


Non-Coding RNA, Volume 8, Issue 4 (August 2022) – 18 articles
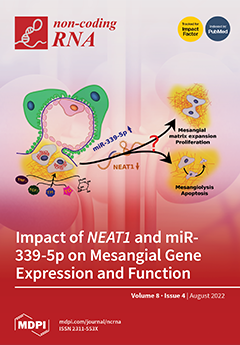
Cover Story (view full-size image):
In diabetic kidney disease when various cellular functions are impaired, mesangial cells are crucial for the maintenance of the glomerular environment. However, diabetes-evoked dysregulation can result in both mesangial proliferation and apoptosis. It still needs to be elucidated which factors cause the decision for one or the other way. Here, we analyzed and discussed the relevance of the interplay of the lncRNA NEAT1 and the miRNA miR-339-5p for mesangial cell physiology in several injury models. While NEAT1 is mainly upregulated in cancer cells promoting, e.g., proliferation, we observed a reduced mesangial NEAT1 expression. Silencing NEAT1 resulted in differential expression of various coding genes and miRNAs, especially miR-339-5p, which was upregulated. Overexpression of miR-339-5p caused a similar phenotype, such as a knockdown of NEAT1. View this paper
- Issues are regarded as officially published after their release is announced to the table of contents alert mailing list.
- You may sign up for e-mail alerts to receive table of contents of newly released issues.
- PDF is the official format for papers published in both, html and pdf forms. To view the papers in pdf format, click on the "PDF Full-text" link, and use the free Adobe Reader to open them.
Previous Issue
Next Issue