
Pan-Cancer Analysis Reveals the Prognostic Potential of the THAP9/THAP9-AS1 Sense–Antisense Gene Pair in Human Cancers


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Characterization of THAP9/THAP9-AS1 Promoter
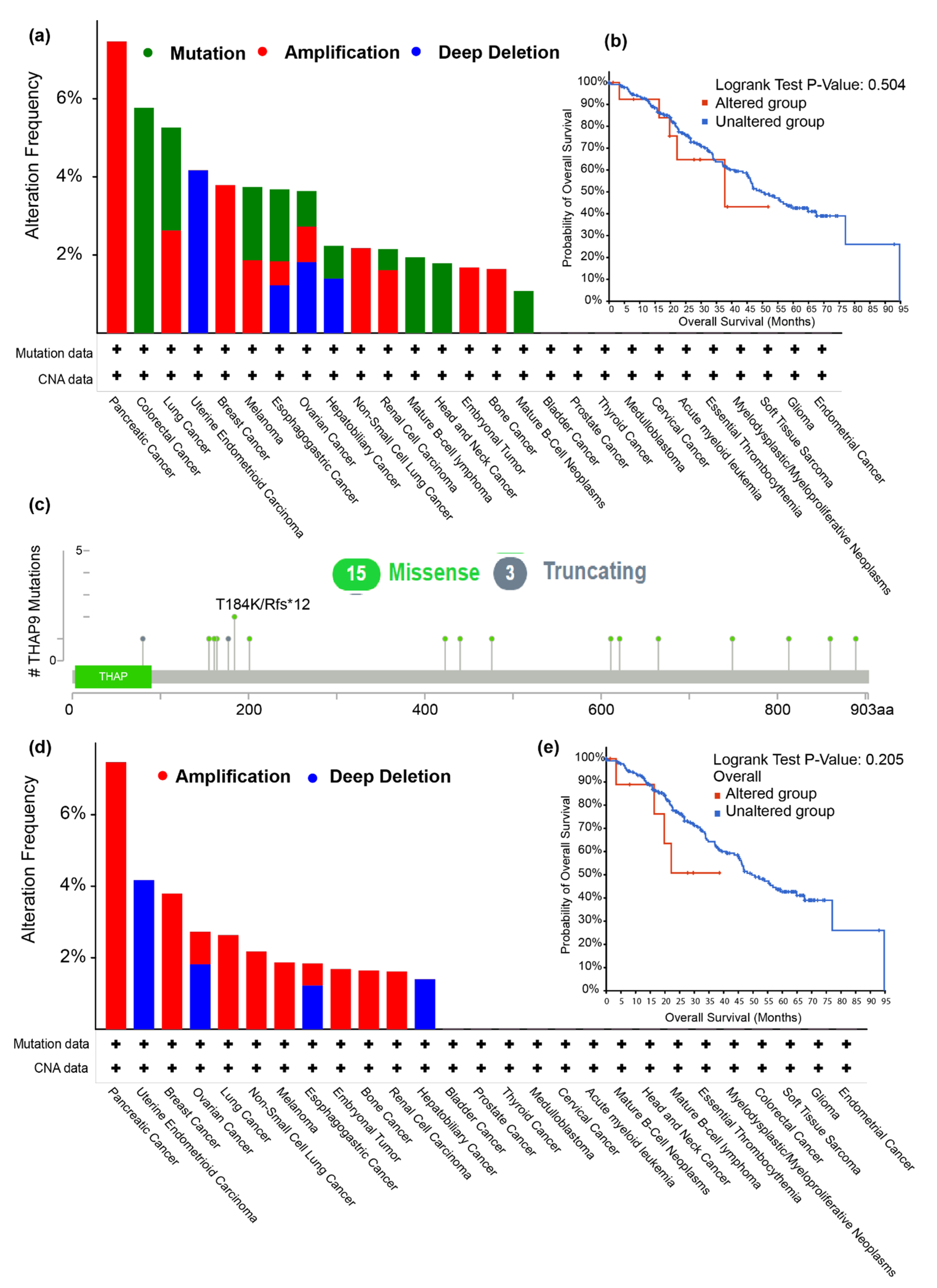
2.2. Analysis of THAP9/THAP9-AS1 Mutations in Various Tumors
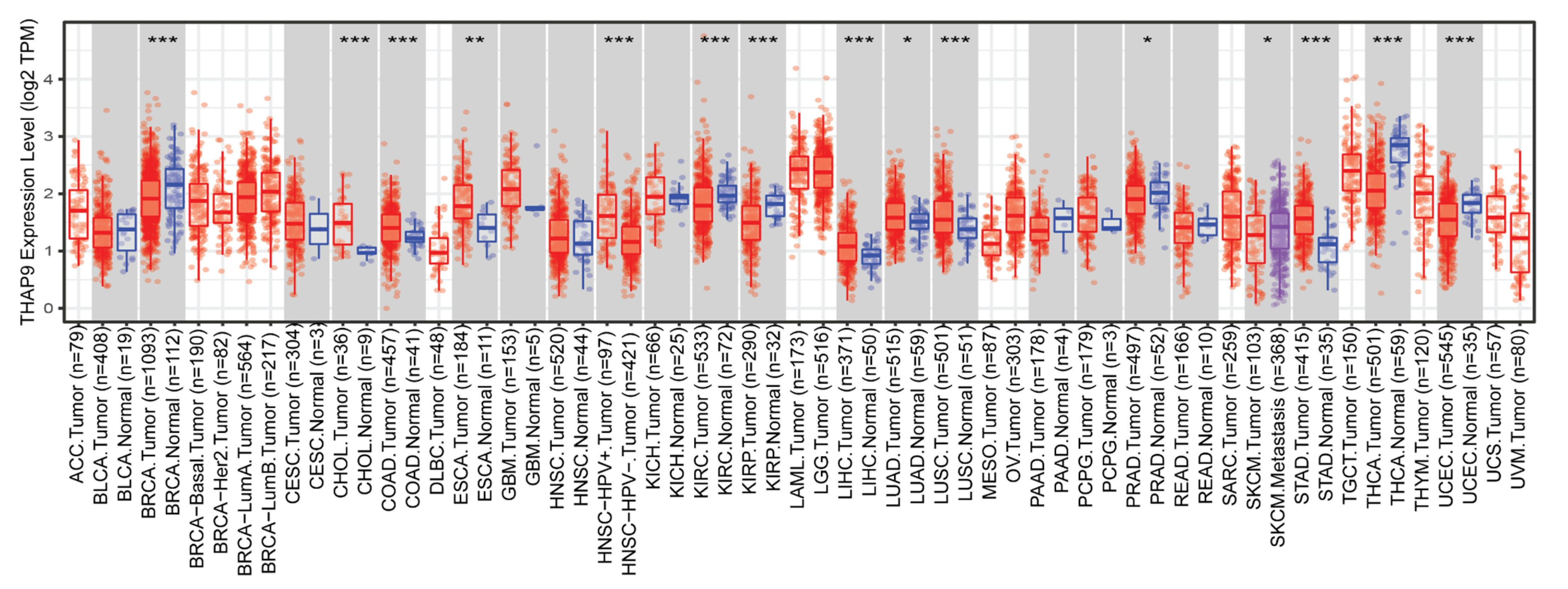
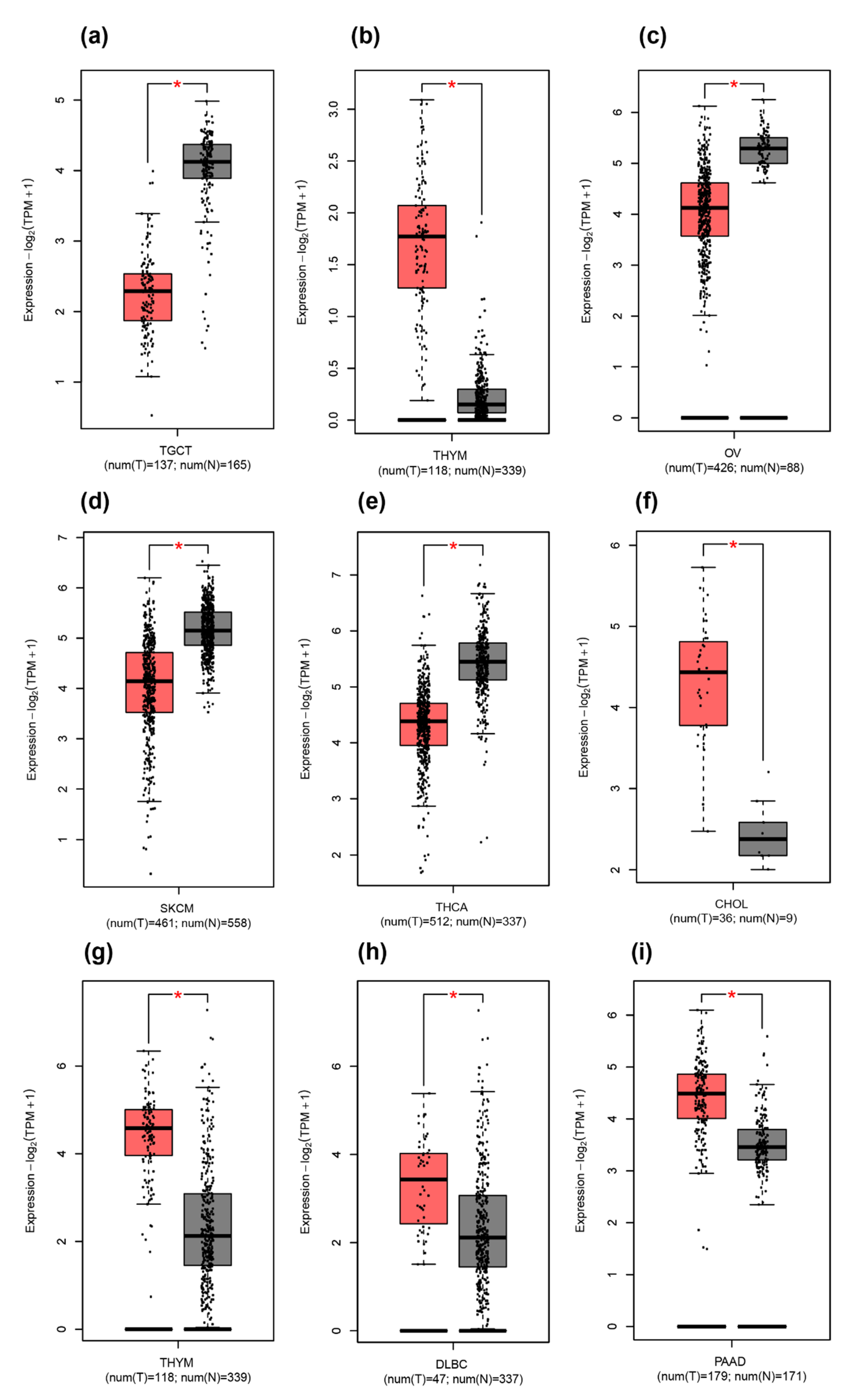
2.3. Difference between THAP9/THAP9-AS1 Expression in Several Cancers
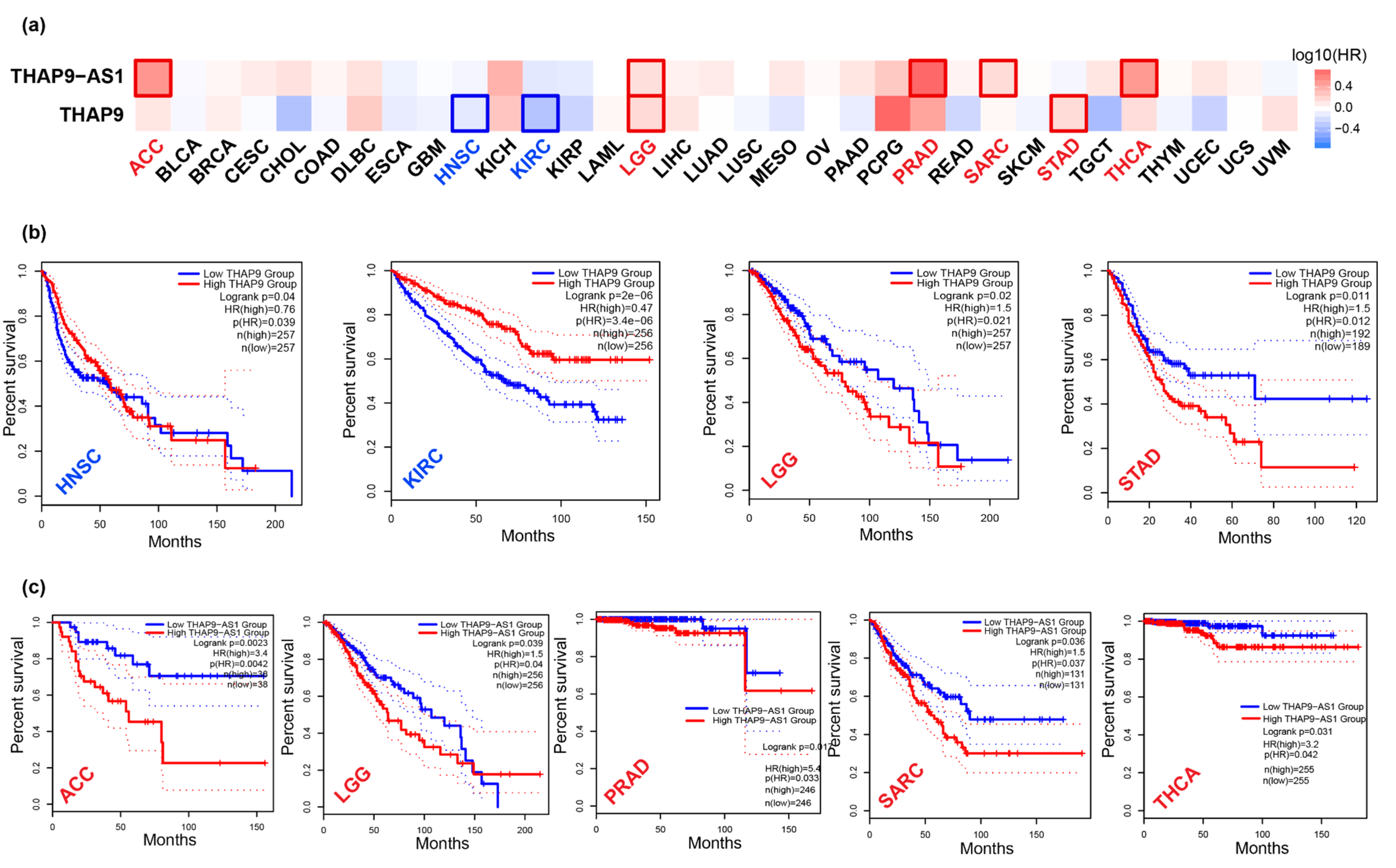
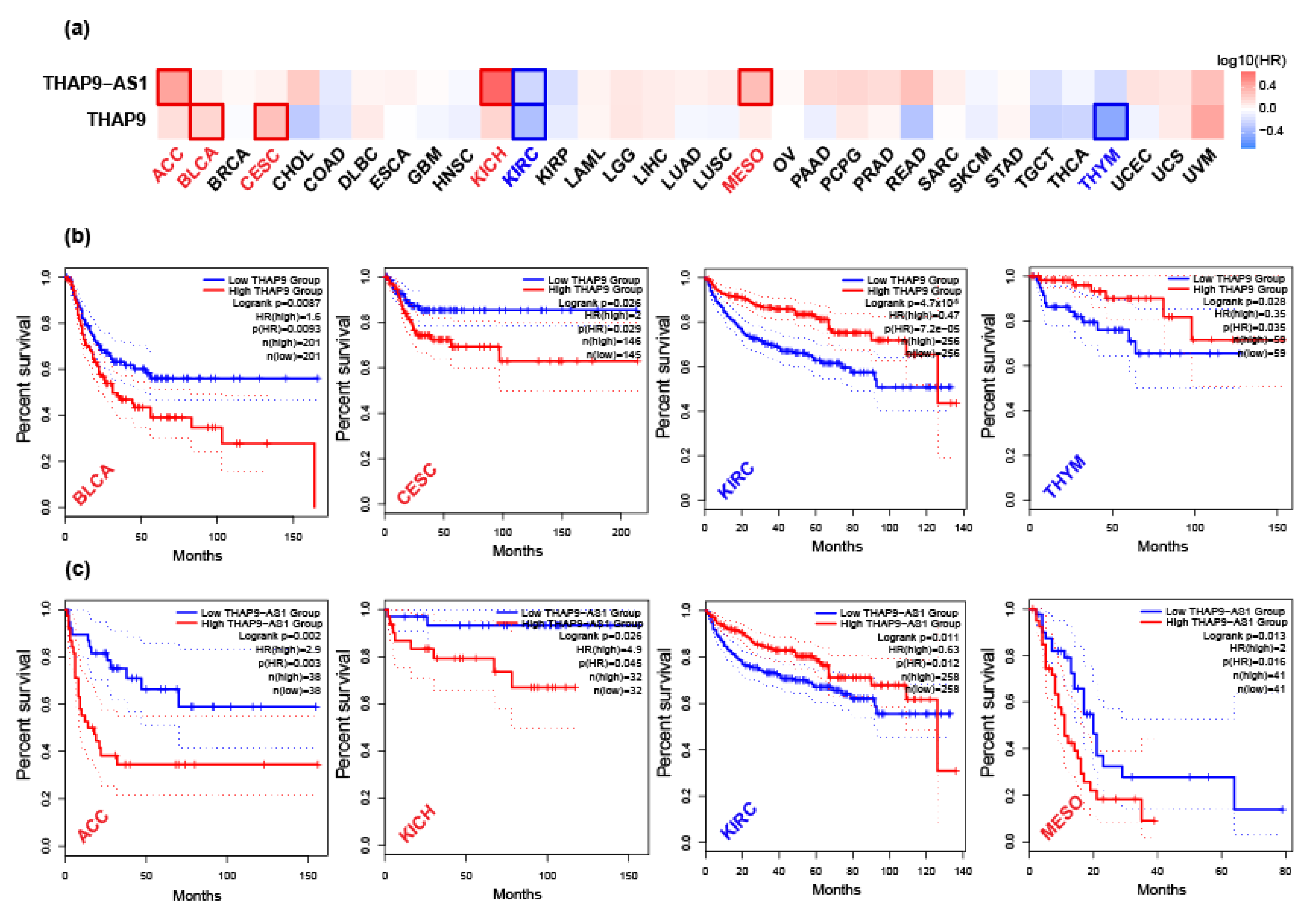
2.4. Prognostic Analysis of THAP9 and THAP9-AS1
2.5. Understanding the Role of THAP9 and THAP9-AS1 Using Guilt-By-Association Analysis
2.5.1. Gene Co-Expression Analysis
2.5.2. Differential Gene Correlation Analysis
3. Discussion
4. Materials and Methods
4.1. Analysis of Promoter Sequence
4.2. Mutation Analysis in Different Types of Tumors
4.3. Gene Expression Analysis
4.3.1. TIMER2.0
4.3.2. GEPIA2
4.4. Prognostic Analysis of THAP9 and THAP9-AS1
4.5. Guilt by Association Analysis
4.5.1. Construction of Weighted Gene Co-Expression Network
4.5.2. Gene Ontology (GO) and KEGG Pathway Enrichment Analysis
4.5.3. Differential Correlation Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ACC | adrenocortical carcinoma; |
| BLCA | bladder urothelial carcinoma; |
| BRCA | breast invasive carcinoma; |
| CESC | cervical and endocervical cancers; |
| CHOL | cholangiocarcinoma; |
| COAD | colon adenocarcinoma; |
| DLBC | lymphoid neoplasm diffuse large B-cell lymphoma; |
| ESCA | esophageal carcinoma; |
| GBM | glioblastoma multiforme; |
| HNSC | head and neck squamous cell carcinoma; |
| KICH | kidney chromophobe; |
| KIRC | kidney renal clear cell carcinoma; |
| KIRP | kidney renal papillary cell carcinoma; |
| LAML | acute myeloid leukemia; |
| LGG | brain lower grade glioma; |
| LIHC | liver hepatocellular carcinoma; |
| LUAD | lung adenocarcinoma; |
| LUSC | lung squamous cell carcinoma; |
| MESO | mesothelioma; |
| OV | ovarian serous cystadenocarcinoma; |
| PAAD | pancreatic adenocarcinoma; |
| PCPG | pheochromocytoma and paraganglioma; |
| PRAD | prostate adenocarcinoma; |
| READ | rectum adenocarcinoma; |
| SARC | sarcoma; |
| SKCM | skin cutaneous melanoma; |
| STAD | stomach adenocarcinoma; |
| STES | stomach and esophageal carcinoma; |
| TGCT | testicular germ cell tumors; |
| THCA | thyroid carcinoma; |
| THYM | thymoma; |
| UCEC | uterine corpus endometrial carcinoma; |
| UCS | uterine carcinosarcoma; |
| UVM | uveal melanoma. |
References
- Hurst, L.D.; Pál, C.; Lercher, M.J. The evolutionary dynamics of eukaryotic gene order. Nat. Rev. Genet. 2004, 5, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Adachi, N.; Lieber, M.R. Bidirectional Gene Organization: A Common Architectural Feature of the Human Genome. Cell 2002, 109, 807–809. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.-Y.; Yu, H.; Guo, Z.-M.; Guo, T.-Q.; Tu, K.; Li, Y.-X. Systematic Analysis of Head-to-Head Gene Organization: Evolutionary Conservation and Potential Biological Relevance. PLoS Comput. Biol. 2006, 2, e74. [Google Scholar] [CrossRef]
- Trinklein, N.D.; Aldred, S.F.; Hartman, S.J.; Schroeder, D.I.; Otillar, R.P.; Myers, R.M. An Abundance of Bidirectional Promoters in the Human Genome. Genome Res. 2004, 14, 62–66. [Google Scholar] [CrossRef] [Green Version]
- Burbelo, P.D.; Martin, G.R.; Yamada, Y. Alpha 1(IV) and alpha 2(IV) collagen genes are regulated by a bidirectional promoter and a shared enhancer. Proc. Natl. Acad. Sci. USA 1988, 85, 9679–9682. [Google Scholar] [CrossRef] [Green Version]
- Heikkilä, P.; Soininen, R.; Tryggvason, K. Directional regulatory activity of cis-acting elements in the bidirectional alpha 1(IV) and alpha 2(IV) collagen gene promoter. J. Biol. Chem. 1993, 268, 24677–24682. [Google Scholar] [CrossRef]
- Schuettengruber, B.; Doetzlhofer, A.; Kroboth, K.; Wintersberger, E.; Seiser, C. Alternate activation of two divergently transcribed mouse genes from a bidirectional promoter is linked to changes in histone modification. J. Biol. Chem. 2003, 278, 1784–1793. [Google Scholar] [CrossRef] [Green Version]
- Hansen, J.J.; Bross, P.; Westergaard, M.; Nielsen, M.N.; Eiberg, H.; Børglum, A.D.; Mogensen, J.; Kristiansen, K.; Bolund, L.; Gregersen, N. Genomic structure of the human mitochondrial chaperonin genes: HSP60 and HSP10 are localised head to head on chromosome 2 separated by a bidirectional promoter. Hum. Genet. 2003, 112, 71–77. [Google Scholar] [CrossRef]
- Balbin, O.A.; Malik, R.; Dhanasekaran, S.M.; Prensner, J.R.; Cao, X.; Wu, Y.-M.; Robinson, D.; Wang, R.; Chen, G.; Beer, D.G.; et al. The landscape of antisense gene expression in human cancers. Genome Res. 2015, 25, 1068–1079. [Google Scholar] [CrossRef] [Green Version]
- Auriol, E.; Billard, L.-M.; Magdinier, F.; Dante, R. Specific binding of the methyl binding domain protein 2 at the BRCA1-NBR2 locus. Nucleic Acids Res. 2005, 33, 4243–4254. [Google Scholar] [CrossRef] [Green Version]
- Luo, L.; Lu, F.M.; Hart, S.; Foroni, L.; Rabbani, H.; Hammarström, L.; Yuille, M.R.; Catovsky, D.; Webster, A.D.; Vorechovský, I. Ataxia-telangiectasia and T-cell leukemias: No evidence for somatic ATM mutation in sporadic T-ALL or for hypermethylation of the ATM-NPAT/E14 bidirectional promoter in T-PLL. Cancer Res. 1998, 58, 2293–2297. [Google Scholar] [PubMed]
- Shinya, E.; Shimada, T. Identification of two initiator elements in the bidirectional promoter of the human dihydrofolate reductase and mismatch repair protein 1 genes. Nucleic Acids Res. 1994, 22, 2143–2149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, P.-Y.; Chang, W.-S.W.; Chou, R.-H.; Lai, Y.-K.; Lin, S.-C.; Chi, C.-Y.; Wu, C.-W. Two non-homologous brain diseases-related genes, SERPINI1 and PDCD10, are tightly linked by an asymmetric bidirectional promoter in an evolutionarily conserved manner. BMC Mol. Biol. 2007, 8, 2. [Google Scholar] [CrossRef]
- Majumdar, S.; Singh, A.; Rio, D.C. The Human THAP9 Gene Encodes an Active P-Element DNA Transposase. Science 2013, 339, 446–448. [Google Scholar] [CrossRef] [Green Version]
- Majumdar, S.; Rio, D.C. P transposable elements in Drosophila and other eukaryotic organisms. Microbiol. Spectr. 2015, 3, MDNA3-0004-2014. [Google Scholar] [CrossRef] [Green Version]
- Campagne, S.; Saurel, O.; Gervais, V.; Milon, A. Structural determinants of specific DNA-recognition by the THAP zinc finger. Nucleic Acids Res. 2010, 38, 3466–3476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabogal, A.; Lyubimov, A.Y.; Corn, J.E.; Berger, J.M.; Rio, D.C. THAP proteins target specific DNA sites through bipartite recognition of adjacent major and minor grooves. Nat. Struct. Mol. Biol. 2010, 17, 117–123. [Google Scholar] [CrossRef] [Green Version]
- Sengel, C.; Gavarini, S.; Sharma, N.; Ozelius, L.J.; Bragg, D.C. Dimerization of the DYT6 dystonia protein, THAP1, requires residues within the coiled-coil domain. J. Neurochem. 2011, 118, 1087–1100. [Google Scholar] [CrossRef] [Green Version]
- Balakrishnan, M.P.; Cilenti, L.; Mashak, Z.; Popat, P.; Alnemri, E.S.; Zervos, A.S. THAP5 is a human cardiac-specific inhibitor of cell cycle that is cleaved by the proapoptotic Omi/HtrA2 protease during cell death. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H643–H653. [Google Scholar] [CrossRef] [Green Version]
- Roussigne, M.; Kossida, S.; Lavigne, A.-C.; Clouaire, T.; Ecochard, V.; Glories, A.; Amalric, F.; Girard, J.-P. The THAP domain: A novel protein motif with similarity to the DNA-binding domain of P element transposase. Trends Biochem. Sci. 2003, 28, 66–69. [Google Scholar] [CrossRef]
- Santos, E.D.; de Bessa, S.A.; Netto, M.M.; Nagai, M.A. Silencing of LRRC49 and THAP10 genes by bidirectional promoter hypermethylation is a frequent event in breast cancer. Int. J. Oncol. 2008, 33, 25–31. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Zhang, H.; Shi, H.; Wang, F.; Du, J.; Wang, Y.; Wei, Y.; Xue, W.; Li, D.; Feng, Y.; et al. THAP11 Functions as a Tumor Suppressor in Gastric Cancer through Regulating c-Myc Signaling Pathways. BioMed Res. Int. 2020, 2020, e7838924. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-X.; Liang, X.-J.; Zhou, L.-Y.; Liu, R.-J.; Bi, W.; Zhang, S.; Li, S.-S.; Yang, W.-H.; Chen, Z.-C.; Yang, X.-M.; et al. Analysis of Differential Expressions of Long Non-coding RNAs in Nasopharyngeal Carcinoma Using Next-generation Deep Sequencing. J. Cancer 2018, 9, 1943–1950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.-X.; Wang, L.-J.; Hou, J.; Liu, H.-Y.; Wang, R.; Wang, C.; Xie, W.-H. Identification of Long Noncoding RNAs as Predictors of Survival in Triple-Negative Breast Cancer Based on Network Analysis. Biomed. Res. Int. 2020, 2020, 8970340. [Google Scholar] [CrossRef]
- Li, N.; Yang, G.; Luo, L.; Ling, L.; Wang, X.; Shi, L.; Lan, J.; Jia, X.; Zhang, Q.; Long, Z.; et al. lncRNA THAP9-AS1 Promotes Pancreatic Ductal Adenocarcinoma Growth and Leads to a Poor Clinical Outcome via Sponging miR-484 and Interacting with YAP. Clin. Cancer Res. 2020, 26, 1736–1748. [Google Scholar] [CrossRef] [Green Version]
- Jia, W.; Zhang, J.; Ma, F.; Hao, S.; Li, X.; Guo, R.; Gao, Q.; Sun, Y.; Jia, J.; Li, W. Long noncoding RNA THAP9-AS1 is induced by Helicobacter pylori and promotes cell growth and migration of gastric cancer. Onco. Targets Ther. 2019, 12, 6653–6663. [Google Scholar] [CrossRef] [Green Version]
- Jiang, N.; Zhang, X.; He, Y.; Luo, B.; He, C.; Liang, Y.; Zeng, J.; Li, W.; Xian, Y.; Zheng, X. Identification of key protein-coding genes and lncRNAs in spontaneous neutrophil apoptosis. Sci. Rep. 2019, 9, 15106. [Google Scholar] [CrossRef] [Green Version]
- Cheng, J.; Ma, H.; Yan, M.; Xing, W. THAP9-AS1/miR-133b/SOX4 positive feedback loop facilitates the progression of esophageal squamous cell carcinoma. Cell Death Dis. 2021, 12, 401. [Google Scholar] [CrossRef]
- Sharma, V.; Thakore, P.; Krishnan, M.; Majumdar, S. Stress induced Differential Expression of THAP9 & THAP9-AS1 in the S-phase of cell cycle. bioRxiv 2021. [Google Scholar] [CrossRef]
- Tomczak, K.; Czerwińska, P.; Wiznerowicz, M. The Cancer Genome Atlas (TCGA): An immeasurable source of knowledge. Contemp. Oncol. 2015, 19, A68–A77. [Google Scholar] [CrossRef]
- GTEx Consortium. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013, 45, 580–585. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Fu, J.; Zeng, Z.; Cohen, D.; Li, J.; Chen, Q.; Li, B.; Liu, X.S. TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res. 2020, 48, W509–W514. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Kang, B.; Li, C.; Chen, T.; Zhang, Z. GEPIA2: An enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res. 2019, 47, W556–W560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- McKenzie, A.T.; Katsyv, I.; Song, W.-M.; Wang, M.; Zhang, B. DGCA: A comprehensive R package for Differential Gene Correlation Analysis. BMC Syst. Biol. 2016, 10, 106. [Google Scholar] [CrossRef] [Green Version]
- Howe, K.L.; Achuthan, P.; Allen, J.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Azov, A.G.; Bennett, R.; Bhai, J.; et al. Ensembl 2021. Nucleic Acids Res. 2021, 49, D884–D891. [Google Scholar] [CrossRef]
- Dreos, R.; Ambrosini, G.; Périer, R.C.; Bucher, P. The Eukaryotic Promoter Database: Expansion of EPDnew and new promoter analysis tools. Nucleic Acids Res. 2015, 43, D92–D96. [Google Scholar] [CrossRef]
- Antequera, F. Structure, function and evolution of CpG island promoters, CMLS. Cell. Mol. Life Sci. 2003, 60, 1647–1658. [Google Scholar] [CrossRef]
- Gardiner-Garden, M.; Frommer, M. CpG islands in vertebrate genomes. J. Mol. Biol. 1987, 196, 261–282. [Google Scholar] [CrossRef]
- Davis, C.A.; Hitz, B.C.; Sloan, C.A.; Chan, E.T.; Davidson, J.M.; Gabdank, I.; Hilton, J.A.; Jain, K.; Baymuradov, U.K.; Narayanan, A.K.; et al. The Encyclopedia of DNA elements (ENCODE): Data portal update. Nucleic Acids Res. 2018, 46, D794–D801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bornelöv, S.; Komorowski, J.; Wadelius, C. Different distribution of histone modifications in genes with unidirectional and bidirectional transcription and a role of CTCF and cohesin in directing transcription. BMC Genom. 2015, 16, 300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant. 2020, 13, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.Q.; Elnitski, L.L. Diversity of core promoter elements comprising human bidirectional promoters. BMC Genom. 2008, 9, S3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sloutskin, A.; Danino, Y.M.; Orenstein, Y.; Zehavi, Y.; Doniger, T.; Shamir, R.; Juven-Gershon, T. ElemeNT: A computational tool for detecting core promoter elements. Transcription 2015, 6, 41–50. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Bolotin, E.; Jiang, T.; Sladek, F.M.; Martinez, E. Prevalence of the initiator over the TATA box in human and yeast genes and identification of DNA motifs enriched in human TATA-less core promoters. Gene 2007, 389, 52–65. [Google Scholar] [CrossRef] [Green Version]
- Smale, S.T.; Kadonaga, J.T. The RNA polymerase II core promoter. Annu. Rev. Biochem. 2003, 72, 449–479. [Google Scholar] [CrossRef] [Green Version]
- Kutach, A.K.; Kadonaga, J.T. The Downstream Promoter Element DPE Appears to Be as Widely Used as the TATA Box in Drosophila Core Promoters. Mol. Cell. Biol. 2000, 20, 4754–4764. [Google Scholar] [CrossRef] [Green Version]
- Lagrange, T.; Kapanidis, A.N.; Tang, H.; Reinberg, D.; Ebright, R.H. New core promoter element in RNA polymerase II-dependent transcription: Sequence-specific DNA binding by transcription factor IIB. Genes Dev. 1998, 12, 34–44. [Google Scholar] [CrossRef] [Green Version]
- Lachance, J. Disease-associated alleles in genome-wide association studies are enriched for derived low frequency alleles relative to HapMap and neutral expectations. BMC Med. Genom. 2010, 3, 57. [Google Scholar] [CrossRef] [Green Version]
- Andiappan, A.K.; Wang, D.Y.; Anantharaman, R.; Parate, P.N.; Suri, B.K.; Low, H.Q.; Li, Y.; Zhao, W.; Castagnoli, P.; Liu, J.; et al. Genome-Wide Association Study for Atopy and Allergic Rhinitis in a Singapore Chinese Population. PLoS ONE 2011, 6, e19719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guarguaglini, G.; Battistoni, A.; Pittoggi, C.; di Matteo, G.; di Fiore, B.; Lavia, P. Expression of the murine RanBP1 and Htf9-c genes is regulated from a shared bidirectional promoter during cell cycle progression. Biochem. J. 1997, 325 Pt 1, 277–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliver, S. Guilt-by-association goes global. Nature 2000, 403, 601–602. [Google Scholar] [CrossRef]
- Van Dam, S.; Võsa, U.; van der Graaf, A.; Franke, L.; de Magalhães, J.P. Gene co-expression analysis for functional classification and gene–disease predictions. Brief. Bioinform. 2017, 19, 575–592. [Google Scholar] [CrossRef] [PubMed]
- Bijnens, A.P.J.J.; Gils, A.; Jutten, B.; Faber, B.C.G.; Heeneman, S.; Kitslaar, P.J.E.H.M.; Tordoir, J.H.M.; de Vries, C.J.M.; Kroon, A.A.; Daemen, M.J.A.P.; et al. Vasculin, a novel vascular protein differentially expressed in human atherogenesis. Blood 2003, 102, 2803–2810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ong, W.-Y.; Ng, M.P.-E.; Loke, S.-Y.; Jin, S.; Wu, Y.-J.; Tanaka, K.; Wong, P.T.-H. Comprehensive Gene Expression Profiling Reveals Synergistic Functional Networks in Cerebral Vessels after Hypertension or Hypercholesterolemia. PLoS ONE 2013, 8, e68335. [Google Scholar] [CrossRef]
- Mathys, H.; Davila-Velderrain, J.; Peng, Z.; Gao, F.; Mohammadi, S.; Young, J.Z.; Menon, M.; He, L.; Abdurrob, F.; Jiang, X.; et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 2019, 570, 332–337. [Google Scholar] [CrossRef]
- Lee, T.; Lee, H. Identification of Disease-Related Genes That Are Common between Alzheimer’s and Cardiovascular Disease Using Blood Genome-Wide Transcriptome Analysis. Biomedicines 2021, 9, 1525. [Google Scholar] [CrossRef]
- Cho, H.; Chung, J.-Y.; Song, K.-H.; Noh, K.H.; Kim, B.W.; Chung, E.J.; Ylaya, K.; Kim, J.H.; Kim, T.W.; Hewitt, S.M.; et al. Apoptosis inhibitor-5 overexpression is associated with tumor progression and poor prognosis in patients with cervical cancer. BMC Cancer 2014, 14, 545. [Google Scholar] [CrossRef] [Green Version]
- Krejci, P.; Pejchalova, K.; Rosenbloom, B.E.; Rosenfelt, F.P.; Tran, E.L.; Laurell, H.; Wilcox, W.R. The antiapoptotic protein Api5 and its partner, high molecular weight FGF2, are up-regulated in B cell chronic lymphoid leukemia. J. Leukoc. Biol. 2007, 82, 1363–1364. [Google Scholar] [CrossRef]
- Mao, C.-P.; Wu, T.; Song, K.-H.; Kim, T.W. Immune-mediated tumor evolution: Nanog links the emergence of a stem like cancer cell state and immune evasion. Oncoimmunology 2014, 3, e947871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, K.-H.; Cho, H.; Kim, S.; Lee, H.-J.; Oh, S.J.; Woo, S.R.; Hong, S.-O.; Jang, H.S.; Noh, K.H.; Choi, C.H.; et al. API5 confers cancer stem cell-like properties through the FGF2-NANOG axis. Oncogenesis 2017, 6, e285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, W.; Swaminathan, G.; Plowey, E.D. GA binding protein augments autophagy via transcriptional activation of BECN1-PIK3C3 complex genes. Autophagy 2014, 10, 1622–1636. [Google Scholar] [CrossRef] [Green Version]
- Morris, S.; Geoghegan, N.D.; Sadler, J.B.A.; Koester, A.M.; Black, H.L.; Laub, M.; Miller, L.; Heffernan, L.; Simpson, J.C.; Mastick, C.C.; et al. Characterisation of GLUT4 trafficking in HeLa cells: Comparable kinetics and orthologous trafficking mechanisms to 3T3-L1 adipocytes. PeerJ 2020, 8, e8751. [Google Scholar] [CrossRef] [PubMed]
- Kagaya, A.; Shimada, H.; Shiratori, T.; Kuboshima, M.; Nakashima-Fujita, K.; Yasuraoka, M.; Nishimori, T.; Kurei, S.; Hachiya, T.; Murakami, A.; et al. Identification of a novel SEREX antigen family, ECSA, in esophageal squamous cell carcinoma. Proteome Sci. 2011, 9, 31. [Google Scholar] [CrossRef] [Green Version]
- Abovich, N.; Rosbash, M. Cross-Intron Bridging Interactions in the Yeast Commitment Complex Are Conserved in Mammals. Cell 1997, 89, 403–412. [Google Scholar] [CrossRef] [Green Version]
- Kao, H.Y.; Siliciano, P.G. Identification of Prp40, a novel essential yeast splicing factor associated with the U1 small nuclear ribonucleoprotein particle. Mol. Cell Biol. 1996, 16, 960–967. [Google Scholar] [CrossRef] [Green Version]
- Buschdorf, J.P.; Strätling, W.H. A WW domain binding region in methyl-CpG-binding protein MeCP2: Impact on Rett syndrome. J. Mol. Med. 2004, 82, 135–143. [Google Scholar] [CrossRef]
- Faber, P.W.; Barnes, G.T.; Srinidhi, J.; Chen, J.; Gusella, J.F.; MacDonald, M.E. Huntingtin Interacts with a Family of WW Domain Proteins. Hum. Mol. Genet. 1998, 7, 1463–1474. [Google Scholar] [CrossRef] [Green Version]
- Huo, Z.; Zhai, S.; Weng, Y.; Qian, H.; Tang, X.; Shi, Y.; Deng, X.; Wang, Y.; Shen, B. PRPF40A as a potential diagnostic and prognostic marker is upregulated in pancreatic cancer tissues and cell lines: An integrated bioinformatics data analysis. Onco. Targets Ther. 2019, 12, 5037–5051. [Google Scholar] [CrossRef] [Green Version]
- Oleksiewicz, U.; Liloglou, T.; Tasopoulou, K.-M.; Daskoulidou, N.; Gosney, J.R.; Field, J.K.; Xinarianos, G. COL1A1, PRPF40A, and UCP2 correlate with hypoxia markers in non-small cell lung cancer. J. Cancer Res. Clin. Oncol. 2017, 143, 1133–1141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Xie, G.; Singh, M.; Ghanbarian, A.T.; Raskó, T.; Szvetnik, A.; Cai, H.; Besser, D.; Prigione, A.; Fuchs, N.V.; et al. Primate-specific endogenous retrovirus-driven transcription defines naive-like stem cells. Nature 2014, 516, 405–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nayler, O.; Hartmann, A.M.; Stamm, S. The ER Repeat Protein Yt521-B Localizes to a Novel Subnuclear Compartment. J. Cell Biol. 2000, 150, 949–962. [Google Scholar] [CrossRef] [PubMed]
- Hirschfeld, M.; Zhang, B.; Jaeger, M.; Stamm, S.; Erbes, T.; Mayer, S.; Tong, X.; Stickeler, E. Hypoxia-dependent mRNA expression pattern of splicing factor YT521 and its impact on oncological important target gene expression. Mol. Carcinog. 2014, 53, 883–892. [Google Scholar] [CrossRef]
- Luxton, H.J.; Simpson, B.S.; Mills, I.G.; Brindle, N.R.; Ahmed, Z.; Stavrinides, V.; Heavey, S.; Stamm, S.; Whitaker, H.C. The Oncogene Metadherin Interacts with the Known Splicing Proteins YTHDC1, Sam68 and T-STAR and Plays a Novel Role in Alternative mRNA Splicing. Cancers 2019, 11, 1233. [Google Scholar] [CrossRef] [Green Version]
- Yoo, B.K.; Emdad, L.; Su, Z.; Villanueva, A.; Chiang, D.Y.; Mukhopadhyay, N.D.; Mills, A.S.; Waxman, S.; Fisher, R.A.; Llovet, J.M.; et al. Astrocyte elevated gene-1 regulates hepatocellular carcinoma development and progression. J. Clin. Investig. 2009, 119, 465–477. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Shao, X.; Zhou, J.; Qiu, J.; Wu, Y.; Cheng, J. YT521 promotes metastases of endometrial cancer by differential splicing of vascular endothelial growth factor A. Tumor Biol. 2016, 37, 15543–15549. [Google Scholar] [CrossRef]
- Feng, Y.; Chen, M.; Manley, J.L. Phosphorylation switches the general splicing repressor SRp38 to a sequence-specific activator. Nat. Struct. Mol. Biol. 2008, 15, 1040–1048. [Google Scholar] [CrossRef]
- Zhou, X.; Li, X.; Cheng, Y.; Wu, W.; Xie, Z.; Xi, Q.; Han, J.; Wu, G.; Fang, J.; Feng, Y. BCLAF1 and its splicing regulator SRSF10 regulate the tumorigenic potential of colon cancer cells. Nat. Commun. 2014, 5, 4581. [Google Scholar] [CrossRef]
- Kasof, G.M.; Goyal, L.; White, E. Btf, a Novel Death-Promoting Transcriptional Repressor That Interacts with Bcl-2-Related Proteins. Mol. Cell. Biol. 1999, 19, 4390–4404. [Google Scholar] [CrossRef] [Green Version]
- McPherson, J.P.; Sarras, H.; Lemmers, B.; Tamblyn, L.; Migon, E.; Matysiak-Zablocki, E.; Hakem, A.; Azami, S.A.; Cardoso, R.; Fish, J.; et al. Essential role for Bclaf1 in lung development and immune system function. Cell Death Differ. 2009, 16, 331–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savage, K.I.; Gorski, J.J.; Barros, E.M.; Irwin, G.W.; Manti, L.; Powell, A.J.; Pellagatti, A.; Lukashchuk, N.; McCance, D.J.; McCluggage, W.G.; et al. Identification of a BRCA1-mRNA Splicing Complex Required for Efficient DNA Repair and Maintenance of Genomic Stability. Mol. Cell. 2014, 54, 445–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, A.-W.; Sun, H.; Geng, Y.; Peng, Q.; Wang, P.; Chen, J.; Xiong, T.; Cao, R.; Tang, J. Bclaf1 is an important NF-κB signaling transducer and C/EBPβ regulator in DNA damage-induced senescence. Cell Death Differ. 2016, 23, 865–875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varia, S.; Potabathula, D.; Deng, Z.; Bubulya, A.; Bubulya, P.A. Btf and TRAP150 have distinct roles in regulating subcellular mRNA distribution. Nucleus 2013, 4, 229–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aiello, C.; Terracciano, A.; Simonati, A.; Discepoli, G.; Cannelli, N.; Claps, D.; Crow, Y.J.; Bianchi, M.; Kitzmuller, C.; Longo, D.; et al. Mutations in MFSD8/CLN7 are a frequent cause of variant-late infantile neuronal ceroid lipofuscinosis. Hum. Mutat. 2009, 30, E530–E540. [Google Scholar] [CrossRef] [PubMed]
- Aldahmesh, M.A.; Al-Hassnan, Z.N.; Aldosari, M.; Alkuraya, F.S. Neuronal ceroid lipofuscinosis caused by MFSD8 mutations: A common theme emerging. Neurogenetics 2009, 10, 307–311. [Google Scholar] [CrossRef]
- Kousi, M.; Siintola, E.; Dvorakova, L.; Vlaskova, H.; Turnbull, J.; Topcu, M.; Yuksel, D.; Gokben, S.; Minassian, B.A.; Elleder, M.; et al. Mutations in CLN7/MFSD8 are a common cause of variant late-infantile neuronal ceroid lipofuscinosis. Brain 2009, 132, 810–819. [Google Scholar] [CrossRef] [Green Version]
- Siintola, E.; Topcu, M.; Aula, N.; Lohi, H.; Minassian, B.A.; Paterson, A.D.; Liu, X.-Q.; Wilson, C.; Lahtinen, U.; Anttonen, A.-K.; et al. The Novel Neuronal Ceroid Lipofuscinosis Gene MFSD8 Encodes a Putative Lysosomal Transporter. Am. J. Hum. Genet. 2007, 81, 136–146. [Google Scholar] [CrossRef] [Green Version]
- Stogmann, E.; El Tawil, S.; Wagenstaller, J.; Gaber, A.; Edris, S.; Abdelhady, A.; Assem-Hilger, E.; Leutmezer, F.; Bonelli, S.; Baumgartner, C.; et al. A novel mutation in the MFSD8 gene in late infantile neuronal ceroid lipofuscinosis. Neurogenetics 2009, 10, 73–77. [Google Scholar] [CrossRef]
- Clifford, R.J.; Zhang, J.; Meerzaman, D.M.; Lyu, M.-S.; Hu, Y.; Cultraro, C.M.; Finney, R.P.; Kelley, J.M.; Efroni, S.; Greenblum, S.I.; et al. Genetic Variations at Loci Involved in the Immune Response Are Risk Factors for Hepatocellular Carcinoma. Hepatology 2010, 52, 2034–2043. [Google Scholar] [CrossRef]
- Ge, S.X.; Jung, D.; Yao, R. ShinyGO: A graphical gene-set enrichment tool for animals and plants. Bioinformatics 2020, 36, 2628–2629. [Google Scholar] [CrossRef] [PubMed]
- Sanghavi, H.M.; Majumdar, S. Oligomerization of THAP9 Transposase via Amino-Terminal Domains. Biochemistry 2021, 60, 1822–1835. [Google Scholar] [CrossRef] [PubMed]
- Amar, D.; Safer, H.; Shamir, R. Dissection of regulatory networks that are altered in disease via differential co-expression. PLoS Comput. Biol. 2013, 9, e1002955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hudson, N.J.; Reverter, A.; Dalrymple, B.P. A differential wiring analysis of expression data correctly identifies the gene containing the causal mutation. PLoS Comput. Biol. 2009, 5, e1000382. [Google Scholar] [CrossRef]
- Kostka, D.; Spang, R. Finding disease specific alterations in the co-expression of genes. Bioinformatics 2004, 1 (Suppl. 20), i194–i199. [Google Scholar] [CrossRef] [Green Version]
- Gao, Q.; Ho, C.; Jia, Y.; Li, J.J.; Huang, H. Biclustering of linear patterns in gene expression data. J. Comput. Biol. 2012, 19, 619–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monaco, G.; van Dam, S.; Ribeiro, J.L.C.N.; Larbi, A.; de Magalhães, J.P. A comparison of human and mouse gene co-expression networks reveals conservation and divergence at the tissue, pathway and disease levels. BMC Evol. Biol. 2015, 15, 259. [Google Scholar] [CrossRef] [Green Version]
- Pierson, E.; Consortium, G.T.E.; Koller, D.; Battle, A.; Mostafavi, S.; Ardlie, K.G.; Getz, G.; Wright, F.A.; Kellis, M.; Volpi, S.; et al. Sharing and Specificity of Co-expression Networks across 35 Human Tissues. PLoS Comput. Biol. 2015, 11, e1004220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeisel, A.; Muñoz-Manchado, A.B.; Codeluppi, S.; Lönnerberg, P.; la Manno, G.; Juréus, A.; Marques, S.; Munguba, H.; He, L.; Betsholtz, C.; et al. Brain structure. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science 2015, 347, 1138–1142. [Google Scholar] [CrossRef]
- Chen, Y.; Li, H.; Li, Y.-Y.; Li, Y. Pan-Cancer Analysis of Head-to-Head Gene Pairs in Terms of Transcriptional Activity, Co-expression and Regulation. Front. Genet. 2021, 11, 1707. [Google Scholar] [CrossRef]
- Shu, J.; Jelinek, J.; Chang, H.; Shen, L.; Qin, T.; Chung, W.; Oki, Y.; Issa, J.-P.J. Silencing of Bidirectional Promoters by DNA Methylation in Tumorigenesis. Cancer Res. 2006, 66, 5077–5084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, M.Q.; Koehly, L.M.; Elnitski, L.L. Comprehensive Annotation of Bidirectional Promoters Identifies Co-Regulation among Breast and Ovarian Cancer Genes. PLOS Comput. Biol. 2007, 3, e72. [Google Scholar] [CrossRef] [PubMed]
- Albig, W.; Kioschis, P.; Poustka, A.; Meergans, K.; Doenecke, D. Human histone gene organization: Nonregular arrangement within a large cluster. Genomics 1997, 40, 314–322. [Google Scholar] [CrossRef]
- Orekhova, A.S.; Rubtsov, P.M. Bidirectional Promoters in the Transcription of Mammalian Genomes. Biochemistry 2013, 78, 7. [Google Scholar] [CrossRef]
- Dovhey, S.E.; Ghosh, N.S.; Wright, K.L. Loss of interferon-gamma inducibility of TAP1 and LMP2 in a renal cell carcinoma cell line. Cancer Res. 2000, 60, 5789–5796. [Google Scholar]
- Rashmi, R.; Nandi, C.; Majumdar, S. Evolutionary analysis of THAP9 transposase: Conserved regions, novel motifs. bioRxiv 2021. [Google Scholar] [CrossRef]
- Parker, J.B.; Palchaudhuri, S.; Yin, H.; Wei, J.; Chakravarti, D. A transcriptional regulatory role of the THAP11-HCF-1 complex in colon cancer cell function. Mol. Cell Biol. 2012, 32, 1654–1670. [Google Scholar] [CrossRef] [Green Version]
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. Studying Gene Expression and Function, Molecular Biology of the Cell, 4th ed.Garland Science: New York, NY, USA, 2002. Available online: https://www.ncbi.nlm.nih.gov/books/NBK26818/ (accessed on 12 January 2022).
- Marcocci, M.E.; Napoletani, G.; Protto, V.; Kolesova, O.; Piacentini, R.; Puma, D.D.L.; Lomonte, P.; Grassi, C.; Palamara, A.T.; de Chiara, G. Herpes Simplex Virus-1 in the Brain: The Dark Side of a Sneaky Infection. Trends Microbiol. 2020, 28, 808–820. [Google Scholar] [CrossRef] [PubMed]
- Kumar, G.S.S.; Venugopal, A.K.; Kashyap, M.K.; Raju, R.; Marimuthu, A.; Palapetta, S.M.; Subbanayya, Y.; Goel, R.; Chawla, A.; Dikshit, J.B.; et al. Gene Expression Profiling of Tuberculous Meningitis Co-infected with HIV. J. Proteom. Bioinform. 2012, 5, 235–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bioconductor. Available online: http://bioconductor.org/packages/org.Hs.eg.db/ (accessed on 12 January 2022).
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]













Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rashmi, R.; Majumdar, S. Pan-Cancer Analysis Reveals the Prognostic Potential of the THAP9/THAP9-AS1 Sense–Antisense Gene Pair in Human Cancers. Non-Coding RNA 2022, 8, 51. https://doi.org/10.3390/ncrna8040051
Rashmi R, Majumdar S. Pan-Cancer Analysis Reveals the Prognostic Potential of the THAP9/THAP9-AS1 Sense–Antisense Gene Pair in Human Cancers. Non-Coding RNA. 2022; 8(4):51. https://doi.org/10.3390/ncrna8040051
Chicago/Turabian StyleRashmi, Richa, and Sharmistha Majumdar. 2022. "Pan-Cancer Analysis Reveals the Prognostic Potential of the THAP9/THAP9-AS1 Sense–Antisense Gene Pair in Human Cancers" Non-Coding RNA 8, no. 4: 51. https://doi.org/10.3390/ncrna8040051
APA StyleRashmi, R., & Majumdar, S. (2022). Pan-Cancer Analysis Reveals the Prognostic Potential of the THAP9/THAP9-AS1 Sense–Antisense Gene Pair in Human Cancers. Non-Coding RNA, 8(4), 51. https://doi.org/10.3390/ncrna8040051