein and refine TYRP1 prognostic value in melanoma. Br. J. Cancer 2015, 113, 91–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, J.; Zandi, R.; Shao, R.; Gu, J.; Ye, Y.; Wang, J.; Zhao, Y.; Pertsemlidis, A.; Wistuba, I.I.; Wu, X.; et al. A miR-SNP biomarker linked to an increased lung cancer survival by miRNA-mediated down-regulation of FZD4 expression and Wnt signaling. Sci. Rep. 2017, 7, 9029. [Google Scholar] [CrossRef] [PubMed]
- Gilam, A.; Conde, J.; Weissglas-Volkov, D.; Oliva, N.; Friedman, E.; Artzi, N.; Shomron, N. Local microRNA delivery targets Palladin and prevents metastatic breast cancer. Nat. Commun. 2016, 7, 12868. [Google Scholar] [CrossRef]
- Prensner, J.R.; Iyer, M.K.; Balbin, O.A.; Dhanasekaran, S.M.; Cao, Q.; Brenner, J.C.; Laxman, B.; Asangani, I.A.; Grasso, C.S.; Kominsky, H.D.; et al. Transcriptome sequencing across a prostate cancer cohort identifies PCAT-1, an unannotated lincRNA implicated in disease progression. Nat. Biotechnol. 2011, 29, 742–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez, N.; Cordiner, R.A.; Young, R.S.; Hug, N.; Macias, S.; Caceres, J.F. Genetic variation and RNA structure regulate microRNA biogenesis. Nat. Commun. 2017, 8, 15114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mu, Y.P.; Su, X.L. Polymorphism in pre-miR-30c contributes to gastric cancer risk in a Chinese population. Med. Oncol. 2012, 29, 1723–1732. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Liu, X.; Meng, F.; Zhang, Y.; Wang, M.; Chen, Y.; Guo, X.; Chen, W.; Wang, W. The rs7911488-T allele promotes the growth and metastasis of colorectal cancer through modulating miR-1307/PRRX1. Cell Death Dis. 2020, 11, 651. [Google Scholar] [CrossRef]
- Yang, Q.; Jie, Z.; Ye, S.; Li, Z.; Han, Z.; Wu, J.; Yang, C.; Jiang, Y. Genetic variations in miR-27a gene decrease mature miR-27a level and reduce gastric cancer susceptibility. Oncogene 2014, 33, 193–202. [Google Scholar] [CrossRef]
- Redis, R.S.; Vela, L.E.; Lu, W.; Ferreira de Oliveira, J.; Ivan, C.; Rodriguez-Aguayo, C.; Adamoski, D.; Pasculli, B.; Taguchi, A.; Chen, Y.; et al. Allele-Specific Reprogramming of Cancer Metabolism by the Long Non-coding RNA CCAT2. Mol. Cell 2016, 61, 520–534. [Google Scholar] [CrossRef] [Green Version]
- Yuan, H.; Liu, H.; Liu, Z.; Owzar, K.; Han, Y.; Su, L.; Wei, Y.; Hung, R.J.; McLaughlin, J.; Brhane, Y.; et al. A Novel Genetic Variant in Long Non-coding RNA Gene NEXN-AS1 is Associated with Risk of Lung Cancer. Sci. Rep. 2016, 6, 34234. [Google Scholar] [CrossRef]
- Wu, S.; Sun, H.; Wang, Y.; Yang, X.; Meng, Q.; Yang, H.; Zhu, H.; Tang, W.; Li, X.; Aschner, M.; et al. MALAT1 rs664589 Polymorphism Inhibits Binding to miR-194-5p, Contributing to Colorectal Cancer Risk, Growth, and Metastasis. Cancer Res. 2019, 79, 5432–5441. [Google Scholar] [CrossRef] [Green Version]
- Shen, C.; Yan, T.; Wang, Z.; Su, H.C.; Zhu, X.; Tian, X.; Fang, J.Y.; Chen, H.; Hong, J. Variant of SNP rs1317082 at CCSlnc362 (RP11-362K14.5) creates a binding site for miR-4658 and diminishes the susceptibility to CRC. Cell Death Dis. 2018, 9, 1177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H.; Zheng, J.; Deng, J.; Hu, M.; You, Y.; Li, N.; Li, W.; Lu, J.; Zhou, Y. A genetic polymorphism in lincRNA-uc003opf.1 is associated with susceptibility to esophageal squamous cell carcinoma in Chinese populations. Carcinogenesis 2013, 34, 2908–2917. [Google Scholar] [CrossRef] [Green Version]
- Zheng, J.; Huang, X.; Tan, W.; Yu, D.; Du, Z.; Chang, J.; Wei, L.; Han, Y.; Wang, C.; Che, X.; et al. Pancreatic cancer risk variant in LINC00673 creates a miR-1231 binding site and interferes with PTPN11 degradation. Nat. Genet. 2016, 48, 747–757. [Google Scholar] [CrossRef] [PubMed]
- Dhamodharan, S.; Rose, M.M.; Chakkarappan, S.R.; Umadharshini, K.V.; Arulmurugan, R.; Subbiah, S.; Inoue, I.; Munirajan, A.K. Genetic variant rs10251977 (G > A) in EGFR-AS1 modulates the expression of EGFR isoforms A and D. Sci. Rep. 2021, 11, 8808. [Google Scholar] [CrossRef]
- Lee, D.S.M.; Park, J.; Kromer, A.; Baras, A.; Rader, D.J.; Ritchie, M.D.; Ghanem, L.R.; Barash, Y. Disrupting upstream translation in mRNAs is associated with human disease. Nat. Commun. 2021, 12, 1515. [Google Scholar] [CrossRef] [PubMed]
- Steri, M.; Idda, M.L.; Whalen, M.B.; Orru, V. Genetic variants in mRNA untranslated regions. Wiley Interdiscip. Rev. RNA 2018, 9, e1474. [Google Scholar] [CrossRef] [PubMed]
- Powers, M.P. The ever-changing world of gene fusions in cancer: A secondary gene fusion and progression. Oncogene 2019, 38, 7197–7199. [Google Scholar] [CrossRef]
- Rashkin, S.R.; Graff, R.E.; Kachuri, L.; Thai, K.K.; Alexeeff, S.E.; Blatchins, M.A.; Cavazos, T.B.; Corley, D.A.; Emami, N.C.; Hoffman, J.D.; et al. Pan-cancer study detects genetic risk variants and shared genetic basis in two large cohorts. Nat. Commun. 2020, 11, 4423. [Google Scholar] [CrossRef] [PubMed]
- Giral, H.; Landmesser, U.; Kratzer, A. Into the Wild: GWAS Exploration of Non-coding RNAs. Front. Cardiovasc. Med. 2018, 5, 181. [Google Scholar] [CrossRef]
- Hrdlickova, B.; de Almeida, R.C.; Borek, Z.; Withoff, S. Genetic variation in the non-coding genome: Involvement of micro-RNAs and long non-coding RNAs in disease. Biochim. Biophys. Acta 2014, 1842, 1910–1922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Consortium, G.T. Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: Multitissue gene regulation in humans. Science 2015, 348, 648–660. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front. Endocrinol. 2018, 9, 402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, J.; Wen, J.; Huang, Z.; Chen, X.P.; Zhang, B.X.; Chu, L. Small Nucleolar RNAs: Insight into Their Function in Cancer. Front. Oncol. 2019, 9, 587. [Google Scholar] [CrossRef] [Green Version]
- Ozata, D.M.; Gainetdinov, I.; Zoch, A.; O’Carroll, D.; Zamore, P.D. PIWI-interacting RNAs: Small RNAs with big functions. Nat. Rev. Genet. 2019, 20, 89–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, K.V.; Mattick, J.S. The rise of regulatory RNA. Nat. Rev. Genet. 2014, 15, 423–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khurana, E.; Fu, Y.; Chakravarty, D.; Demichelis, F.; Rubin, M.A.; Gerstein, M. Role of non-coding sequence variants in cancer. Nat. Rev. Genet. 2016, 17, 93–108. [Google Scholar] [CrossRef]
- Zhu, H.; Uuskula-Reimand, L.; Isaev, K.; Wadi, L.; Alizada, A.; Shuai, S.; Huang, V.; Aduluso-Nwaobasi, D.; Paczkowska, M.; Abd-Rabbo, D.; et al. Candidate Cancer Driver Mutations in Distal Regulatory Elements and Long-Range Chromatin Interaction Networks. Mol. Cell 2020, 77, 1307.e1310–1321.e1310. [Google Scholar] [CrossRef]
- Cheng, Y.; Quinn, J.F.; Weiss, L.A. An eQTL mapping approach reveals that rare variants in the SEMA5A regulatory network impact autism risk. Hum. Mol. Genet. 2013, 22, 2960–2972. [Google Scholar] [CrossRef]
- Li, X.; Kim, Y.; Tsang, E.K.; Davis, J.R.; Damani, F.N.; Chiang, C.; Hess, G.T.; Zappala, Z.; Strober, B.J.; Scott, A.J.; et al. The impact of rare variation on gene expression across tissues. Nature 2017, 550, 239–243. [Google Scholar] [CrossRef] [Green Version]
- Huyghe, J.R.; Bien, S.A.; Harrison, T.A.; Kang, H.M.; Chen, S.; Schmit, S.L.; Conti, D.V.; Qu, C.; Jeon, J.; Edlund, C.K.; et al. Discovery of common and rare genetic risk variants for colorectal cancer. Nat. Genet. 2019, 51, 76–87. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Luo, Y.; Sun, Y.; Guo, W.; Zhao, X.; Xi, Y.; Ma, Y.; Shao, M.; Tan, W.; Gao, G.; et al. Genomic and transcriptomic alterations associated with drug vulnerabilities and prognosis in adenocarcinoma at the gastroesophageal junction. Nat. Commun. 2020, 11, 6091. [Google Scholar] [CrossRef] [PubMed]
- Bi, H.; Tian, T.; Zhu, L.; Zhou, H.; Hu, H.; Liu, Y.; Li, X.; Hu, F.; Zhao, Y.; Wang, G. Copy number variation of E3 ubiquitin ligase genes in peripheral blood leukocyte and colorectal cancer. Sci. Rep. 2016, 6, 29869. [Google Scholar] [CrossRef] [Green Version]
- Mamlouk, S.; Childs, L.H.; Aust, D.; Heim, D.; Melching, F.; Oliveira, C.; Wolf, T.; Durek, P.; Schumacher, D.; Blaker, H.; et al. DNA copy number changes define spatial patterns of heterogeneity in colorectal cancer. Nat. Commun. 2017, 8, 14093. [Google Scholar] [CrossRef] [PubMed]
- Fagny, M.; Platig, J.; Kuijjer, M.L.; Lin, X.; Quackenbush, J. Nongenic cancer-risk SNPs affect oncogenes, tumour-suppressor genes, and immune function. Br. J. Cancer 2020, 122, 569–577. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.Q.; Liu, X.S.; Rong, J.Y.; Gao, F.; Wu, Y.D.; Deng, C.H.; Jiang, H.Y.; Li, X.F.; Chen, Y.Q.; Zhao, Z.G.; et al. Precise diagnosis of three top cancers using dbGaP data. Sci. Rep. 2021, 11, 823. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Song, J.; Lu, Y.; Fall, K.; Chen, X.; Fang, F.; Landen, M.; Hultman, C.M.; Czene, K.; Sullivan, P.; et al. A shared genetic contribution to breast cancer and schizophrenia. Nat. Commun. 2020, 11, 4637. [Google Scholar] [CrossRef]
- Zhong, Q.; Lu, M.; Yuan, W.; Cui, Y.; Ouyang, H.; Fan, Y.; Wang, Z.; Wu, C.; Qiao, J.; Hang, J. Eight-lncRNA signature of cervical cancer were identified by integrating DNA methylation, copy number variation and transcriptome data. J. Transl. Med. 2021, 19, 58. [Google Scholar] [CrossRef]
- Diederichs, S.; Bartsch, L.; Berkmann, J.C.; Frose, K.; Heitmann, J.; Hoppe, C.; Iggena, D.; Jazmati, D.; Karschnia, P.; Linsenmeier, M.; et al. The dark matter of the cancer genome: Aberrations in regulatory elements, untranslated regions, splice sites, non-coding RNA and synonymous mutations. EMBO Mol. Med. 2016, 8, 442–457. [Google Scholar] [CrossRef] [PubMed]
- Poulos, R.C.; Sloane, M.A.; Hesson, L.B.; Wong, J.W. The search for cis-regulatory driver mutations in cancer genomes. Oncotarget 2015, 6, 32509–32525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haberle, V.; Stark, A. Eukaryotic core promoters and the functional basis of transcription initiation. Nat. Rev. Mol. Cell Biol. 2018, 19, 621–637. [Google Scholar] [CrossRef] [PubMed]
- Muratani, M.; Deng, N.; Ooi, W.F.; Lin, S.J.; Xing, M.; Xu, C.; Qamra, A.; Tay, S.T.; Malik, S.; Wu, J.; et al. Nanoscale chromatin profiling of gastric adenocarcinoma reveals cancer-associated cryptic promoters and somatically acquired regulatory elements. Nat. Commun. 2014, 5, 4361. [Google Scholar] [CrossRef]
- Liu, J.; Adhav, R.; Miao, K.; Su, S.M.; Mo, L.; Chan, U.I.; Zhang, X.; Xu, J.; Li, J.; Shu, X.; et al. Characterization of BRCA1-deficient premalignant tissues and cancers identifies Plekha5 as a tumor metastasis suppressor. Nat. Commun. 2020, 11, 4875. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Estecio, M.R.; Chen, R.; Reuben, A.; Wang, L.; Fujimoto, J.; Carrot-Zhang, J.; McGranahan, N.; Ying, L.; Fukuoka, J.; et al. Evolution of DNA methylome from precancerous lesions to invasive lung adenocarcinomas. Nat. Commun. 2021, 12, 687. [Google Scholar] [CrossRef]
- Chen, H.; Li, C.; Peng, X.; Zhou, Z.; Weinstein, J.N.; Cancer Genome Atlas Research, N.; Liang, H. A Pan-Cancer Analysis of Enhancer Expression in Nearly 9000 Patient Samples. Cell 2018, 173, 386.e312–399.e312. [Google Scholar] [CrossRef] [Green Version]
- Lidschreiber, K.; Jung, L.A.; von der Emde, H.; Dave, K.; Taipale, J.; Cramer, P.; Lidschreiber, M. Transcriptionally active enhancers in human cancer cells. Mol. Syst. Biol. 2021, 17, e9873. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Liao, Z.; Gritsch, D.; Hadzhiev, Y.; Bai, Y.; Locascio, J.J.; Guennewig, B.; Liu, G.; Blauwendraat, C.; Wang, T.; et al. Enhancers active in dopamine neurons are a primary link between genetic variation and neuropsychiatric disease. Nat. Neurosci. 2018, 21, 1482–1492. [Google Scholar] [CrossRef] [PubMed]
- Garieri, M.; Delaneau, O.; Santoni, F.; Fish, R.J.; Mull, D.; Carninci, P.; Dermitzakis, E.T.; Antonarakis, S.E.; Fort, A. The effect of genetic variation on promoter usage and enhancer activity. Nat. Commun. 2017, 8, 1358. [Google Scholar] [CrossRef]
- Elkon, R.; Agami, R. Characterization of noncoding regulatory DNA in the human genome. Nat. Biotechnol. 2017, 35, 732–746. [Google Scholar] [CrossRef]
- Thandapani, P. Super-enhancers in cancer. Pharmacol. Ther. 2019, 199, 129–138. [Google Scholar] [CrossRef]
- Li, Y.; He, Y.; Peng, J.; Su, Z.; Li, Z.; Zhang, B.; Ma, J.; Zhuo, M.; Zou, D.; Liu, X.; et al. Mutant Kras co-opts a proto-oncogenic enhancer network in inflammation-induced metaplastic progenitor cells to initiate pancreatic cancer. Nature Cancer 2021, 2, 49–65. [Google Scholar] [CrossRef]
- Chen, R.; Ren, S.; Sun, Y. Genome-wide association studies on prostate cancer: The end or the beginning? Protein Cell 2013, 4, 677–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amin Al Olama, A.; Kote-Jarai, Z.; Schumacher, F.R.; Wiklund, F.; Berndt, S.I.; Benlloch, S.; Giles, G.G.; Severi, G.; Neal, D.E.; Hamdy, F.C.; et al. A meta-analysis of genome-wide association studies to identify prostate cancer susceptibility loci associated with aggressive and non-aggressive disease. Hum. Mol. Genet. 2013, 22, 408–415. [Google Scholar] [CrossRef] [Green Version]
- Shui, I.M.; Lindstrom, S.; Kibel, A.S.; Berndt, S.I.; Campa, D.; Gerke, T.; Penney, K.L.; Albanes, D.; Berg, C.; Bueno-de-Mesquita, H.B.; et al. Prostate cancer (PCa) risk variants and risk of fatal PCa in the National Cancer Institute Breast and Prostate Cancer Cohort Consortium. Eur. Urol. 2014, 65, 1069–1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, P.; Xia, J.H.; Sipeky, C.; Dong, X.M.; Zhang, Q.; Yang, Y.; Zhang, P.; Cruz, S.P.; Zhang, K.; Zhu, J.; et al. Biology and Clinical Implications of the 19q13 Aggressive Prostate Cancer Susceptibility Locus. Cell 2018, 174, 576.e518–589.e518. [Google Scholar] [CrossRef] [Green Version]
- Shang, Z.; Yu, J.; Sun, L.; Tian, J.; Zhu, S.; Zhang, B.; Dong, Q.; Jiang, N.; Flores-Morales, A.; Chang, C.; et al. LncRNA PCAT1 activates AKT and NF-kappaB signaling in castration-resistant prostate cancer by regulating the PHLPP/FKBP51/IKKalpha complex. Nucleic Acids Res. 2019, 47, 4211–4225. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.H.; Shiota, M.; Kuroiwa, K.; Naito, S.; Oda, Y. The important role of glycine N-methyltransferase in the carcinogenesis and progression of prostate cancer. Mod. Pathol 2011, 24, 1272–1280. [Google Scholar] [CrossRef] [Green Version]
- Bonaccorsi, L.; Luciani, P.; Nesi, G.; Mannucci, E.; Deledda, C.; Dichiara, F.; Paglierani, M.; Rosati, F.; Masieri, L.; Serni, S.; et al. Androgen receptor regulation of the seladin-1/DHCR24 gene: Altered expression in prostate cancer. Lab. Investig. 2008, 88, 1049–1056. [Google Scholar] [CrossRef] [Green Version]
- Wasserman, N.F.; Aneas, I.; Nobrega, M.A. An 8q24 gene desert variant associated with prostate cancer risk confers differential in vivo activity to a MYC enhancer. Genome Res. 2010, 20, 1191–1197. [Google Scholar] [CrossRef] [Green Version]
- Wright, J.B.; Brown, S.J.; Cole, M.D. Upregulation of c-MYC in cis through a large chromatin loop linked to a cancer risk-associated single-nucleotide polymorphism in colorectal cancer cells. Mol. Cell Biol. 2010, 30, 1411–1420. [Google Scholar] [CrossRef] [Green Version]
- Doni Jayavelu, N.; Jajodia, A.; Mishra, A.; Hawkins, R.D. Candidate silencer elements for the human and mouse genomes. Nat. Commun. 2020, 11, 1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, Y.; Zhang, Y.; Loh, Y.P.; Tng, J.Q.; Lim, M.C.; Cao, Z.; Raju, A.; Lieberman Aiden, E.; Li, S.; Manikandan, L.; et al. H3K27me3-rich genomic regions can function as silencers to repress gene expression via chromatin interactions. Nat. Commun. 2021, 12, 719. [Google Scholar] [CrossRef] [PubMed]
- Pang, B.; Snyder, M.P. Systematic identification of silencers in human cells. Nat. Genet. 2020, 52, 254–263. [Google Scholar] [CrossRef] [PubMed]
- Dunning, A.M.; Michailidou, K.; Kuchenbaecker, K.B.; Thompson, D.; French, J.D.; Beesley, J.; Healey, C.S.; Kar, S.; Pooley, K.A.; Lopez-Knowles, E.; et al. Breast cancer risk variants at 6q25 display different phenotype associations and regulate ESR1, RMND1 and CCDC170. Nat. Genet. 2016, 48, 374–386. [Google Scholar] [CrossRef]
- Jing, H.; Vakoc, C.R.; Ying, L.; Mandat, S.; Wang, H.; Zheng, X.; Blobel, G.A. Exchange of GATA factors mediates transitions in looped chromatin organization at a developmentally regulated gene locus. Mol. Cell 2008, 29, 232–242. [Google Scholar] [CrossRef] [Green Version]
- Grotsch, B.; Brachs, S.; Lang, C.; Luther, J.; Derer, A.; Schlotzer-Schrehardt, U.; Bozec, A.; Fillatreau, S.; Berberich, I.; Hobeika, E.; et al. The AP-1 transcription factor Fra1 inhibits follicular B cell differentiation into plasma cells. J. Exp. Med. 2014, 211, 2199–2212. [Google Scholar] [CrossRef]
- Hadjiagapiou, C.; Borthakur, A.; Dahdal, R.Y.; Gill, R.K.; Malakooti, J.; Ramaswamy, K.; Dudeja, P.K. Role of USF1 and USF2 as potential repressor proteins for human intestinal monocarboxylate transporter 1 promoter. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 288, G1118–G1126. [Google Scholar] [CrossRef] [Green Version]
- Nakayama, A.; Murakami, H.; Maeyama, N.; Yamashiro, N.; Sakakibara, A.; Mori, N.; Takahashi, M. Role for RFX transcription factors in non-neuronal cell-specific inactivation of the microtubule-associated protein MAP1A promoter. J. Biol. Chem 2003, 278, 233–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Timblin, G.A.; Schlissel, M.S. Ebf1 and c-Myb repress rag transcription downstream of Stat5 during early B cell development. J. Immunol. 2013, 191, 4676–4687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsukumo, S.; Unno, M.; Muto, A.; Takeuchi, A.; Kometani, K.; Kurosaki, T.; Igarashi, K.; Saito, T. Bach2 maintains T cells in a naive state by suppressing effector memory-related genes. Proc. Natl. Acad. Sci. USA 2013, 110, 10735–10740. [Google Scholar] [CrossRef] [Green Version]
- Jolma, A.; Yan, J.; Whitington, T.; Toivonen, J.; Nitta, K.R.; Rastas, P.; Morgunova, E.; Enge, M.; Taipale, M.; Wei, G.; et al. DNA-binding specificities of human transcription factors. Cell 2013, 152, 327–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, Y.; Morgunova, E.; Jolma, A.; Kaasinen, E.; Sahu, B.; Khund-Sayeed, S.; Das, P.K.; Kivioja, T.; Dave, K.; Zhong, F.; et al. Impact of cytosine methylation on DNA binding specificities of human transcription factors. Science 2017, 356. [Google Scholar] [CrossRef]
- Gaszner, M.; Felsenfeld, G. Insulators: Exploiting transcriptional and epigenetic mechanisms. Nat. Rev. Genet. 2006, 7, 703–713. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Maurano, M.T.; Wang, H.; Qi, H.; Song, C.Z.; Navas, P.A.; Emery, D.W.; Stamatoyannopoulos, J.A.; Stamatoyannopoulos, G. Genomic discovery of potent chromatin insulators for human gene therapy. Nat. Biotechnol. 2015, 33, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Lou, D.; Wang, Z. Crosstalk of Genetic Variants, Allele-Specific DNA Methylation, and Environmental Factors for Complex Disease Risk. Front. Genet. 2018, 9, 695. [Google Scholar] [CrossRef] [PubMed]
- Deng, N.; Zhou, H.; Fan, H.; Yuan, Y. Single nucleotide polymorphisms and cancer susceptibility. Oncotarget 2017, 8, 110635–110649. [Google Scholar] [CrossRef] [Green Version]
- Dai, J.; Zhu, M.; Wang, C.; Shen, W.; Zhou, W.; Sun, J.; Liu, J.; Jin, G.; Ma, H.; Hu, Z.; et al. Systematical analyses of variants in CTCF-binding sites identified a novel lung cancer susceptibility locus among Chinese population. Sci. Rep. 2015, 5, 7833. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Guan, X.; Zhang, Y.; Ge, S.; Zhang, L.; Li, H.; Wang, X.; Liu, R.; Ning, T.; Deng, T.; et al. Exosomal miR-27a Derived from Gastric Cancer Cells Regulates the Transformation of Fibroblasts into Cancer-Associated Fibroblasts. Cell Physiol. Biochem. 2018, 49, 869–883. [Google Scholar] [CrossRef]
- Zhou, L.; Liang, X.; Zhang, L.; Yang, L.; Nagao, N.; Wu, H.; Liu, C.; Lin, S.; Cai, G.; Liu, J. MiR-27a-3p functions as an oncogene in gastric cancer by targeting BTG2. Oncotarget 2016, 7, 51943–51954. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H.; Hwang, J.; Jung, J.H.; Lee, H.J.; Lee, D.Y.; Kim, S.H. Molecular networks of FOXP family: Dual biologic functions, interplay with other molecules and clinical implications in cancer progression. Mol. Cancer 2019, 18, 180. [Google Scholar] [CrossRef] [PubMed]
- Statello, L.; Guo, C.J.; Chen, L.L.; Huarte, M. Gene regulation by long non-coding RNAs and its biological functions. Nat. Rev. Mol. Cell Biol. 2021, 22, 96–118. [Google Scholar] [CrossRef] [PubMed]
- Barbieri, I.; Kouzarides, T. Role of RNA modifications in cancer. Nat. Rev. Cancer 2020, 20, 303–322. [Google Scholar] [CrossRef]
- Peng, Y.; Croce, C.M. The role of MicroRNAs in human cancer. Signal Transduct. Target. Ther. 2016, 1, 15004. [Google Scholar] [CrossRef] [Green Version]
- Qi, Y.; Lai, Y.L.; Shen, P.C.; Chen, F.H.; Lin, L.J.; Wu, H.H.; Peng, P.H.; Hsu, K.W.; Cheng, W.C. Identification and validation of a miRNA-based prognostic signature for cervical cancer through an integrated bioinformatics approach. Sci. Rep. 2020, 10, 22270. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Getz, G.; Miska, E.A.; Alvarez-Saavedra, E.; Lamb, J.; Peck, D.; Sweet-Cordero, A.; Ebert, B.L.; Mak, R.H.; Ferrando, A.A.; et al. MicroRNA expression profiles classify human cancers. Nature 2005, 435, 834–838. [Google Scholar] [CrossRef] [PubMed]
- Helwak, A.; Kudla, G.; Dudnakova, T.; Tollervey, D. Mapping the human miRNA interactome by CLASH reveals frequent noncanonical binding. Cell 2013, 153, 654–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryan, B.M.; Robles, A.I.; Harris, C.C. Genetic variation in microRNA networks: The implications for cancer research. Nat. Rev. Cancer 2010, 10, 389–402. [Google Scholar] [CrossRef] [PubMed]
- Dhawan, A.; Scott, J.G.; Harris, A.L.; Buffa, F.M. Pan-cancer characterisation of microRNA across cancer hallmarks reveals microRNA-mediated downregulation of tumour suppressors. Nat. Commun. 2018, 9, 5228. [Google Scholar] [CrossRef]
- Lin, W.C.; Shih, P.H.; Wang, W.; Wu, C.H.; Hsia, S.M.; Wang, H.J.; Hwang, P.A.; Wang, C.Y.; Chen, S.H.; Kuo, Y.T. Inhibitory effects of high stability fucoxanthin on palmitic acid-induced lipid accumulation in human adipose-derived stem cells through modulation of long non-coding RNA. Food Funct. 2015, 6, 2215–2223. [Google Scholar] [CrossRef]
- Lee, A.R.; Park, J.; Jung, K.J.; Jee, S.H.; Kim-Yoon, S. Genetic variation rs7930 in the miR-4273-5p target site is associated with a risk of colorectal cancer. Onco. Targets Ther. 2016, 9, 6885–6895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Du, M.; Ma, L.; Chu, H.; Lv, Q.; Ye, D.; Guo, J.; Gu, C.; Xia, G.; Zhu, Y.; et al. A functional variant in TP63 at 3q28 associated with bladder cancer risk by creating an miR-140-5p binding site. Int. J. Cancer 2016, 139, 65–74. [Google Scholar] [CrossRef]
- Wynendaele, J.; Bohnke, A.; Leucci, E.; Nielsen, S.J.; Lambertz, I.; Hammer, S.; Sbrzesny, N.; Kubitza, D.; Wolf, A.; Gradhand, E.; et al. An illegitimate microRNA target site within the 3’ UTR of MDM4 affects ovarian cancer progression and chemosensitivity. Cancer Res. 2010, 70, 9641–9649. [Google Scholar] [CrossRef] [Green Version]
- Jacinta-Fernandes, A.; Xavier, J.M.; Magno, R.; Lage, J.G.; Maia, A.T. Allele-specific miRNA-binding analysis identifies candidate target genes for breast cancer risk. NPJ Genom. Med. 2020, 5, 4. [Google Scholar] [CrossRef] [Green Version]
- Hua, K.T.; Liu, Y.F.; Hsu, C.L.; Cheng, T.Y.; Yang, C.Y.; Chang, J.S.; Lee, W.J.; Hsiao, M.; Juan, H.F.; Chien, M.H.; et al. 3’UTR polymorphisms of carbonic anhydrase IX determine the miR-34a targeting efficiency and prognosis of hepatocellular carcinoma. Sci. Rep. 2017, 7, 4466. [Google Scholar] [CrossRef] [Green Version]
- Hogg, D.R.; Harries, L.W. Human genetic variation and its effect on miRNA biogenesis, activity and function. Biochem. Soc. Trans. 2014, 42, 1184–1189. [Google Scholar] [CrossRef]
- Song, X.; Wan, X.; Huang, T.; Zeng, C.; Sastry, N.; Wu, B.; James, C.D.; Horbinski, C.; Nakano, I.; Zhang, W.; et al. SRSF3-Regulated RNA Alternative Splicing Promotes Glioblastoma Tumorigenicity by Affecting Multiple Cellular Processes. Cancer Res. 2019, 79, 5288–5301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mori, M.; Triboulet, R.; Mohseni, M.; Schlegelmilch, K.; Shrestha, K.; Camargo, F.D.; Gregory, R.I. Hippo signaling regulates microprocessor and links cell-density-dependent miRNA biogenesis to cancer. Cell 2014, 156, 893–906. [Google Scholar] [CrossRef] [Green Version]
- Gould, P.S.; Bird, H.; Easton, A.J. Translation toeprinting assays using fluorescently labeled primers and capillary electrophoresis. Biotechniques 2005, 38, 397–400. [Google Scholar] [CrossRef]
- Wilkinson, K.A.; Merino, E.J.; Weeks, K.M. Selective 2’-hydroxyl acylation analyzed by primer extension (SHAPE): Quantitative RNA structure analysis at single nucleotide resolution. Nat. Protoc. 2006, 1, 1610–1616. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Ambrosone, C.B.; Zhao, H. Novel genetic variants in microRNA genes and familial breast cancer. Int. J. Cancer 2009, 124, 1178–1182. [Google Scholar] [CrossRef] [PubMed]
- Bockhorn, J.; Dalton, R.; Nwachukwu, C.; Huang, S.; Prat, A.; Yee, K.; Chang, Y.F.; Huo, D.; Wen, Y.; Swanson, K.E.; et al. MicroRNA-30c inhibits human breast tumour chemotherapy resistance by regulating TWF1 and IL-11. Nat. Commun. 2013, 4, 1393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huarte, M. The emerging role of lncRNAs in cancer. Nat. Med. 2015, 21, 1253–1261. [Google Scholar] [CrossRef] [PubMed]
- Yousefi, H.; Maheronnaghsh, M.; Molaei, F.; Mashouri, L.; Reza Aref, A.; Momeny, M.; Alahari, S.K. Long noncoding RNAs and exosomal lncRNAs: Classification, and mechanisms in breast cancer metastasis and drug resistance. Oncogene 2020, 39, 953–974. [Google Scholar] [CrossRef] [PubMed]
- Chiu, H.S.; Somvanshi, S.; Patel, E.; Chen, T.W.; Singh, V.P.; Zorman, B.; Patil, S.L.; Pan, Y.; Chatterjee, S.S.; Cancer Genome Atlas Research, N.; et al. Pan-Cancer Analysis of lncRNA Regulation Supports Their Targeting of Cancer Genes in Each Tumor Context. Cell Rep. 2018, 23, 297.e212–312.e212. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.Z.; Liu, H.; Chen, S.R. Mechanisms of Long Non-Coding RNAs in Cancers and Their Dynamic Regulations. Cancers 2020, 12, 1245. [Google Scholar] [CrossRef]
- Begolli, R.; Sideris, N.; Giakountis, A. LncRNAs as Chromatin Regulators in Cancer: From Molecular Function to Clinical Potential. Cancers 2019, 11, 1524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, C.; Kanhere, A. The Missing Link Between Cancer-Associated Variants and LncRNAs. Trends Genet. 2021, 37, 410–413. [Google Scholar] [CrossRef]
- Weinhold, N.; Jacobsen, A.; Schultz, N.; Sander, C.; Lee, W. Genome-wide analysis of noncoding regulatory mutations in cancer. Nat. Genet. 2014, 46, 1160–1165. [Google Scholar] [CrossRef]
- Johnsson, P.; Lipovich, L.; Grander, D.; Morris, K.V. Evolutionary conservation of long non-coding RNAs; sequence, structure, function. Biochim. Biophys. Acta 2014, 1840, 1063–1071. [Google Scholar] [CrossRef] [Green Version]
- Kapusta, A.; Feschotte, C. Volatile evolution of long noncoding RNA repertoires: Mechanisms and biological implications. Trends Genet. 2014, 30, 439–452. [Google Scholar] [CrossRef] [Green Version]
- Jonas, K.; Calin, G.A.; Pichler, M. RNA-Binding Proteins as Important Regulators of Long Non-Coding RNAs in Cancer. Int. J. Mol. Sci. 2020, 21, 2969. [Google Scholar] [CrossRef] [Green Version]
- Pisignano, G.; Ladomery, M. Post-Transcriptional Regulation through Long Non-Coding RNAs (lncRNAs). Noncoding RNA 2021, 7, 29. [Google Scholar] [CrossRef]
- Liz, J.; Portela, A.; Soler, M.; Gomez, A.; Ling, H.; Michlewski, G.; Calin, G.A.; Guil, S.; Esteller, M. Regulation of pri-miRNA processing by a long noncoding RNA transcribed from an ultraconserved region. Mol. Cell 2014, 55, 138–147. [Google Scholar] [CrossRef] [Green Version]
- Dhir, A.; Dhir, S.; Proudfoot, N.J.; Jopling, C.L. Microprocessor mediates transcriptional termination of long noncoding RNA transcripts hosting microRNAs. Nat. Struct. Mol. Biol. 2015, 22, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Gil, N.; Ulitsky, I. Regulation of gene expression by cis-acting long non-coding RNAs. Nat. Rev. Genet. 2020, 21, 102–117. [Google Scholar] [CrossRef] [PubMed]
- Zampetaki, A.; Albrecht, A.; Steinhofel, K. Long Non-coding RNA Structure and Function: Is There a Link? Front. Physiol. 2018, 9, 1201. [Google Scholar] [CrossRef] [PubMed]
- Aznaourova, M.; Schmerer, N.; Schmeck, B.; Schulte, L.N. Disease-Causing Mutations and Rearrangements in Long Non-coding RNA Gene Loci. Front. Genet. 2020, 11, 527484. [Google Scholar] [CrossRef]
- Gao, P.; Wei, G.H. Genomic Insight into the Role of lncRNA in Cancer Susceptibility. Int. J. Mol. Sci. 2017, 18, 1239. [Google Scholar] [CrossRef] [Green Version]
- Tang, X.; Feng, D.; Li, M.; Zhou, J.; Li, X.; Zhao, D.; Hao, B.; Li, D.; Ding, K. Transcriptomic Analysis of mRNA-lncRNA-miRNA Interactions in Hepatocellular Carcinoma. Sci. Rep. 2019, 9, 16096. [Google Scholar] [CrossRef]
- Xia, T.; Liao, Q.; Jiang, X.; Shao, Y.; Xiao, B.; Xi, Y.; Guo, J. Long noncoding RNA associated-competing endogenous RNAs in gastric cancer. Sci. Rep. 2014, 4, 6088. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Liu, S.; Cai, G.; Kong, L.; Zhang, T.; Ren, Y.; Wu, Y.; Mei, M.; Zhang, L.; Wang, X. Long Non Coding RNA MALAT1 Promotes Tumor Growth and Metastasis by inducing Epithelial-Mesenchymal Transition in Oral Squamous Cell Carcinoma. Sci. Rep. 2015, 5, 15972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.; Li, X.; Qiao, L.; Liu, W.; Xu, C.; Wang, X. MALAT1 regulates miR-34a expression in melanoma cells. Cell Death Dis. 2019, 10, 389. [Google Scholar] [CrossRef]
- Han, Y.; Wu, Z.; Wu, T.; Huang, Y.; Cheng, Z.; Li, X.; Sun, T.; Xie, X.; Zhou, Y.; Du, Z. Tumor-suppressive function of long noncoding RNA MALAT1 in glioma cells by downregulation of MMP2 and inactivation of ERK/MAPK signaling. Cell Death Dis. 2016, 7, e2123. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Piao, H.L.; Kim, B.J.; Yao, F.; Han, Z.; Wang, Y.; Xiao, Z.; Siverly, A.N.; Lawhon, S.E.; Ton, B.N.; et al. Long noncoding RNA MALAT1 suppresses breast cancer metastasis. Nat. Genet. 2018, 50, 1705–1715. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Guo, Z.; Zhao, Y.; Jin, Y.; An, L.; Wu, B.; Liu, Z.; Chen, X.; Chen, X.; Zhou, H.; et al. Genetic polymorphisms of lncRNA-p53 regulatory network genes are associated with concurrent chemoradiotherapy toxicities and efficacy in nasopharyngeal carcinoma patients. Sci. Rep. 2017, 7, 8320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, B.; Liu, C.; Li, H.; Zhang, L.; Luo, G.; Liang, S.; Lu, M. Research progress on the interactions between long non-coding RNAs and microRNAs in human cancer. Oncol. Lett. 2020, 19, 595–605. [Google Scholar] [CrossRef] [Green Version]
- Jin, N.; Bi, A.; Lan, X.; Xu, J.; Wang, X.; Liu, Y.; Wang, T.; Tang, S.; Zeng, H.; Chen, Z.; et al. Identification of metabolic vulnerabilities of receptor tyrosine kinases-driven cancer. Nat. Commun. 2019, 10, 2701. [Google Scholar] [CrossRef] [Green Version]
- Lo, R.S. Receptor tyrosine kinases in cancer escape from BRAF inhibitors. Cell Res. 2012, 22, 945–947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, Z.; Lovly, C.M. Mechanisms of receptor tyrosine kinase activation in cancer. Mol. Cancer 2018, 17, 58. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.; Brown, B.P.; Kim, S.; Ferguson, D.; Pavlick, D.C.; Jayakumaran, G.; Benayed, R.; Gallant, J.N.; Zhang, Y.K.; Yan, Y.; et al. Structure-function analysis of oncogenic EGFR Kinase Domain Duplication reveals insights into activation and a potential approach for therapeutic targeting. Nat. Commun. 2021, 12, 1382. [Google Scholar] [CrossRef] [PubMed]
- Banys-Paluchowski, M.; Witzel, I.; Riethdorf, S.; Rack, B.; Janni, W.; Fasching, P.A.; Solomayer, E.F.; Aktas, B.; Kasimir-Bauer, S.; Pantel, K.; et al. Evaluation of serum epidermal growth factor receptor (EGFR) in correlation to circulating tumor cells in patients with metastatic breast cancer. Sci. Rep. 2017, 7, 17307. [Google Scholar] [CrossRef] [Green Version]
- Nakagawa, T.; Takeuchi, S.; Yamada, T.; Nanjo, S.; Ishikawa, D.; Sano, T.; Kita, K.; Nakamura, T.; Matsumoto, K.; Suda, K.; et al. Combined therapy with mutant-selective EGFR inhibitor and Met kinase inhibitor for overcoming erlotinib resistance in EGFR-mutant lung cancer. Mol. Cancer Ther. 2012, 11, 2149–2157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandes Neto, J.M.; Nadal, E.; Bosdriesz, E.; Ooft, S.N.; Farre, L.; McLean, C.; Klarenbeek, S.; Jurgens, A.; Hagen, H.; Wang, L.; et al. Multiple low dose therapy as an effective strategy to treat EGFR inhibitor-resistant NSCLC tumours. Nat. Commun. 2020, 11, 3157. [Google Scholar] [CrossRef]
- Gyorffy, B.; Pongor, L.; Bottai, G.; Li, X.; Budczies, J.; Szabo, A.; Hatzis, C.; Pusztai, L.; Santarpia, L. An integrative bioinformatics approach reveals coding and non-coding gene variants associated with gene expression profiles and outcome in breast cancer molecular subtypes. Br. J. Cancer 2018, 118, 1107–1114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soltis, A.R.; Dalgard, C.L.; Pollard, H.B.; Wilkerson, M.D. MutEnricher: A flexible toolset for somatic mutation enrichment analysis of tumor whole genomes. BMC Bioinform. 2020, 21, 338. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef] [Green Version]
- Firth, H.V.; Richards, S.M.; Bevan, A.P.; Clayton, S.; Corpas, M.; Rajan, D.; Van Vooren, S.; Moreau, Y.; Pettett, R.M.; Carter, N.P. DECIPHER: Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources. Am. J. Hum. Genet. 2009, 84, 524–533. [Google Scholar] [CrossRef] [Green Version]
- Fulco, C.P.; Nasser, J.; Jones, T.R.; Munson, G.; Bergman, D.T.; Subramanian, V.; Grossman, S.R.; Anyoha, R.; Doughty, B.R.; Patwardhan, T.A.; et al. Activity-by-contact model of enhancer-promoter regulation from thousands of CRISPR perturbations. Nat. Genet. 2019, 51, 1664–1669. [Google Scholar] [CrossRef]
- Sheffield, N.C.; Furey, T.S. Identifying and characterizing regulatory sequences in the human genome with chromatin accessibility assays. Genes 2012, 3, 651–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fadason, T.; Schierding, W.; Lumley, T.; O’Sullivan, J.M. Chromatin interactions and expression quantitative trait loci reveal genetic drivers of multimorbidities. Nat. Commun. 2018, 9, 5198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keele, G.R.; Quach, B.C.; Israel, J.W.; Chappell, G.A.; Lewis, L.; Safi, A.; Simon, J.M.; Cotney, P.; Crawford, G.E.; Valdar, W.; et al. Integrative QTL analysis of gene expression and chromatin accessibility identifies multi-tissue patterns of genetic regulation. PLoS Genet. 2020, 16, e1008537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Rendon, A.; Wernisch, L. Transcription factor and chromatin features predict genes associated with eQTLs. Nucleic Acids Res. 2013, 41, 1450–1463. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.; Jang, I.; Kim, M.; Choi, J.; Kim, M.S.; Lee, B.; Jung, I. 3DIV update for 2021: A comprehensive resource of 3D genome and 3D cancer genome. Nucleic Acids Res. 2021, 49, D38–D46. [Google Scholar] [CrossRef] [PubMed]
- Wu, C. The 5′ ends of Drosophila heat shock genes in chromatin are hypersensitive to DNase I. Nature 1980, 286, 854–860. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.; Chen, D.; Chen, Y. Advances of DNase-seq for mapping active gene regulatory elements across the genome in animals. Gene 2018, 667, 83–94. [Google Scholar] [CrossRef]
- Kumar, V.; Muratani, M.; Rayan, N.A.; Kraus, P.; Lufkin, T.; Ng, H.H.; Prabhakar, S. Uniform, optimal signal processing of mapped deep-sequencing data. Nat. Biotechnol. 2013, 31, 615–622. [Google Scholar] [CrossRef] [Green Version]
- Lu, F.; Liu, Y.; Inoue, A.; Suzuki, T.; Zhao, K.; Zhang, Y. Establishing Chromatin Regulatory Landscape during Mouse Preimplantation Development. Cell 2016, 165, 1375–1388. [Google Scholar] [CrossRef] [Green Version]
- Yan, H.; Tian, S.; Slager, S.L.; Sun, Z.; Ordog, T. Genome-Wide Epigenetic Studies in Human Disease: A Primer on -Omic Technologies. Am. J. Epidemiol. 2016, 183, 96–109. [Google Scholar] [CrossRef] [Green Version]
- Zentner, G.E.; Henikoff, S. Surveying the epigenomic landscape, one base at a time. Genome Biol. 2012, 13, 250. [Google Scholar] [CrossRef]
- Guo, F.; Li, L.; Li, J.; Wu, X.; Hu, B.; Zhu, P.; Wen, L.; Tang, F. Single-cell multi-omics sequencing of mouse early embryos and embryonic stem cells. Cell Res. 2017, 27, 967–988. [Google Scholar] [CrossRef] [PubMed]
- D’antonio, M.; Weghorn, D.; D’antonio-Chronowska, A.; Coulet, F.; Olson, K.M.; DeBoever, C.; Drees, F.; Arias, A.; Alakus, H.; Richardson, A.L.; et al. Identifying DNase I hypersensitive sites as driver distal regulatory elements in breast cancer. Nat. Commun. 2017, 8, 436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, W.; Tang, Q.; Wan, M.; Cui, K.; Zhang, Y.; Ren, G.; Ni, B.; Sklar, J.; Przytycka, T.M.; Childs, R.; et al. Genome-wide detection of DNase I hypersensitive sites in single cells and FFPE tissue samples. Nature 2015, 528, 142–146. [Google Scholar] [CrossRef] [Green Version]
- Simon, J.M.; Giresi, P.G.; Davis, I.J.; Lieb, J.D. Using formaldehyde-assisted isolation of regulatory elements (FAIRE) to isolate active regulatory DNA. Nat. Protoc. 2012, 7, 256–267. [Google Scholar] [CrossRef] [Green Version]
- Schones, D.E.; Cui, K.; Cuddapah, S.; Roh, T.Y.; Barski, A.; Wang, Z.; Wei, G.; Zhao, K. Dynamic regulation of nucleosome positioning in the human genome. Cell 2008, 132, 887–898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, D.C.; Hainer, S.J. Genomic methods in profiling DNA accessibility and factor localization. Chromosome Res. 2020, 28, 69–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buenrostro, J.D.; Giresi, P.G.; Zaba, L.C.; Chang, H.Y.; Greenleaf, W.J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 2013, 10, 1213–1218. [Google Scholar] [CrossRef]
- Cui, K.; Zhao, K. Genome-wide approaches to determining nucleosome occupancy in metazoans using MNase-Seq. Methods Mol. Biol. 2012, 833, 413–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shashikant, T.; Ettensohn, C.A. Genome-wide analysis of chromatin accessibility using ATAC-seq. Methods Cell Biol. 2019, 151, 219–235. [Google Scholar] [CrossRef]
- Bryois, J.; Garrett, M.E.; Song, L.; Safi, A.; Giusti-Rodriguez, P.; Johnson, G.D.; Shieh, A.W.; Buil, A.; Fullard, J.F.; Roussos, P.; et al. Evaluation of chromatin accessibility in prefrontal cortex of individuals with schizophrenia. Nat. Commun. 2018, 9, 3121. [Google Scholar] [CrossRef]
- Kheradpour, P.; Ernst, J.; Melnikov, A.; Rogov, P.; Wang, L.; Zhang, X.; Alston, J.; Mikkelsen, T.S.; Kellis, M. Systematic dissection of regulatory motifs in 2000 predicted human enhancers using a massively parallel reporter assay. Genome Res. 2013, 23, 800–811. [Google Scholar] [CrossRef] [Green Version]
- Kwasnieski, J.C.; Mogno, I.; Myers, C.A.; Corbo, J.C.; Cohen, B.A. Complex effects of nucleotide variants in a mammalian cis-regulatory element. Proc. Natl. Acad. Sci. USA 2012, 109, 19498–19503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birnbaum, R.Y.; Patwardhan, R.P.; Kim, M.J.; Findlay, G.M.; Martin, B.; Zhao, J.; Bell, R.J.; Smith, R.P.; Ku, A.A.; Shendure, J.; et al. Systematic dissection of coding exons at single nucleotide resolution supports an additional role in cell-specific transcriptional regulation. PLoS Genet. 2014, 10, e1004592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melnikov, A.; Murugan, A.; Zhang, X.; Tesileanu, T.; Wang, L.; Rogov, P.; Feizi, S.; Gnirke, A.; Callan, C.G., Jr.; Kinney, J.B.; et al. Systematic dissection and optimization of inducible enhancers in human cells using a massively parallel reporter assay. Nat. Biotechnol. 2012, 30, 271–277. [Google Scholar] [CrossRef] [Green Version]
- Patwardhan, R.P.; Hiatt, J.B.; Witten, D.M.; Kim, M.J.; Smith, R.P.; May, D.; Lee, C.; Andrie, J.M.; Lee, S.I.; Cooper, G.M.; et al. Massively parallel functional dissection of mammalian enhancers in vivo. Nat. Biotechnol. 2012, 30, 265–270. [Google Scholar] [CrossRef] [Green Version]
- White, M.A.; Myers, C.A.; Corbo, J.C.; Cohen, B.A. Massively parallel in vivo enhancer assay reveals that highly local features determine the cis-regulatory function of ChIP-seq peaks. Proc. Natl. Acad. Sci. USA 2013, 110, 11952–11957. [Google Scholar] [CrossRef] [Green Version]
- Arnold, C.D.; Gerlach, D.; Stelzer, C.; Boryn, L.M.; Rath, M.; Stark, A. Genome-wide quantitative enhancer activity maps identified by STARR-seq. Science 2013, 339, 1074–1077. [Google Scholar] [CrossRef]
- Muerdter, F.; Boryn, L.M.; Arnold, C.D. STARR-seq - principles and applications. Genomics 2015, 106, 145–150. [Google Scholar] [CrossRef] [Green Version]
- Klein, J.C.; Keith, A.; Rice, S.J.; Shepherd, C.; Agarwal, V.; Loughlin, J.; Shendure, J. Functional testing of thousands of osteoarthritis-associated variants for regulatory activity. Nat. Commun. 2019, 10, 2434. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Xia, J.H.; Zhu, J.; Gao, P.; Tian, Y.J.; Du, M.; Guo, Y.C.; Suleman, S.; Zhang, Q.; Kohli, M.; et al. High-throughput screening of prostate cancer risk loci by single nucleotide polymorphisms sequencing. Nat. Commun. 2018, 9, 2022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanhille, L.; Griffon, A.; Maqbool, M.A.; Zacarias-Cabeza, J.; Dao, L.T.; Fernandez, N.; Ballester, B.; Andrau, J.C.; Spicuglia, S. High-throughput and quantitative assessment of enhancer activity in mammals by CapStarr-seq. Nat. Commun. 2015, 6, 6905. [Google Scholar] [CrossRef] [Green Version]
- Arnold, C.D.; Gerlach, D.; Spies, D.; Matts, J.A.; Sytnikova, Y.A.; Pagani, M.; Lau, N.C.; Stark, A. Quantitative genome-wide enhancer activity maps for five Drosophila species show functional enhancer conservation and turnover during cis-regulatory evolution. Nat. Genet. 2014, 46, 685–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, G.D.; Barrera, A.; McDowell, I.C.; D’Ippolito, A.M.; Majoros, W.H.; Vockley, C.M.; Wang, X.; Allen, A.S.; Reddy, T.E. Human genome-wide measurement of drug-responsive regulatory activity. Nat. Commun. 2018, 9, 5317. [Google Scholar] [CrossRef]
- Gong, H.; Yang, Y.; Zhang, S.; Li, M.; Zhang, X. Application of Hi-C and other omics data analysis in human cancer and cell differentiation research. Comput. Struct. Biotechnol. J. 2021, 19, 2070–2083. [Google Scholar] [CrossRef]
- Fullwood, M.J.; Ruan, Y. ChIP-based methods for the identification of long-range chromatin interactions. J. Cell Biochem. 2009, 107, 30–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Zhao, K. The Toolbox for Untangling Chromosome Architecture in Immune Cells. Front. Immunol. 2021, 12, 670884. [Google Scholar] [CrossRef]
- Li, G.; Cai, L.; Chang, H.; Hong, P.; Zhou, Q.; Kulakova, E.V.; Kolchanov, N.A.; Ruan, Y. Chromatin Interaction Analysis with Paired-End Tag (ChIA-PET) sequencing technology and application. BMC Genom. 2014, 15 (Suppl. S12), S11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Luo, O.J.; Wang, P.; Zheng, M.; Wang, D.; Piecuch, E.; Zhu, J.J.; Tian, S.Z.; Tang, Z.; Li, G.; et al. Long-read ChIA-PET for base-pair-resolution mapping of haplotype-specific chromatin interactions. Nat. Protoc. 2017, 12, 899–915. [Google Scholar] [CrossRef] [Green Version]
- Al Bkhetan, Z.; Plewczynski, D. Three-dimensional Epigenome Statistical Model: Genome-wide Chromatin Looping Prediction. Sci. Rep. 2018, 8, 5217. [Google Scholar] [CrossRef] [PubMed]
- Fullwood, M.J.; Liu, M.H.; Pan, Y.F.; Liu, J.; Xu, H.; Mohamed, Y.B.; Orlov, Y.L.; Velkov, S.; Ho, A.; Mei, P.H.; et al. An oestrogen-receptor-alpha-bound human chromatin interactome. Nature 2009, 462, 58–64. [Google Scholar] [CrossRef] [Green Version]
- Xi, W.; Beer, M.A. Loop competition and extrusion model predicts CTCF interaction specificity. Nat. Commun. 2021, 12, 1046. [Google Scholar] [CrossRef] [PubMed]
- Couch, F.J.; Kuchenbaecker, K.B.; Michailidou, K.; Mendoza-Fandino, G.A.; Nord, S.; Lilyquist, J.; Olswold, C.; Hallberg, E.; Agata, S.; Ahsan, H.; et al. Identification of four novel susceptibility loci for oestrogen receptor negative breast cancer. Nat. Commun. 2016, 7, 11375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milne, R.L.; Kuchenbaecker, K.B.; Michailidou, K.; Beesley, J.; Kar, S.; Lindstrom, S.; Hui, S.; Lemacon, A.; Soucy, P.; Dennis, J.; et al. Identification of ten variants associated with risk of estrogen-receptor-negative breast cancer. Nat. Genet. 2017, 49, 1767–1778. [Google Scholar] [CrossRef] [Green Version]
- Cai, L.; Chang, H.; Fang, Y.; Li, G. A Comprehensive Characterization of the Function of LincRNAs in Transcriptional Regulation Through Long-Range Chromatin Interactions. Sci. Rep. 2016, 6, 36572. [Google Scholar] [CrossRef] [PubMed]
- Mumbach, M.R.; Rubin, A.J.; Flynn, R.A.; Dai, C.; Khavari, P.A.; Greenleaf, W.J.; Chang, H.Y. HiChIP: Efficient and sensitive analysis of protein-directed genome architecture. Nat. Methods 2016, 13, 919–922. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharyya, S.; Chandra, V.; Vijayanand, P.; Ay, F. Identification of significant chromatin contacts from HiChIP data by FitHiChIP. Nat. Commun. 2019, 10, 4221. [Google Scholar] [CrossRef] [Green Version]
- Ray, J.P.; de Boer, C.G.; Fulco, C.P.; Lareau, C.A.; Kanai, M.; Ulirsch, J.C.; Tewhey, R.; Ludwig, L.S.; Reilly, S.K.; Bergman, D.T.; et al. Prioritizing disease and trait causal variants at the TNFAIP3 locus using functional and genomic features. Nat. Commun. 2020, 11, 1237. [Google Scholar] [CrossRef]
- O’Mara, T.A.; Spurdle, A.B.; Glubb, D.M.; Endometrial Cancer Association, C. Analysis of Promoter-Associated Chromatin Interactions Reveals Biologically Relevant Candidate Target Genes at Endometrial Cancer Risk Loci. Cancers 2019, 11, 1440. [Google Scholar] [CrossRef] [Green Version]
- Fang, R.; Yu, M.; Li, G.; Chee, S.; Liu, T.; Schmitt, A.D.; Ren, B. Mapping of long-range chromatin interactions by proximity ligation-assisted ChIP-seq. Cell Res. 2016, 26, 1345–1348. [Google Scholar] [CrossRef] [Green Version]
- Corces, M.R.; Shcherbina, A.; Kundu, S.; Gloudemans, M.J.; Fresard, L.; Granja, J.M.; Louie, B.H.; Eulalio, T.; Shams, S.; Bagdatli, S.T.; et al. Single-cell epigenomic analyses implicate candidate causal variants at inherited risk loci for Alzheimer’s and Parkinson’s diseases. Nat. Genet. 2020, 52, 1158–1168. [Google Scholar] [CrossRef]
- Deng, X.; Kong, F.; Li, S.; Jiang, H.; Dong, L.; Xu, X.; Zhang, X.; Yuan, H.; Xu, Y.; Chu, Y.; et al. A KLF4/PiHL/EZH2/HMGA2 regulatory axis and its function in promoting oxaliplatin-resistance of colorectal cancer. Cell Death Dis. 2021, 12, 485. [Google Scholar] [CrossRef]
- Feng, Y.C.; Liu, X.Y.; Teng, L.; Ji, Q.; Wu, Y.; Li, J.M.; Gao, W.; Zhang, Y.Y.; La, T.; Tabatabaee, H.; et al. c-Myc inactivation of p53 through the pan-cancer lncRNA MILIP drives cancer pathogenesis. Nat. Commun. 2020, 11, 4980. [Google Scholar] [CrossRef]
- Xu, L.; Huan, L.; Guo, T.; Wu, Y.; Liu, Y.; Wang, Q.; Huang, S.; Xu, Y.; Liang, L.; He, X. LncRNA SNHG11 facilitates tumor metastasis by interacting with and stabilizing HIF-1alpha. Oncogene 2020, 39, 7005–7018. [Google Scholar] [CrossRef] [PubMed]
- Mondal, T.; Subhash, S.; Vaid, R.; Enroth, S.; Uday, S.; Reinius, B.; Mitra, S.; Mohammed, A.; James, A.R.; Hoberg, E.; et al. MEG3 long noncoding RNA regulates the TGF-beta pathway genes through formation of RNA-DNA triplex structures. Nat. Commun. 2015, 6, 7743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feretzaki, M.; Pospisilova, M.; Valador Fernandes, R.; Lunardi, T.; Krejci, L.; Lingner, J. RAD51-dependent recruitment of TERRA lncRNA to telomeres through R-loops. Nature 2020, 587, 303–308. [Google Scholar] [CrossRef] [PubMed]
- Laffleur, B.; Lim, J.; Zhang, W.; Chen, Y.; Pefanis, E.; Bizarro, J.; Batista, C.R.; Wu, L.; Economides, A.N.; Wang, J.; et al. Noncoding RNA processing by DIS3 regulates chromosomal architecture and somatic hypermutation in B cells. Nat. Genet. 2021, 53, 230–242. [Google Scholar] [CrossRef]
- Chu, C.; Qu, K.; Zhong, F.L.; Artandi, S.E.; Chang, H.Y. Genomic maps of long noncoding RNA occupancy reveal principles of RNA-chromatin interactions. Mol. Cell 2011, 44, 667–678. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.C.; Zaleta-Rivera, K.; Huang, X.; Dai, X.; Zhong, S. RNA, Action through Interactions. Trends Genet. 2018, 34, 867–882. [Google Scholar] [CrossRef]
- Kato, M.; Carninci, P. Genome-Wide Technologies to Study RNA-Chromatin Interactions. Noncoding RNA 2020, 6, 20. [Google Scholar] [CrossRef]
- Machyna, M.; Simon, M.D. Catching RNAs on chromatin using hybridization capture methods. Brief. Funct. Genom. 2018, 17, 96–103. [Google Scholar] [CrossRef]
- Sridhar, B.; Rivas-Astroza, M.; Nguyen, T.C.; Chen, W.; Yan, Z.; Cao, X.; Hebert, L.; Zhong, S. Systematic Mapping of RNA-Chromatin Interactions In Vivo. Curr. Biol. 2017, 27, 602–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, W.; Yan, Z.; Nguyen, T.C.; Bouman Chen, Z.; Chien, S.; Zhong, S. Mapping RNA-chromatin interactions by sequencing with iMARGI. Nat. Protoc. 2019, 14, 3243–3272. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhou, B.; Chen, L.; Gou, L.T.; Li, H.; Fu, X.D. GRID-seq reveals the global RNA-chromatin interactome. Nat. Biotechnol. 2017, 35, 940–950. [Google Scholar] [CrossRef]
- Zhou, B.; Li, X.; Luo, D.; Lim, D.H.; Zhou, Y.; Fu, X.D. GRID-seq for comprehensive analysis of global RNA-chromatin interactions. Nat. Protoc. 2019, 14, 2036–2068. [Google Scholar] [CrossRef] [PubMed]
- Bell, J.C.; Jukam, D.; Teran, N.A.; Risca, V.I.; Smith, O.K.; Johnson, W.L.; Skotheim, J.M.; Greenleaf, W.J.; Straight, A.F. Chromatin-associated RNA sequencing (ChAR-seq) maps genome-wide RNA-to-DNA contacts. eLife 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Engreitz, J.; Lander, E.S.; Guttman, M. RNA antisense purification (RAP) for mapping RNA interactions with chromatin. Methods Mol. Biol. 2015, 1262, 183–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Antonio, M.; D’Onorio De Meo, P.; Pallocca, M.; Picardi, E.; D’Erchia, A.M.; Calogero, R.A.; Castrignano, T.; Pesole, G. RAP: RNA-Seq Analysis Pipeline, a new cloud-based NGS web application. BMC Genom. 2015, 16, S3. [Google Scholar] [CrossRef] [Green Version]
- Grillone, K.; Riillo, C.; Scionti, F.; Rocca, R.; Tradigo, G.; Guzzi, P.H.; Alcaro, S.; Di Martino, M.T.; Tagliaferri, P.; Tassone, P. Non-coding RNAs in cancer: Platforms and strategies for investigating the genomic “dark matter”. J. Exp. Clin. Cancer Res. 2020, 39, 117. [Google Scholar] [CrossRef]
- Weidmann, C.A.; Mustoe, A.M.; Jariwala, P.B.; Calabrese, J.M.; Weeks, K.M. Analysis of RNA-protein networks with RNP-MaP defines functional hubs on RNA. Nat. Biotechnol. 2021, 39, 347–356. [Google Scholar] [CrossRef]
- Yi, W.; Li, J.; Zhu, X.; Wang, X.; Fan, L.; Sun, W.; Liao, L.; Zhang, J.; Li, X.; Ye, J.; et al. CRISPR-assisted detection of RNA-protein interactions in living cells. Nat. Methods 2020, 17, 685–688. [Google Scholar] [CrossRef] [PubMed]
- Cottrell, K.A.; Chaudhari, H.G.; Cohen, B.A.; Djuranovic, S. PTRE-seq reveals mechanism and interactions of RNA binding proteins and miRNAs. Nat. Commun. 2018, 9, 301. [Google Scholar] [CrossRef] [Green Version]
- Wan, Y.; Qu, K.; Ouyang, Z.; Chang, H.Y. Genome-wide mapping of RNA structure using nuclease digestion and high-throughput sequencing. Nat. Protoc. 2013, 8, 849–869. [Google Scholar] [CrossRef]
- Kertesz, M.; Wan, Y.; Mazor, E.; Rinn, J.L.; Nutter, R.C.; Chang, H.Y.; Segal, E. Genome-wide measurement of RNA secondary structure in yeast. Nature 2010, 467, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Solomon, O.; Di Segni, A.; Cesarkas, K.; Porath, H.T.; Marcu-Malina, V.; Mizrahi, O.; Stern-Ginossar, N.; Kol, N.; Farage-Barhom, S.; Glick-Saar, E.; et al. RNA editing by ADAR1 leads to context-dependent transcriptome-wide changes in RNA secondary structure. Nat. Commun. 2017, 8, 1440. [Google Scholar] [CrossRef] [PubMed]
- Doudna, J.A.; Charpentier, E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 2014, 346, 1258096. [Google Scholar] [CrossRef] [PubMed]
- Sander, J.D.; Joung, J.K. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat. Biotechnol. 2014, 32, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Bortesi, L.; Fischer, R. The CRISPR/Cas9 system for plant genome editing and beyond. Biotechnol. Adv. 2015, 33, 41–52. [Google Scholar] [CrossRef]
- Wang, H.; La Russa, M.; Qi, L.S. CRISPR/Cas9 in Genome Editing and Beyond. Annu. Rev. Biochem. 2016, 85, 227–264. [Google Scholar] [CrossRef] [Green Version]
- Larson, M.H.; Gilbert, L.A.; Wang, X.; Lim, W.A.; Weissman, J.S.; Qi, L.S. CRISPR interference (CRISPRi) for sequence-specific control of gene expression. Nat. Protoc. 2013, 8, 2180–2196. [Google Scholar] [CrossRef] [Green Version]
- Hou, G.; Harley, I.T.W.; Lu, X.; Zhou, T.; Xu, N.; Yao, C.; Qin, Y.; Ouyang, Y.; Ma, J.; Zhu, X.; et al. SLE non-coding genetic risk variant determines the epigenetic dysfunction of an immune cell specific enhancer that controls disease-critical microRNA expression. Nat. Commun. 2021, 12, 135. [Google Scholar] [CrossRef]
- Nakamura, M.; Gao, Y.; Dominguez, A.A.; Qi, L.S. CRISPR technologies for precise epigenome editing. Nat. Cell Biol. 2021, 23, 11–22. [Google Scholar] [CrossRef]
- Palin, K.; Pitkanen, E.; Turunen, M.; Sahu, B.; Pihlajamaa, P.; Kivioja, T.; Kaasinen, E.; Valimaki, N.; Hanninen, U.A.; Cajuso, T.; et al. Contribution of allelic imbalance to colorectal cancer. Nat. Commun. 2018, 9, 3664. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.; Soares, F.; Xia, J.H.; Yang, Y.; Li, J.; Guo, H.; Su, P.; Tian, Y.; Lee, H.J.; Wang, M.; et al. CRISPRi screens reveal a DNA methylation-mediated 3D genome dependent causal mechanism in prostate cancer. Nat. Commun. 2021, 12, 1781. [Google Scholar] [CrossRef]
- Nichols, C.A.; Gibson, W.J.; Brown, M.S.; Kosmicki, J.A.; Busanovich, J.P.; Wei, H.; Urbanski, L.M.; Curimjee, N.; Berger, A.C.; Gao, G.F.; et al. Loss of heterozygosity of essential genes represents a widespread class of potential cancer vulnerabilities. Nat. Commun. 2020, 11, 2517. [Google Scholar] [CrossRef] [PubMed]
- Pickar-Oliver, A.; Gersbach, C.A. The next generation of CRISPR-Cas technologies and applications. Nat. Rev. Mol. Cell Biol. 2019, 20, 490–507. [Google Scholar] [CrossRef]
- Smargon, A.A.; Shi, Y.J.; Yeo, G.W. RNA-targeting CRISPR systems from metagenomic discovery to transcriptomic engineering. Nat. Cell Biol. 2020, 22, 143–150. [Google Scholar] [CrossRef]
- Guo, Y.; Perez, A.A.; Hazelett, D.J.; Coetzee, G.A.; Rhie, S.K.; Farnham, P.J. CRISPR-mediated deletion of prostate cancer risk-associated CTCF loop anchors identifies repressive chromatin loops. Genome Biol. 2018, 19, 160. [Google Scholar] [CrossRef]
- Hasin, Y.; Seldin, M.; Lusis, A. Multi-omics approaches to disease. Genome Biol. 2017, 18, 83. [Google Scholar] [CrossRef] [PubMed]
- Sims, D.; Sudbery, I.; Ilott, N.E.; Heger, A.; Ponting, C.P. Sequencing depth and coverage: Key considerations in genomic analyses. Nat. Rev. Genet. 2014, 15, 121–132. [Google Scholar] [CrossRef]
- Wagner, A.H.; Walsh, B.; Mayfield, G.; Tamborero, D.; Sonkin, D.; Krysiak, K.; Deu-Pons, J.; Duren, R.P.; Gao, J.; McMurry, J.; et al. A harmonized meta-knowledgebase of clinical interpretations of somatic genomic variants in cancer. Nat. Genet. 2020, 52, 448–457. [Google Scholar] [CrossRef] [Green Version]
- Piraino, S.W.; Furney, S.J. Beyond the exome: The role of non-coding somatic mutations in cancer. Ann. Oncol. 2016, 27, 240–248. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Li, C.; Shen, S.; Chen, X.; Szlachta, K.; Edmonson, M.N.; Shao, Y.; Ma, X.; Hyle, J.; Wright, S.; et al. Discovery of regulatory noncoding variants in individual cancer genomes by using cis-X. Nat. Genet. 2020, 52, 811–818. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Drubay, D.; Michiels, S.; Gautheret, D. Mining the coding and non-coding genome for cancer drivers. Cancer Lett. 2015, 369, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Zou, H.; Wu, L.X.; Tan, L.; Shang, F.F.; Zhou, H.H. Significance of Single-Nucleotide Variants in Long Intergenic Non-protein Coding RNAs. Front. Cell Dev. Biol. 2020, 8, 347. [Google Scholar] [CrossRef]
- Kikutake, C.; Yoshihara, M.; Suyama, M. Pan-cancer analysis of non-coding recurrent mutations and their possible involvement in cancer pathogenesis. NAR Cancer 2021, 3. [Google Scholar] [CrossRef]
- Pamula-Pilat, J.; Tecza, K.; Kalinowska-Herok, M.; Grzybowska, E. Genetic 3’UTR variations and clinical factors significantly contribute to survival prediction and clinical response in breast cancer patients. Sci. Rep. 2020, 10, 5736. [Google Scholar] [CrossRef] [PubMed]
- Ye, D.; Hu, Y.; Jing, F.; Li, Y.; Gu, S.; Jiang, X.; Mao, Y.; Li, Q.; Jin, M.; Chen, K. A novel SNP in promoter region of RP11-3N2.1 is associated with reduced risk of colorectal cancer. J. Hum. Genet. 2018, 63, 47–54. [Google Scholar] [CrossRef]
- Wu, E.R.; Chou, Y.E.; Liu, Y.F.; Hsueh, K.C.; Lee, H.L.; Yang, S.F.; Su, S.C. Association of lncRNA H19 Gene Polymorphisms with the Occurrence of Hepatocellular Carcinoma. Genes 2019, 10, 506. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.G.; Jiang, L.Y.; Xu, Q. Comprehensive assessment for miRNA polymorphisms in hepatocellular cancer risk: A systematic review and meta-analysis. BioSci. Rep. 2018, 38. [Google Scholar] [CrossRef]
- Endo, S.; Oguri, H.; Segawa, J.; Kawai, M.; Hu, D.; Xia, S.; Okada, T.; Irie, K.; Fujii, S.; Gouda, H.; et al. Development of Novel AKR1C3 Inhibitors as New Potential Treatment for Castration-Resistant Prostate Cancer. J. Med. Chem. 2020, 63, 10396–10411. [Google Scholar] [CrossRef] [PubMed]
- Kaur, H.; Mao, S.; Li, Q.; Sameni, M.; Krawetz, S.A.; Sloane, B.F.; Mattingly, R.R. RNA-Seq of human breast ductal carcinoma in situ models reveals aldehyde dehydrogenase isoform 5A1 as a novel potential target. PLoS ONE 2012, 7, e50249. [Google Scholar] [CrossRef] [Green Version]
- Bray, J.; Sludden, J.; Griffin, M.J.; Cole, M.; Verrill, M.; Jamieson, D.; Boddy, A.V. Influence of pharmacogenetics on response and toxicity in breast cancer patients treated with doxorubicin and cyclophosphamide. Br. J. Cancer 2010, 102, 1003–1009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rawlings-Goss, R.A.; Campbell, M.C.; Tishkoff, S.A. Global population-specific variation in miRNA associated with cancer risk and clinical biomarkers. BMC Med. Genom. 2014, 7, 53. [Google Scholar] [CrossRef] [Green Version]
- Hoffman, A.E.; Liu, R.; Fu, A.; Zheng, T.; Slack, F.; Zhu, Y. Targetome profiling, pathway analysis and genetic association study implicate miR-202 in lymphomagenesis. Cancer Epidemiol. Biomark. Prev. 2013, 22, 327–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pipan, V.; Zorc, M.; Kunej, T. MicroRNA Polymorphisms in Cancer: A Literature Analysis. Cancers 2015, 7, 1806–1814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, F.; Feng, Y.; Zheng, Y.; Ogundiran, T.O.; Ojengbede, O.; Zheng, W.; Blot, W.; Ambrosone, C.B.; John, E.M.; Bernstein, L.; et al. Genetic variants in microRNA and microRNA biogenesis pathway genes and breast cancer risk among women of African ancestry. Hum. Genet. 2016, 135, 1145–1159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersson, R.; Gebhard, C.; Miguel-Escalada, I.; Hoof, I.; Bornholdt, J.; Boyd, M.; Chen, Y.; Zhao, X.; Schmidl, C.; Suzuki, T.; et al. An atlas of active enhancers across human cell types and tissues. Nature 2014, 507, 455–461. [Google Scholar] [CrossRef]
- Topalian, S.L.; Taube, J.M.; Anders, R.A.; Pardoll, D.M. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat. Rev. Cancer 2016, 16, 275–287. [Google Scholar] [CrossRef]
- Flaherty, K.T.; Gray, R.J.; Chen, A.P.; Li, S.; McShane, L.M.; Patton, D.; Hamilton, S.R.; Williams, P.M.; Iafrate, A.J.; Sklar, J.; et al. Molecular Landscape and Actionable Alterations in a Genomically Guided Cancer Clinical Trial: National Cancer Institute Molecular Analysis for Therapy Choice (NCI-MATCH). J. Clin. Oncol. 2020, 38, 3883–3894. [Google Scholar] [CrossRef] [PubMed]
- Belin, L.; Kamal, M.; Mauborgne, C.; Plancher, C.; Mulot, F.; Delord, J.P.; Goncalves, A.; Gavoille, C.; Dubot, C.; Isambert, N.; et al. Randomized phase II trial comparing molecularly targeted therapy based on tumor molecular profiling versus conventional therapy in patients with refractory cancer: Cross-over analysis from the SHIVA trial. Ann. Oncol 2017, 28, 590–596. [Google Scholar] [CrossRef]
- Gambardella, V.; Tarazona, N.; Cejalvo, J.M.; Lombardi, P.; Huerta, M.; Rosello, S.; Fleitas, T.; Roda, D.; Cervantes, A. Personalized Medicine: Recent Progress in Cancer Therapy. Cancers 2020, 12, 1009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasconcellos, V.F.; Colli, L.M.; Awada, A.; de Castro Junior, G. Precision oncology: As much expectations as limitations. Ecancermedicalscience 2018, 12, ed86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cowie, P.; Hay, E.A.; MacKenzie, A. The noncoding human genome and the future of personalised medicine. Expert Rev. Mol. Med. 2015, 17, e4. [Google Scholar] [CrossRef]
- Zhang, Z.; Gu, M.; Gu, Z.; Lou, Y.R. Role of Long Non-Coding RNA Polymorphisms in Cancer Chemotherapeutic Response. J. Pers. Med. 2021, 11, 513. [Google Scholar] [CrossRef]
- Lin, A.; Hu, Q.; Li, C.; Xing, Z.; Ma, G.; Wang, C.; Li, J.; Ye, Y.; Yao, J.; Liang, K.; et al. The LINK-A lncRNA interacts with PtdIns(3,4,5)P3 to hyperactivate AKT and confer resistance to AKT inhibitors. Nat. Cell Biol. 2017, 19, 238–251. [Google Scholar] [CrossRef]
- Meddens, C.A.; van der List, A.C.J.; Nieuwenhuis, E.E.S.; Mokry, M. Non-coding DNA in IBD: From sequence variation in DNA regulatory elements to novel therapeutic potential. Gut 2019, 68, 928–941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Liu, Q.; Yuan, S.; Xie, W.; Liu, Y.; Xiang, Y.; Wu, N.; Wu, L.; Ma, X.; Cai, T.; et al. Genetic predisposition to lung cancer: Comprehensive literature integration, meta-analysis, and multiple evidence assessment of candidate-gene association studies. Sci. Rep. 2017, 7, 8371. [Google Scholar] [CrossRef]
- Yan, T.; Shen, C.; Jiang, P.; Yu, C.; Guo, F.; Tian, X.; Zhu, X.; Lu, S.; Han, B.; Zhong, M.; et al. Risk SNP-induced lncRNA-SLCC1 drives colorectal cancer through activating glycolysis signaling. Signal Transduct. Target. Ther. 2021, 6, 70. [Google Scholar] [CrossRef]
- Dentro, S.C.; Leshchiner, I.; Haase, K.; Tarabichi, M.; Wintersinger, J.; Deshwar, A.G.; Yu, K.; Rubanova, Y.; Macintyre, G.; Demeulemeester, J.; et al. Characterizing genetic intra-tumor heterogeneity across 2,658 human cancer genomes. Cell 2021, 184, 2239.e2239–2254.e2239. [Google Scholar] [CrossRef]
- Lu, Y.; Kweon, S.S.; Tanikawa, C.; Jia, W.H.; Xiang, Y.B.; Cai, Q.; Zeng, C.; Schmit, S.L.; Shin, A.; Matsuo, K.; et al. Large-Scale Genome-Wide Association Study of East Asians Identifies Loci Associated with Risk for Colorectal Cancer. Gastroenterology 2019, 156, 1455–1466. [Google Scholar] [CrossRef] [Green Version]
- Pon, J.R.; Marra, M.A. Driver and passenger mutations in cancer. Annu. Rev. Pathol. 2015, 10, 25–50. [Google Scholar] [CrossRef]
- Lin, Y.; Nakatochi, M.; Hosono, Y.; Ito, H.; Kamatani, Y.; Inoko, A.; Sakamoto, H.; Kinoshita, F.; Kobayashi, Y.; Ishii, H.; et al. Genome-wide association meta-analysis identifies GP2 gene risk variants for pancreatic cancer. Nat. Commun. 2020, 11, 3175. [Google Scholar] [CrossRef]
- Pashayan, N.; Antoniou, A.C.; Ivanus, U.; Esserman, L.J.; Easton, D.F.; French, D.; Sroczynski, G.; Hall, P.; Cuzick, J.; Evans, D.G.; et al. Personalized early detection and prevention of breast cancer: ENVISION consensus statement. Nat. Rev. Clin. Oncol. 2020, 17, 687–705. [Google Scholar] [CrossRef] [PubMed]
- Erichsen, H.C.; Chanock, S.J. SNPs in cancer research and treatment. Br. J. Cancer 2004, 90, 747–751. [Google Scholar] [CrossRef] [Green Version]
- Fu, X.; Shi, Y.; Qi, T.; Qiu, S.; Huang, Y.; Zhao, X.; Sun, Q.; Lin, G. Precise design strategies of nanomedicine for improving cancer therapeutic efficacy using subcellular targeting. Signal Transduct. Target. Ther. 2020, 5, 262. [Google Scholar] [CrossRef] [PubMed]
- Muthuirulan, P.; Zhao, D.; Young, M.; Richard, D.; Liu, Z.; Emami, A.; Portilla, G.; Hosseinzadeh, S.; Cao, J.; Maridas, D.; et al. Joint disease-specificity at the regulatory base-pair level. Nat. Commun. 2021, 12, 4161. [Google Scholar] [CrossRef]
- Russ, A.P.; Lampel, S. The druggable genome: An update. Drug Discov. Today 2005, 10, 1607–1610. [Google Scholar] [CrossRef]
- Geary, R.S.; Norris, D.; Yu, R.; Bennett, C.F. Pharmacokinetics, biodistribution and cell uptake of antisense oligonucleotides. Adv. Drug Deliv. Rev. 2015, 87, 46–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, W.; Rogge, M. Targeting RNA: A Transformative Therapeutic Strategy. Clin. Transl. Sci. 2019, 12, 98–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopechek, J.A.; McTiernan, C.F.; Chen, X.; Zhu, J.; Mburu, M.; Feroze, R.; Whitehurst, D.A.; Lavery, L.; Cyriac, J.; Villanueva, F.S. Ultrasound and Microbubble-targeted Delivery of a microRNA Inhibitor to the Heart Suppresses Cardiac Hypertrophy and Preserves Cardiac Function. Theranostics 2019, 9, 7088–7098. [Google Scholar] [CrossRef] [PubMed]
- Winkle, M.; El-Daly, S.M.; Fabbri, M.; Calin, G.A. Noncoding RNA therapeutics—Challenges and potential solutions. Nat. Rev. Drug Discov. 2021. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Wang, J.; He, Z.; Chen, P.; Jiang, X.; Chen, Y.; Liu, X.; Jiang, J. LncRNA CERS6-AS1 promotes proliferation and metastasis through the upregulation of YWHAG and activation of ERK signaling in pancreatic cancer. Cell Death Dis. 2021, 12, 648. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Wang, R.; Shen, D.; Cheng, S.; Wang, H.; Lu, Z.; Zheng, Q.; Wang, L.; Xia, L.; Li, G. Role of noncoding RNA in drug resistance of prostate cancer. Cell Death Dis. 2021, 12, 590. [Google Scholar] [CrossRef]




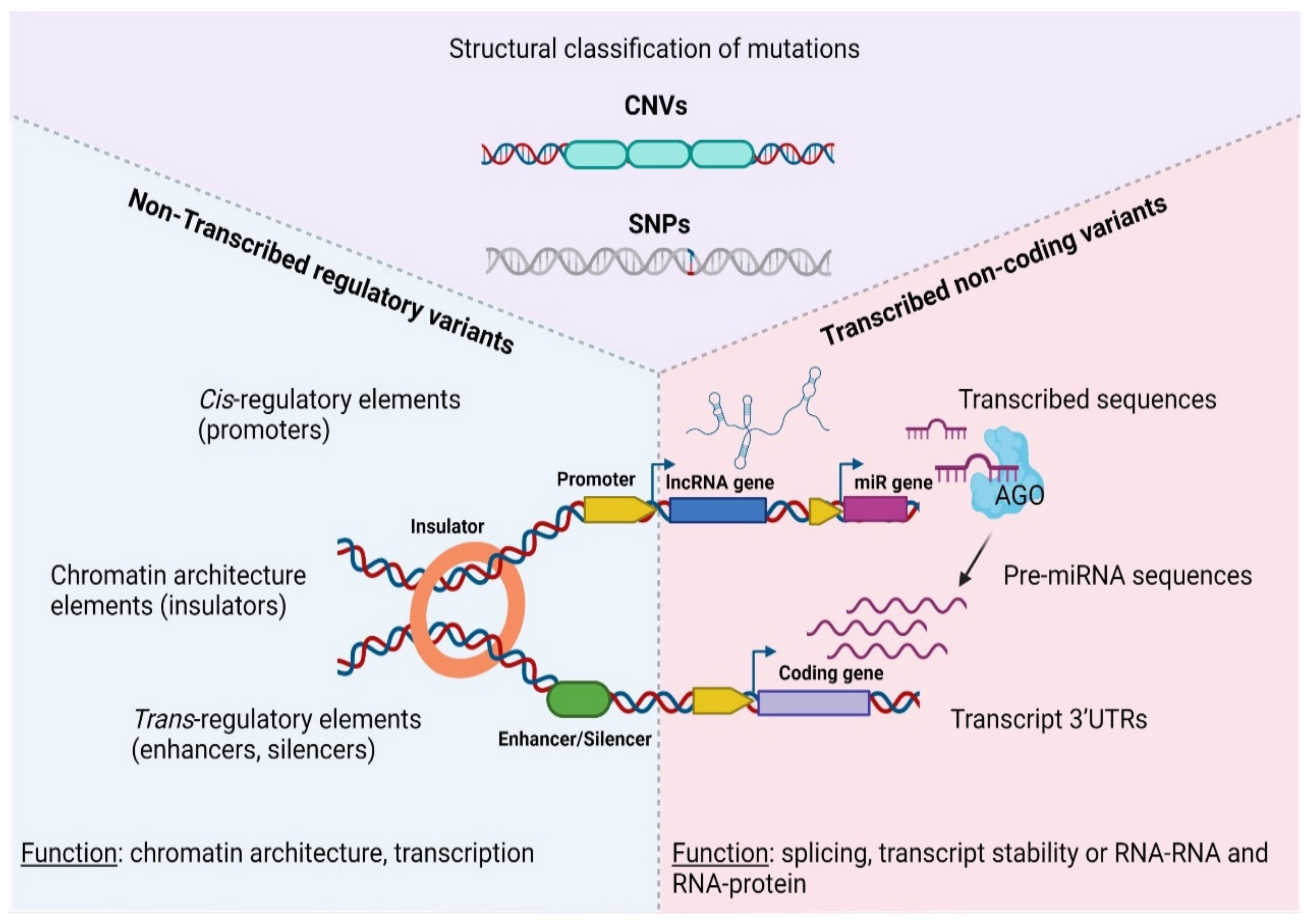{kind=link}
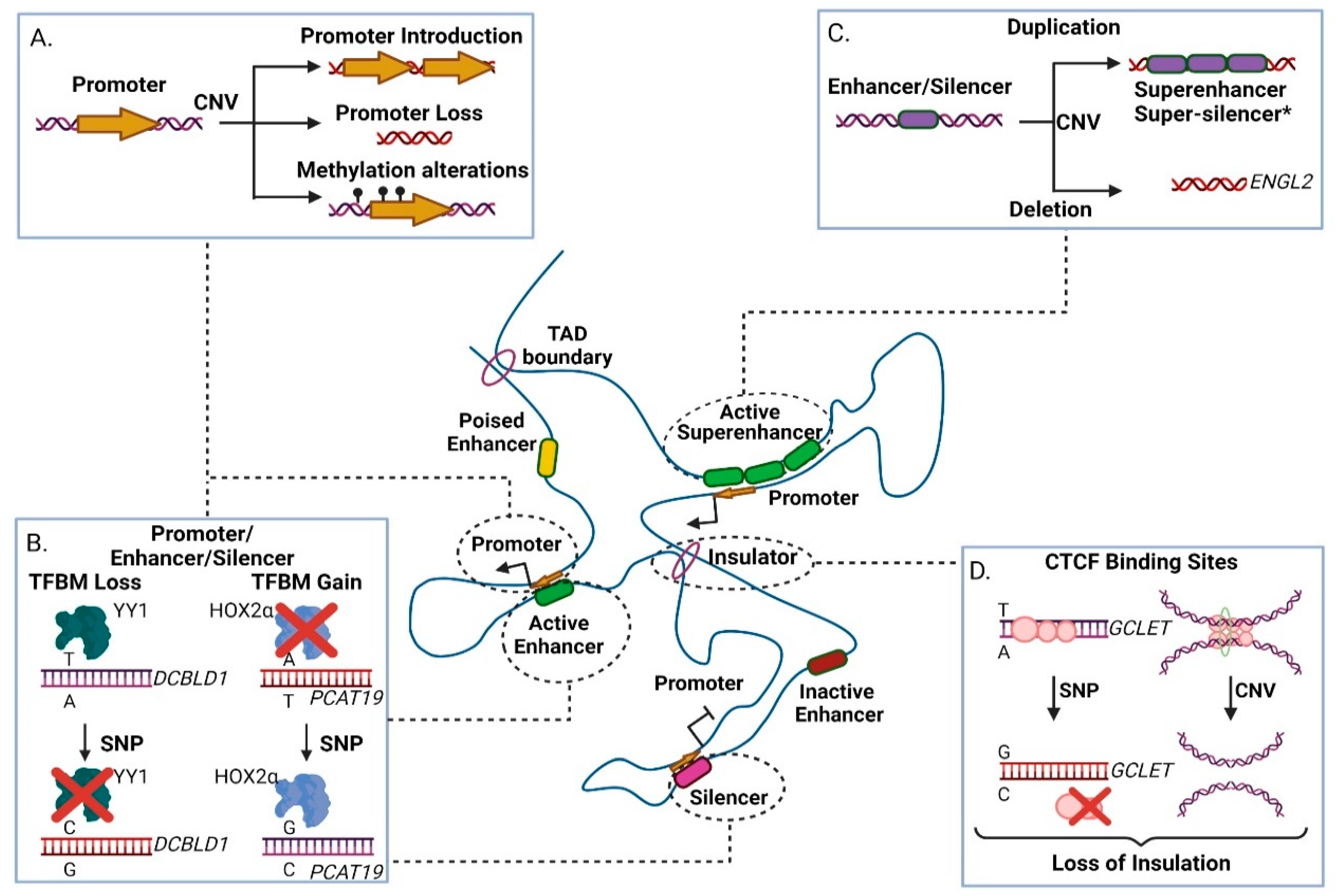{kind=link}
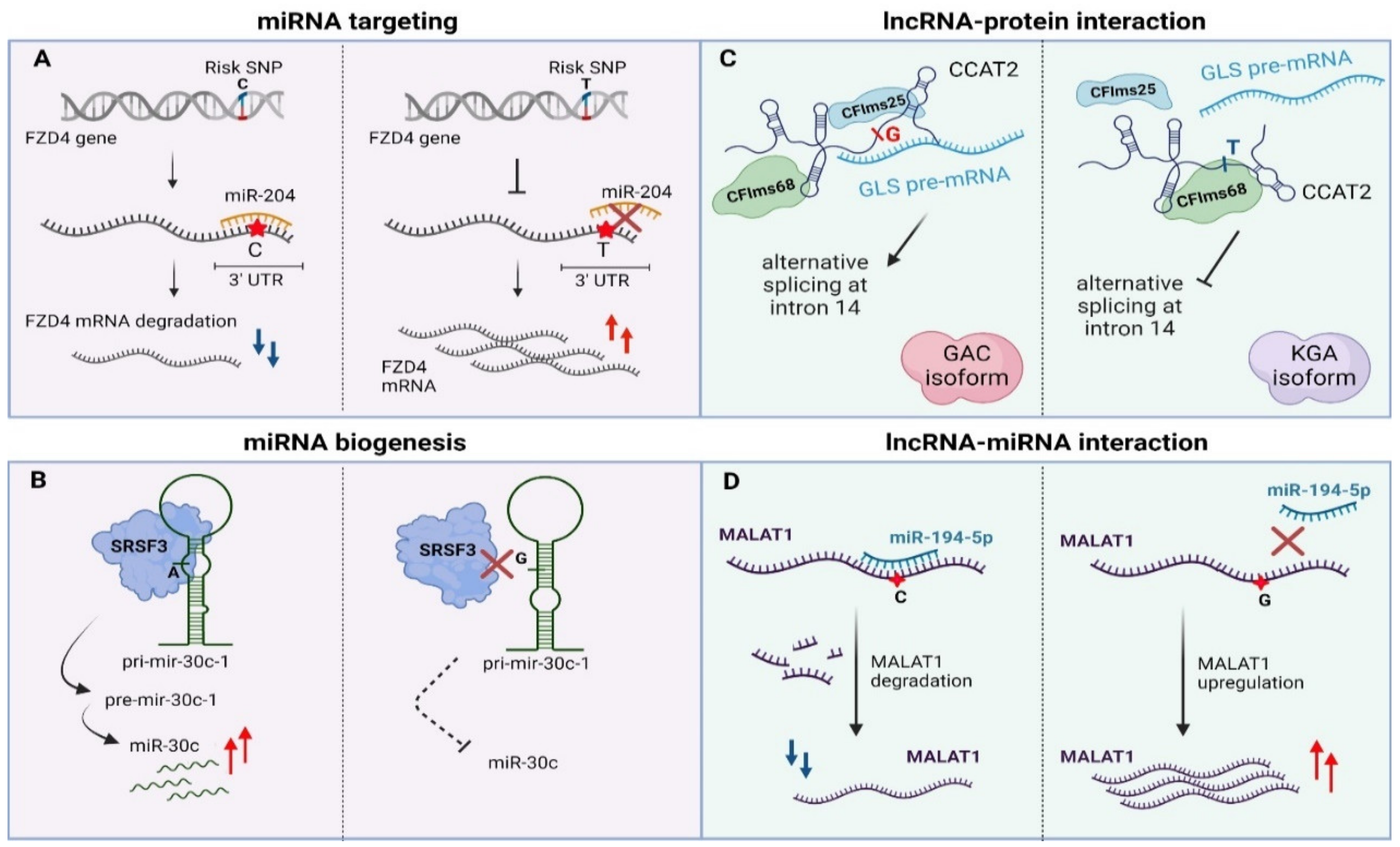{kind=link}
| Non-Coding Type | Variant ID | Target Locus | Mechanism | Cancer Type | Citation |
|---|---|---|---|---|---|
| Non-transcribed regulatory variants | |||||
| Promoters | rs11672691 | PCAT19 promoter | NKX3.1, YY1 binding | Prostate | [68] |
| rs887391 | PCAT19 promoter | NKX3.1, YY1 binding | Prostate | ||
| rs17079281 | DCBLD1 promoter | YY1 binding | Lung | [69] | |
| rs2267531 | Glypican-3 promoter | - | HCC | [70] | |
| rs2280059 | HSPH1 promoter | HSPH1 increased expression | NSCLC | [71] | |
| Enhancer | rs11672691 | PCAT19 Enhancer | NKX3.1, YY1, HOXA2 interaction with PCAT19 | Prostate | [68] |
| rs7463708 | PCAT1 Enhancer | ONECUT, AR interaction with PCAT1 | Prostate | [72] | |
| rs35252396 | Enhancer between MYC and PVT1 genes | Binding of HIFs | RCC | [73] | |
| rs6983267 | Enhancer between MYC and PVT1 | Binding of HIFs | Prostate, Colorectal | [74] | |
| EGLN2 CNV | Enhancer | Genomic deletion | Ovaries | [75] | |
| rs67311347 | Enhancer of ENTPD3-AS1 | Binding of ZNF8 | RCC | [76] | |
| rs4693608 | Enhancer of HPSE | Regulation of HPSE | ALL | [77] | |
| Silencer | rs249473 | Silencer in AKT locus | Binding of AKT | Endometrial | [78] |
| Insulator | rs3850997 | Insulator at GCLET intron | CTCF binding | Gastric | [79] |
| MYCN CNV | Insulator of MYCN | Deletion, Loss of CTCF binding | Neuroblastoma | [80] | |
| Transcribed regulatory variants | |||||
| miRNA | rs683/rs910 SNPs | 3′UTR region of TYRP1 | miRNA targeting | Μelanoma | [81] |
| rs713065 | miR-204 | miRNA targeting of FZD4 | NSCLC | [82] | |
| rs1071738 | 3′UTR of Palladin | miR-96/miR-182 targeting of Palladin | Breast | [83] | |
| rs1048638 | 3′UTR of CA9 | miR-34a targeting of CA9 | HCC | [84] | |
| rs928508 | miR-30c | pri-mir-30c-1 biogenesis miR-30c interaction with SRSF3 | Breast, Gastric | [85,86] | |
| rs6983267 | Pre-miR-1307 | pre-miR-1307 maturation | Colorectal | [87] | |
| rs11671784 | Maturation process of miR-27a | miR-27a HOXA | Gastric | [88] | |
| lncRNA | rs6983267 | CCAT2 | lncRNA interaction with CFIms25 | Colorectal | [89] |
| rs114020893 | lncRNA NEXN-AS1 | LncRNA secondary structure | Lung | [90] | |
| rs664589 | miR-194-5p | miR-194-5p interaction with MALAT1 | Colorectal | [91] | |
| rs1317082 | CCSlnc362 | miR-4658 interaction with CCSlnc362 | Colorectal | [92] | |
| rs11752942 | LINC00951 | miRNA-149 interaction with LINC00951 | ESCC | [93] | |
| rs11655237 | LINC00673 | miR-1231 interaction with LINC00673 | PDCA | [94] | |
| rs10251977 | EGFR-AS1 | Isoform selection via miR-891b and EGFR-AS interaction | Oral | [95] | |
| Experimental Approach | Advantages | Disadvantages | Publicly Available Databases/Software |
|---|---|---|---|
| Methodologies to study genomic areas in open-chromatin state | |||
| DNase-seq | • Enrich in cis-acting Res • No need for specific TF targeting • scDNase-seq improves sensitivity | • Biased in favor of promoters | • HOMER (Hypergeometric Optimization of Motif EnRichment) http://homer.ucsd.edu/homer/download.html |
| FAIRE-seq | • Simple application • Low bias • Sensitivity for intronic | • Low signal-to-noise ratio. • Requires high fixation efficiency. | • ENCODE: Wiggler https://sites.google.com/site/anshulkundaje/projects/wiggler |
| MNase-seq | • Less noise from mtDNA | • Laborious protocol • Digestion-based | • http://compbio-zhanglab.org/NUCOME/ |
| ATAC-seq | • Efficiency • Simple, cost-efficient application • Nucleosome and TF occupancy | • Demands coupling with other techniques | • ENCODE-DCC version 10 https://github.com/ENCODE-DCC/encoded/releases/tag/v101.0 |
| Methodologies for non-transcribed functional variant identification | |||
| MPRAs/CRE-seq | • High-throughput examination of enhancer activity • Allows multiple independent examinations | • Episomal assay • Cell-type specific enhancer activation profile • False-positive ratio | • Shendurelab/MPRAflow https://github.com/shendurelab/MPRAflow |
| STARR-seq | • High-throughput examination of enhancer activity • Reduced false-positive ratio • No barcoding | • Episomal assay • Cell-type specific enhancer activation profile • Reporter transcript stability | • Gersteinlab/starrpeaker https://github.com/gersteinlab/starrpeaker • hyulab/eSTARR https://github.com/hyulab/eSTARR |
| ChIA-PET | • Precise global interaction map • Long-read ChIA-PETS has improved mapping efficiency | • Complex data analysis • Inefficient • Demands coupling with RNA-targeted methodology | • ChIA-PET Utilities-CPU https://github.com/cheehongsg/CPU • Mango https://github.com/dphansti/mango • TheJacksonLaboratory/ChIA-PIPE https://github.com/TheJacksonLaboratory/ChIA-PIPE |
| HiChIP | • Efficiency • Low false-positive ratio • Simple workflow | • Not available | • FitHiChIP https://github.com/ay-lab/FitHiChIP |
| PLAC-seq | • Efficiency • Specificity • Simple workflow | • Not available | • HPRep https://github.com/yunliUNC/HPRep • MAPS https://github.com/HuMingLab/MAPS |
| ChIRP-seq | • Commonly used | • Increased false-positive ratio • Targets known RNA | • Not available |
| Experimental Approach | Advantages | Disadvantages | Publicly Available Databases/Software |
|---|---|---|---|
| Methodologies for transcribed functional variant identification | |||
| RAP-seq | • Genome-wide RNA:DNA interaction maps • Low background noise | • Known RNA sequence | • SPRITE https://github.com/GuttmanLab/sprite-pipeline |
| RNP-MaP | • Efficient • Resolution • Unbiased • Study protein networks | • Coupling with protein-targeted methodology | • Not available |
| CARPID | • Specificity • Low background noise • Determine allelic expression | • Coupling with protein-targeted methodology | • Not available |
| PTRE-seq | • High-throughput examination of 3′UTR regulatory activity | • Not available | • Not available |
| PARS-seq | • RNA structural information • Distinguish paired/unpaired bases • Alternative to MS, NMR, crystallography | • Non-specific enzyme digestion • RNA over-digestion • Need for optimization • Only in vitro applications | • RNAFramework https://github.com/dincarnato/RNAFramework |
| Experimental Approach | Advantages | Disadvantages | Publicly Available Databases/Software |
|---|---|---|---|
| CRISPR-Cas Systems | • Allows variant correction/creation • Low off-target effects (especially when using nickase Cas9) • Activation and inhibition of regulatory element function • Alteration in methylation status • RNA targeting | • Needs fine-tuning to avoid off-target effects • Efficiency differs between systems | • Design http://www.rgenome.net/be-designer/ http://zifit.partners.org/ZiFiT/ http://www.e-crisp.org/E-CRISP/ https://chopchop.cbu.uib.no/ http://crispr-era.stanford.edu/ https://portals.broadinstitute.org/gpp/public/analysis-tools/sgrna-design • Analysis http://www.rgenome.net/be-analyzer/# |
| SNP ID | Target Locus | Clinical Trait | Cancer Type | Citation |
|---|---|---|---|---|
| rs291593 | DPYD 3′UTR | Drug toxicity | Breast cancer | [316] |
| rs3209896, rs1824125 | AKR1C3 3′UTR, PGR 3′UTR | Progression-free Survival | ||
| rs1054899 | ALDH5A1 3′UTR | Chemotherapeutic response to FAC | ||
| rs7756222, rs9487402 | SLC22A16 3′UTR | Overall survival | ||
| rs13230517 | RP11-3N2.1 promoter | Cancer Risk | Colorectal cancer | [317] |
| rs531564 | pri-miR-124 | Lymph node metastasis | ||
| rs3741219, rs2910164, rs4938723 | H19 lncRNA, hsa-mir-146a, hsa-mir-34b/c | Cancer risk | Hepatocellular carcinoma | [318] |
| rs11614913, rs2292832 | hsa-mir-196a-2, hsa-mir-149 | Cancer risk | HBV-related HCC | [319] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lange, M.; Begolli, R.; Giakountis, A. Non-Coding Variants in Cancer: Mechanistic Insights and Clinical Potential for Personalized Medicine. Non-Coding RNA 2021, 7, 47. https://doi.org/10.3390/ncrna7030047
Lange M, Begolli R, Giakountis A. Non-Coding Variants in Cancer: Mechanistic Insights and Clinical Potential for Personalized Medicine. Non-Coding RNA. 2021; 7(3):47. https://doi.org/10.3390/ncrna7030047
Chicago/Turabian StyleLange, Marios, Rodiola Begolli, and Antonis Giakountis. 2021. "Non-Coding Variants in Cancer: Mechanistic Insights and Clinical Potential for Personalized Medicine" Non-Coding RNA 7, no. 3: 47. https://doi.org/10.3390/ncrna7030047
APA StyleLange, M., Begolli, R., & Giakountis, A. (2021). Non-Coding Variants in Cancer: Mechanistic Insights and Clinical Potential for Personalized Medicine. Non-Coding RNA, 7(3), 47. https://doi.org/10.3390/ncrna7030047