
Systematic Analysis of Long Non-Coding RNA Genes in Nonalcoholic Fatty Liver Disease

 ,
,  , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
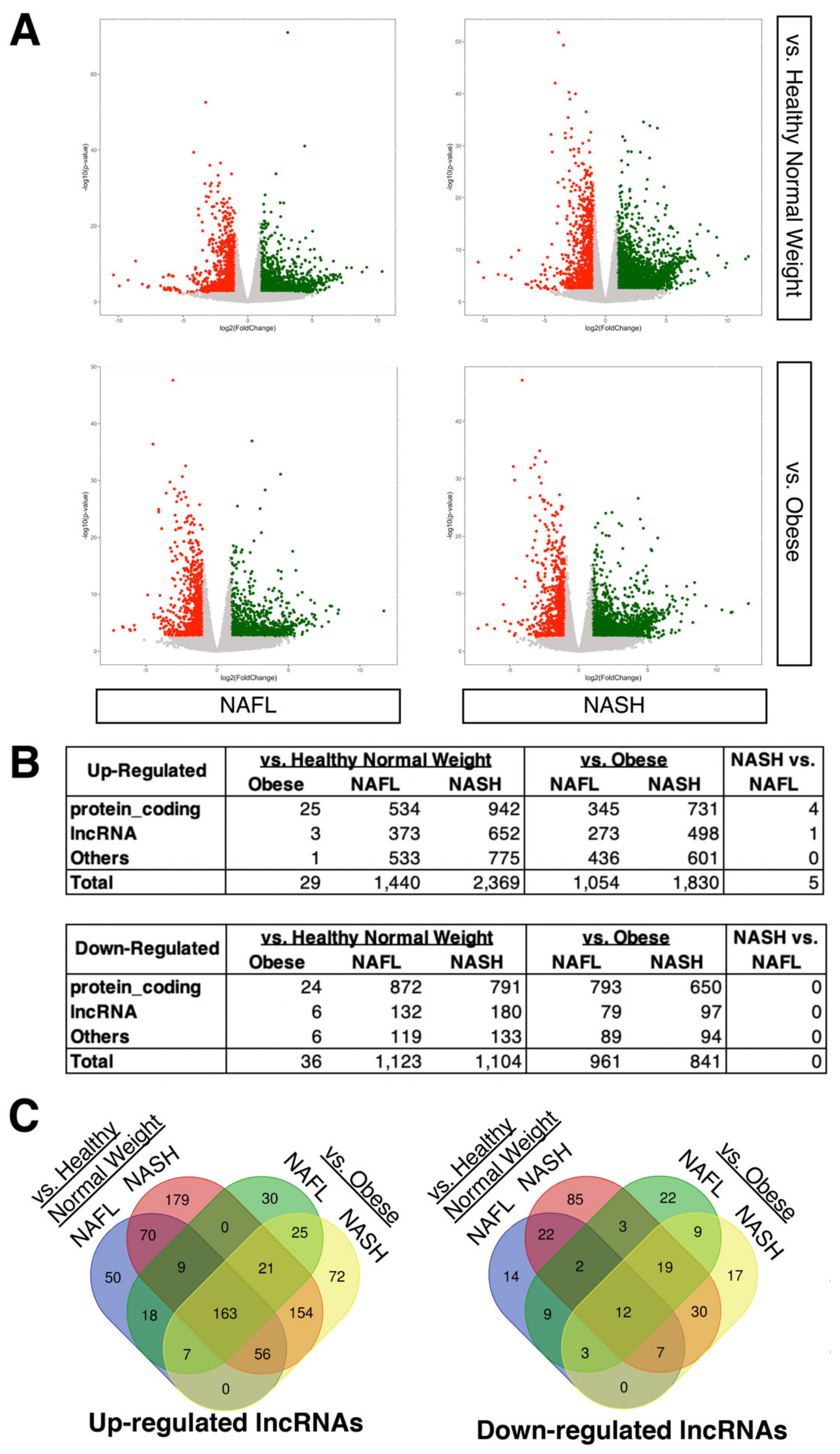
2.1. Expression Profiling of LncRNAs in NAFLD Patients Compared to Healthy Donors
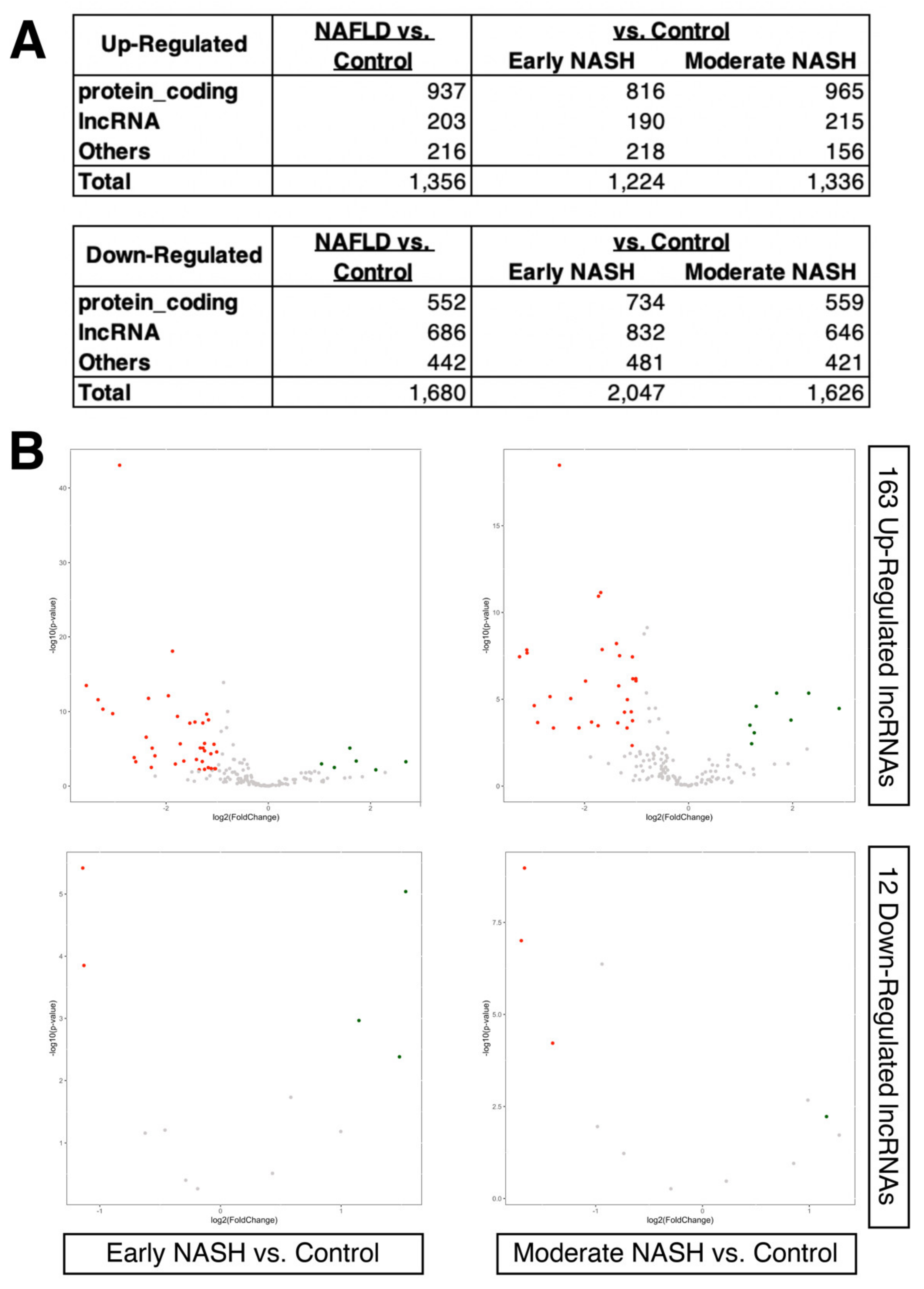
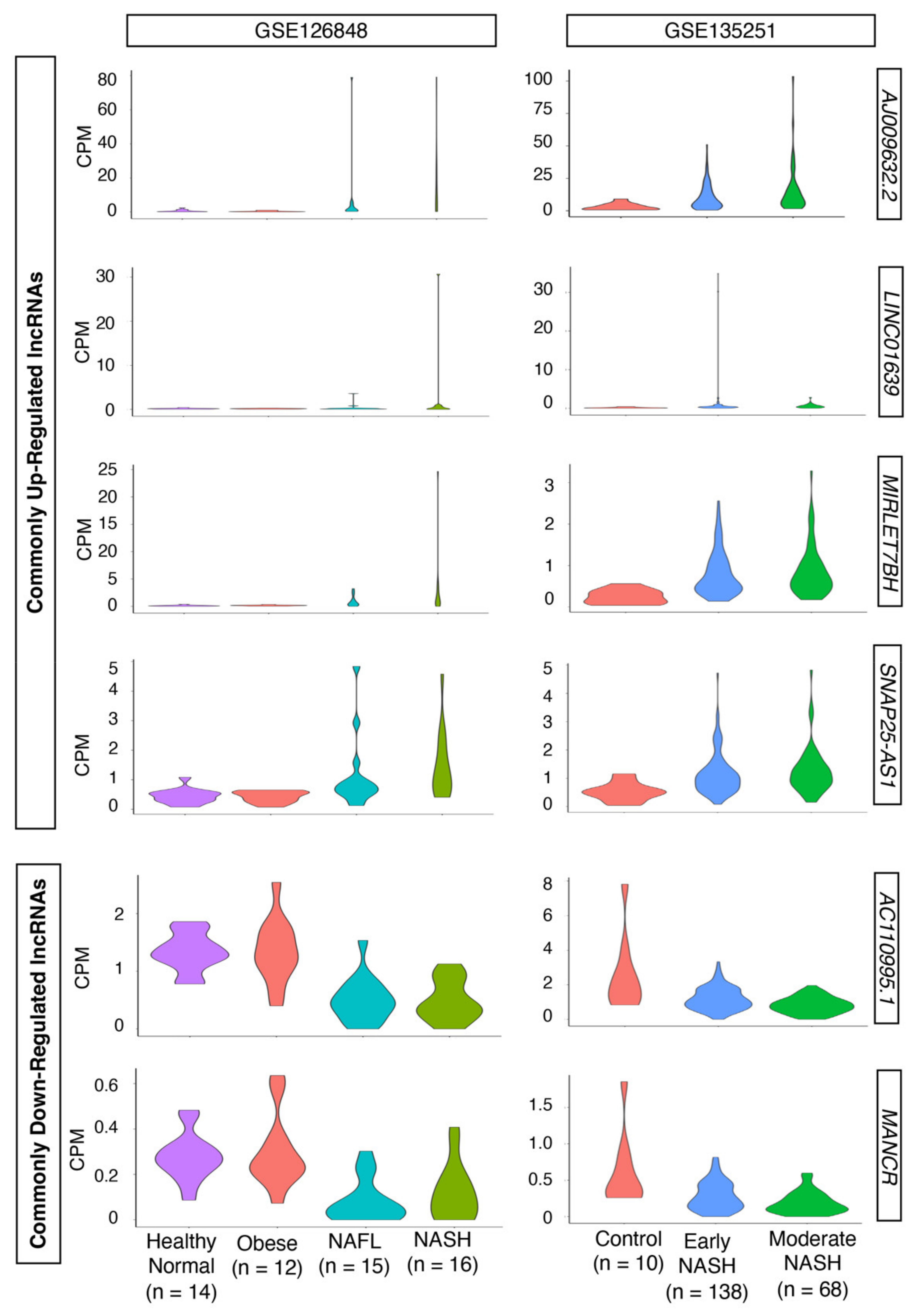
2.2. Dysregulated LncRNAs during the Progression of NASH
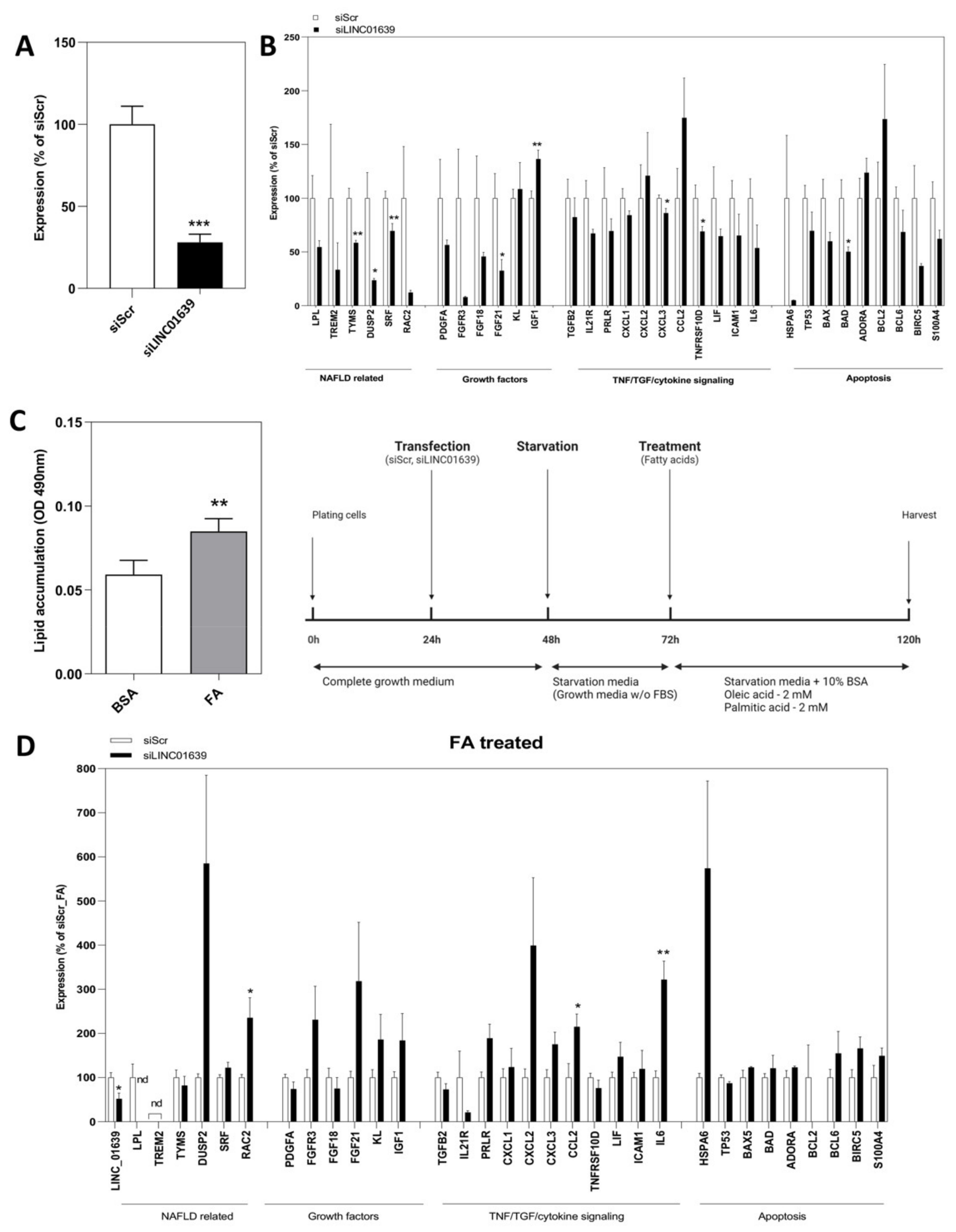
2.3. Loss-of-Function Experiments to Uncover the Roles of Differentially Expressed LncRNAs
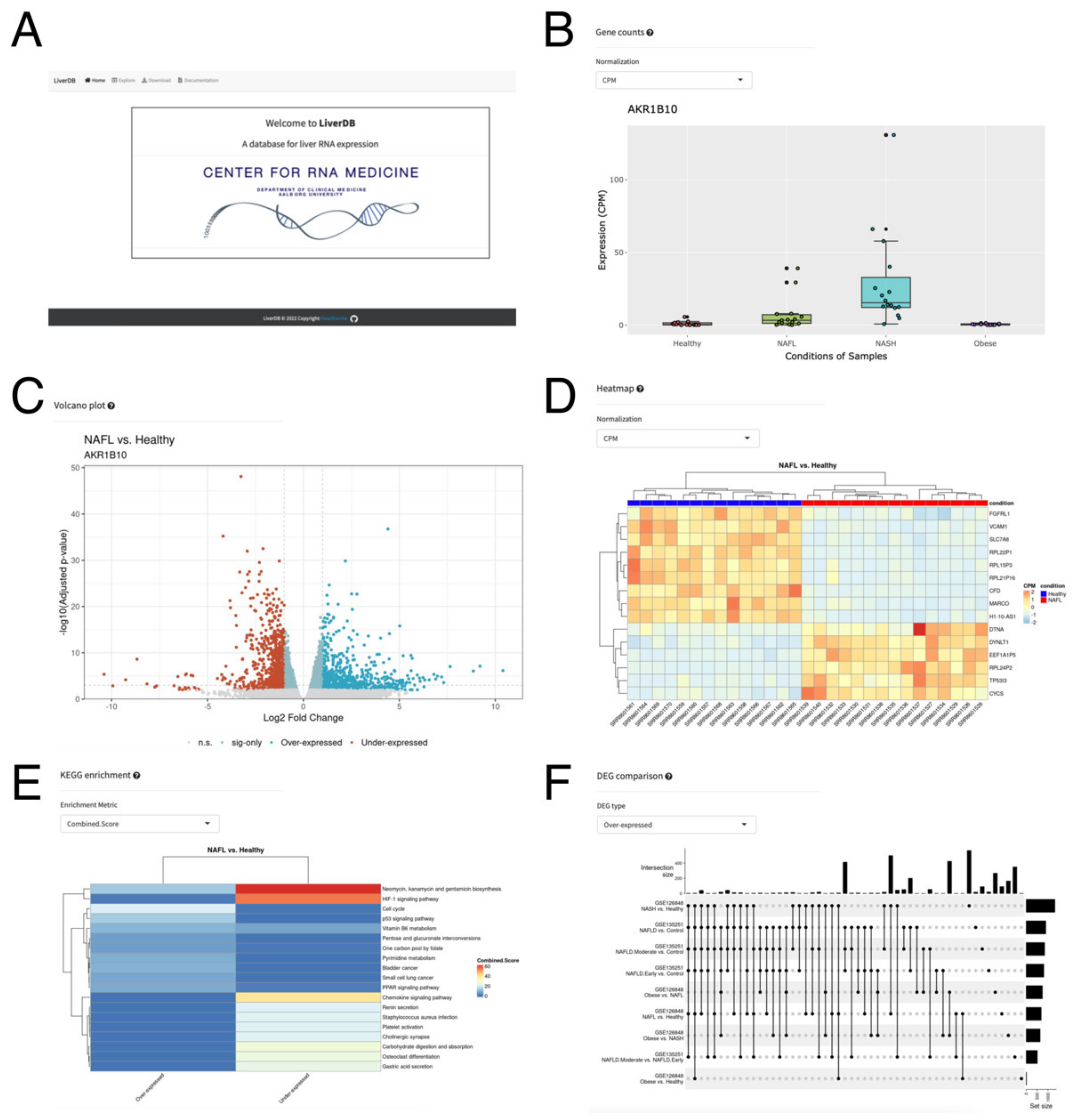
2.4. A Web Database, LiverDB, for Exploration of NAFLD-Related Genes
3. Discussion
4. Materials and Methods
4.1. RNA-Seq Data Analysis
4.2. Data analysis and Visualization
4.3. Interactive Web Database, LiverDB
4.4. Cell Culture
4.5. Transfection with siRNA and Treatment with Fatty Acids (FA)
4.6. Lipid Accumulation Assay
4.7. Isolation of Total RNA and RT-PCR
4.8. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zabaleta, N.; Hommel, M.; Salas, D.; Gonzalez-Aseguinolaza, G. Genetic-Based Approaches to Inherited Metabolic Liver Diseases. Hum. Gene Ther. 2019, 30, 1190–1203. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Montal, R.; Sia, D.; Finn, R.S. Molecular therapies and precision medicine for hepatocellular carcinoma. Nat. Rev. Clin. Oncol. 2018, 15, 599–616. [Google Scholar] [CrossRef] [PubMed]
- El-Serag, H.B. Hepatocellular carcinoma. N. Engl. J. Med. 2011, 365, 1118–1127. [Google Scholar] [CrossRef] [PubMed]
- Michalopoulos, G.K. Liver regeneration. J. Cell. Physiol. 2007, 213, 286–300. [Google Scholar] [CrossRef] [PubMed]
- Trotter, J.F. Current Issues in Liver Transplantation. Gastroenterol. Hepatol. 2016, 12, 214–219. [Google Scholar]
- Asrani, S.K.; Devarbhavi, H.; Eaton, J.; Kamath, P.S. Burden of liver diseases in the world. J. Hepatol. 2019, 70, 151–171. [Google Scholar] [CrossRef]
- Weirick, T.; Militello, G.; Muller, R.; John, D.; Dimmeler, S.; Uchida, S. The identification and characterization of novel transcripts from RNA-seq data. Brief. Bioinform. 2016, 17, 678–685. [Google Scholar] [CrossRef]
- Uchida, S.; Adams, J.C. Physiological roles of non-coding RNAs. Am. J. Physiol. Cell. Physiol. 2019, 317, C1–C2. [Google Scholar] [CrossRef]
- Rinn, J.L.; Chang, H.Y. Genome regulation by long noncoding RNAs. Annu. Rev. Biochem. 2012, 81, 145–166. [Google Scholar] [CrossRef] [Green Version]
- Katz, K.; Shutov, O.; Lapoint, R.; Kimelman, M.; Brister, J.R.; O’Sullivan, C. The Sequence Read Archive: A decade more of explosive growth. Nucleic Acids Res. 2022, 50, D387–D390. [Google Scholar] [CrossRef]
- De Vincentis, A.; Rahmani, Z.; Muley, M.; Vespasiani-Gentilucci, U.; Ruggiero, S.; Zamani, P.; Jamialahmadi, T.; Sahebkar, A. Long noncoding RNAs in nonalcoholic fatty liver disease and liver fibrosis: State-of-the-art and perspectives in diagnosis and treatment. Drug Discov. Today 2020, 25, 1277–1286. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.A.; Park, K.K.; Lee, S.J. LncRNAs Act as a Link between Chronic Liver Disease and Hepatocellular Carcinoma. Int. J. Mol. Sci. 2020, 21, 2883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfister, D.; Nunez, N.G.; Pinyol, R.; Govaere, O.; Pinter, M.; Szydlowska, M.; Gupta, R.; Qiu, M.; Deczkowska, A.; Weiner, A.; et al. NASH limits anti-tumour surveillance in immunotherapy-treated HCC. Nature 2021, 592, 450–456. [Google Scholar] [CrossRef] [PubMed]
- Parry, S.A.; Hodson, L. Managing NAFLD in Type 2 Diabetes: The Effect of Lifestyle Interventions, a Narrative Review. Adv. Ther. 2020, 37, 1381–1406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eslam, M.; Sanyal, A.J.; George, J. MAFLD: A Consensus-Driven Proposed Nomenclature for Metabolic Associated Fatty Liver Disease. Gastroenterology 2020, 158, 1999–2014.e1991. [Google Scholar] [CrossRef]
- Byrne, C.D.; Targher, G. NAFLD: A multisystem disease. J. Hepatol. 2015, 62, S47–S64. [Google Scholar] [CrossRef] [Green Version]
- Diehl, A.M.; Day, C. Cause, Pathogenesis, and Treatment of Nonalcoholic Steatohepatitis. N. Engl. J Med. 2017, 377, 2063–2072. [Google Scholar] [CrossRef]
- Rinella, M.E. Nonalcoholic fatty liver disease: A systematic review. JAMA 2015, 313, 2263–2273. [Google Scholar] [CrossRef]
- Younossi, Z.M. Non-alcoholic fatty liver disease—A global public health perspective. J. Hepatol. 2019, 70, 531–544. [Google Scholar] [CrossRef] [Green Version]
- Huang, P.; Tu, B.; Liao, H.J.; Huang, F.Z.; Li, Z.Z.; Zhu, K.Y.; Dai, F.; Liu, H.Z.; Zhang, T.Y.; Sun, C.Z. Elevation of plasma tRNA fragments as a promising biomarker for liver fibrosis in nonalcoholic fatty liver disease. Sci. Rep. 2021, 11, 5886. [Google Scholar] [CrossRef]
- Govaere, O.; Cockell, S.; Tiniakos, D.; Queen, R.; Younes, R.; Vacca, M.; Alexander, L.; Ravaioli, F.; Palmer, J.; Petta, S.; et al. Transcriptomic profiling across the nonalcoholic fatty liver disease spectrum reveals gene signatures for steatohepatitis and fibrosis. Sci. Transl. Med. 2020, 12, eaba4448. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.; Hu, H.; Liu, S.; Zhang, Z.; Li, Y.; Zhou, L. Comprehensive analysis of the translatome reveals the relationship between the translational and transcriptional control in high fat diet-induced liver steatosis. RNA Biol. 2020, 18, 863–874. [Google Scholar] [CrossRef] [PubMed]
- Veyel, D.; Wenger, K.; Broermann, A.; Bretschneider, T.; Luippold, A.H.; Krawczyk, B.; Rist, W.; Simon, E. Biomarker discovery for chronic liver diseases by multi-omics—A preclinical case study. Sci. Rep. 2020, 10, 1314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haas, J.T.; Vonghia, L.; Mogilenko, D.A.; Verrijken, A.; Molendi-Coste, O.; Fleury, S.; Deprince, A.; Nikitin, A.; Woitrain, E.; Ducrocq-Geoffroy, L.; et al. Transcriptional Network Analysis Implicates Altered Hepatic Immune Function in NASH development and resolution. Nat. Metab. 2019, 1, 604–614. [Google Scholar] [CrossRef] [PubMed]
- Jia, X.; Zhai, T. Integrated Analysis of Multiple Microarray Studies to Identify Novel Gene Signatures in Non-alcoholic Fatty Liver Disease. Front. Endocrinol. 2019, 10, 599. [Google Scholar] [CrossRef] [Green Version]
- Lefebvre, P.; Lalloyer, F.; Bauge, E.; Pawlak, M.; Gheeraert, C.; Dehondt, H.; Vanhoutte, J.; Woitrain, E.; Hennuyer, N.; Mazuy, C.; et al. Interspecies NASH disease activity whole-genome profiling identifies a fibrogenic role of PPARalpha-regulated dermatopontin. JCI Insight 2017, 2, e92264. [Google Scholar] [CrossRef]
- Teufel, A.; Itzel, T.; Erhart, W.; Brosch, M.; Wang, X.Y.; Kim, Y.O.; von Schonfels, W.; Herrmann, A.; Bruckner, S.; Stickel, F.; et al. Comparison of Gene Expression Patterns Between Mouse Models of Nonalcoholic Fatty Liver Disease and Liver Tissues From Patients. Gastroenterology 2016, 151, 513–525.E0. [Google Scholar] [CrossRef]
- Arendt, B.M.; Comelli, E.M.; Ma, D.W.; Lou, W.; Teterina, A.; Kim, T.; Fung, S.K.; Wong, D.K.; McGilvray, I.; Fischer, S.E.; et al. Altered hepatic gene expression in nonalcoholic fatty liver disease is associated with lower hepatic n-3 and n-6 polyunsaturated fatty acids. Hepatology 2015, 61, 1565–1578. [Google Scholar] [CrossRef] [Green Version]
- Zhao, S.; Fung-Leung, W.P.; Bittner, A.; Ngo, K.; Liu, X. Comparison of RNA-Seq and microarray in transcriptome profiling of activated T cells. PLoS ONE 2014, 9, e78644. [Google Scholar] [CrossRef]
- Moylan, C.A.; Pang, H.; Dellinger, A.; Suzuki, A.; Garrett, M.E.; Guy, C.D.; Murphy, S.K.; Ashley-Koch, A.E.; Choi, S.S.; Michelotti, G.A.; et al. Hepatic gene expression profiles differentiate presymptomatic patients with mild versus severe nonalcoholic fatty liver disease. Hepatology 2014, 59, 471–482. [Google Scholar] [CrossRef]
- Starmann, J.; Falth, M.; Spindelbock, W.; Lanz, K.L.; Lackner, C.; Zatloukal, K.; Trauner, M.; Sultmann, H. Gene expression profiling unravels cancer-related hepatic molecular signatures in steatohepatitis but not in steatosis. PLoS ONE 2012, 7, e46584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suppli, M.P.; Rigbolt, K.T.G.; Veidal, S.S.; Heeboll, S.; Eriksen, P.L.; Demant, M.; Bagger, J.I.; Nielsen, J.C.; Oro, D.; Thrane, S.W.; et al. Hepatic transcriptome signatures in patients with varying degrees of nonalcoholic fatty liver disease compared with healthy normal-weight individuals. Am. J. Physiol. Gastrointest. Liver Physiol. 2019, 316, G462–G472. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Wang, Y.; Xue, H. Long non-coding RNA GAS5 contributes to the progression of nonalcoholic fatty liver disease by targeting the microRNA-29a-3p/NOTCH2 axis. Bioengineered 2022, 13, 8370–8381. [Google Scholar] [CrossRef] [PubMed]
- Guo, B.; Cheng, Y.; Yao, L.; Zhang, J.; Lu, J.; Qi, H.; Chen, H. LncRNA HOTAIR regulates the lipid accumulation in non-alcoholic fatty liver disease via miR-130b-3p/ROCK1 axis. Cell. Signal. 2022, 90, 110190. [Google Scholar] [CrossRef]
- Ge, X.; Sun, T.; Zhang, Y.; Li, Y.; Gao, P.; Zhang, D.; Zhang, B.; Wang, P.; Ma, W.; Lu, S. The role and possible mechanism of the long noncoding RNA LINC01260 in nonalcoholic fatty liver disease. Nutr. Metab. 2022, 19, 3. [Google Scholar] [CrossRef]
- Zhang, H.; Niu, Q.; Liang, K.; Li, X.; Jiang, J.; Bian, C. Effect of LncPVT1/miR-20a-5p on Lipid Metabolism and Insulin Resistance in NAFLD. Diabetes Metab. Syndr. Obes. 2021, 14, 4599–4608. [Google Scholar] [CrossRef]
- Kneeman, J.M.; Misdraji, J.; Corey, K.E. Secondary causes of nonalcoholic fatty liver disease. Ther. Adv. Gastroenterol. 2012, 5, 199–207. [Google Scholar] [CrossRef] [Green Version]
- Pal, P.; Palui, R.; Ray, S. Heterogeneity of non-alcoholic fatty liver disease: Implications for clinical practice and research activity. World J. Hepatol. 2021, 13, 1584–1610. [Google Scholar] [CrossRef]
- Arun, J.; Jhala, N.; Lazenby, A.J.; Clements, R.; Abrams, G.A. Influence of liver biopsy heterogeneity and diagnosis of nonalcoholic steatohepatitis in subjects undergoing gastric bypass. Obes. Surg. 2007, 17, 155–161. [Google Scholar] [CrossRef]
- Shi, L.; Guo, S.; Zhang, S.; Gao, X.; Liu, A.; Wang, Q.; Zhang, T.; Zhang, Y.; Wen, A. Glycyrrhetinic acid attenuates disturbed vitamin a metabolism in non-alcoholic fatty liver disease through AKR1B10. Eur. J. Pharmacol. 2020, 883, 173167. [Google Scholar] [CrossRef]
- Kanno, M.; Kawaguchi, K.; Honda, M.; Horii, R.; Takatori, H.; Shimakami, T.; Kitamura, K.; Arai, K.; Yamashita, T.; Sakai, Y.; et al. Serum aldo-keto reductase family 1 member B10 predicts advanced liver fibrosis and fatal complications of nonalcoholic steatohepatitis. J. Gastroenterol. 2019, 54, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Sans, A.; Bonnafous, S.; Rousseau, D.; Patouraux, S.; Canivet, C.M.; Leclere, P.S.; Tran-Van-Nhieu, J.; Luci, C.; Bailly-Maitre, B.; Xu, X.; et al. The Differential Expression of Cide Family Members is Associated with Nafld Progression from Steatosis to Steatohepatitis. Sci. Rep. 2019, 9, 7501. [Google Scholar] [CrossRef] [PubMed]
- Sahini, N.; Borlak, J. Genomics of human fatty liver disease reveal mechanistically linked lipid droplet-associated gene regulations in bland steatosis and nonalcoholic steatohepatitis. Transl. Res. 2016, 177, 41–69. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, N.; Takahashi, S.; Matsubara, T.; Jiang, C.; Sakamoto, W.; Chanturiya, T.; Teng, R.; Gavrilova, O.; Gonzalez, F.J. Adipocyte-specific disruption of fat-specific protein 27 causes hepatosteatosis and insulin resistance in high-fat diet-fed mice. J. Biol. Chem. 2015, 290, 3092–3105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruce, K.D.; Dobrinskikh, E.; Wang, H.; Rudenko, I.; Gao, H.; Libby, A.E.; Gorkhali, S.; Yu, T.; Zsombok, A.; Eckel, R.H. Neuronal Lipoprotein Lipase Deficiency Alters Neuronal Function and Hepatic Metabolism. Metabolites 2020, 10, 385. [Google Scholar] [CrossRef]
- Pardina, E.; Baena-Fustegueras, J.A.; Llamas, R.; Catalan, R.; Galard, R.; Lecube, A.; Fort, J.M.; Llobera, M.; Allende, H.; Vargas, V.; et al. Lipoprotein lipase expression in livers of morbidly obese patients could be responsible for liver steatosis. Obes. Surg. 2009, 19, 608–616. [Google Scholar] [CrossRef]
- Abderrahmani, A.; Yengo, L.; Caiazzo, R.; Canouil, M.; Cauchi, S.; Raverdy, V.; Plaisance, V.; Pawlowski, V.; Lobbens, S.; Maillet, J.; et al. Increased Hepatic PDGF-AA Signaling Mediates Liver Insulin Resistance in Obesity-Associated Type 2 Diabetes. Diabetes 2018, 67, 1310–1321. [Google Scholar] [CrossRef] [Green Version]
- Hou, J.; Zhang, J.; Cui, P.; Zhou, Y.; Liu, C.; Wu, X.; Ji, Y.; Wang, S.; Cheng, B.; Ye, H.; et al. TREM2 sustains macrophage-hepatocyte metabolic coordination in nonalcoholic fatty liver disease and sepsis. J. Clin. Investig. 2021, 131, e135197. [Google Scholar] [CrossRef]
- Seidman, J.S.; Troutman, T.D.; Sakai, M.; Gola, A.; Spann, N.J.; Bennett, H.; Bruni, C.M.; Ouyang, Z.; Li, R.Z.; Sun, X.; et al. Niche-Specific Reprogramming of Epigenetic Landscapes Drives Myeloid Cell Diversity in Nonalcoholic Steatohepatitis. Immunity 2020, 52, 1057–1074.e7. [Google Scholar] [CrossRef]
- Xiong, X.; Kuang, H.; Ansari, S.; Liu, T.; Gong, J.; Wang, S.; Zhao, X.Y.; Ji, Y.; Li, C.; Guo, L.; et al. Landscape of Intercellular Crosstalk in Healthy and NASH Liver Revealed by Single-Cell Secretome Gene Analysis. Mol. Cell 2019, 75, 644–660.e5. [Google Scholar] [CrossRef]
- Maguire, M.; Bushkofsky, J.R.; Larsen, M.C.; Foong, Y.H.; Tanumihardjo, S.A.; Jefcoate, C.R. Diet-dependent retinoid effects on liver gene expression include stellate and inflammation markers and parallel effects of the nuclear repressor Shp. J. Nutr. Biochem. 2017, 47, 63–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Liu, J.; Xie, T.; Jiang, Q.; Ding, L.; Zhu, J.; Ye, Q. Oleate acid-stimulated HMMR expression by CEBPalpha is associated with nonalcoholic steatohepatitis and hepatocellular carcinoma. Int. J. Biol. Sci. 2020, 16, 2812–2827. [Google Scholar] [CrossRef] [PubMed]
- Cao, Q.; Dong, Z.; Liu, S.; An, G.; Yan, B.; Lei, L. Construction of a metastasis-associated ceRNA network reveals a prognostic signature in lung cancer. Cancer Cell Int. 2020, 20, 208. [Google Scholar] [CrossRef] [PubMed]
- Tahmouresi, F.; Razmara, E.; Pakravan, K.; Mossahebi-Mohammadi, M.; Rouhollah, F.; Montazeri, M.; Sarrafzadeh, A.; Fahimi, H.; Babashah, S. Upregulation of the long noncoding RNAs DSCAM-AS1 and MANCR is a potential diagnostic marker for breast carcinoma. Biotechnol. Appl. Biochem. 2020, 68, 1250–1256. [Google Scholar] [CrossRef]
- Tracy, K.M.; Tye, C.E.; Ghule, P.N.; Malaby, H.L.H.; Stumpff, J.; Stein, J.L.; Stein, G.S.; Lian, J.B. Mitotically-Associated lncRNA (MANCR) Affects Genomic Stability and Cell Division in Aggressive Breast Cancer. Mol. Cancer Res. 2018, 16, 587–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, L.; Yan, J.; Gan, L.; Huang, S.; Cheng, F.; Fang, N. Upregulation of MANCR predicts poor survival in patients with gastric cancer. Oncol. Lett. 2019, 18, 6801–6806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, N.S.; Lei, B.W.; Tan, L.C.; Yu, P.C.; Shi, X.; Wang, Y.; Ji, Q.H.; Wei, W.J.; Lu, Z.W.; Wang, Y.L. Mitotically associated long non-coding RNA is a tumor promoter in anaplastic thyroid cancer. Ann. Transl. Med. 2020, 8, 1226. [Google Scholar] [CrossRef]
- Zhang, X.; Li, H.; Mao, M.; Wang, X.; Zheng, J.; Yang, S. Mitotically-associated long non-coding RNA promotes cancer cell proliferation in hepatocellular carcinoma by downregulating miR-122a. Oncol. Lett. 2019, 18, 6237–6242. [Google Scholar] [CrossRef] [Green Version]
- Nagasawa, M.; Tomimatsu, K.; Terada, K.; Kondo, K.; Miyazaki, K.; Miyazaki, M.; Motooka, D.; Okuzaki, D.; Yoshida, T.; Kageyama, S.; et al. Long non-coding RNA MANCR is a target of BET bromodomain protein BRD4 and plays a critical role in cellular migration and invasion abilities of prostate cancer. Biochem. Biophys. Res. Commun. 2020, 526, 128–134. [Google Scholar] [CrossRef]
- Wen, S.; Zeng, M.; Li, Y.; Hu, X.; Li, S.; Liang, X.; Zhu, L.; Yang, S. Downregulation of MANCR inhibits cancer cell proliferation in mantle cell lymphoma possibly by interacting with RUNX2. Acta Biochim. Biophys. Sin. 2019, 51, 1142–1147. [Google Scholar] [CrossRef]
- Xia, Y.; Zhen, L.; Li, H.; Wang, S.; Chen, S.; Wang, C.; Yang, X. MIRLET7BHG promotes hepatocellular carcinoma progression by activating hepatic stellate cells through exosomal SMO to trigger Hedgehog pathway. Cell Death Dis. 2021, 12, 326. [Google Scholar] [CrossRef] [PubMed]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets—Update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stelzer, G.; Rosen, N.; Plaschkes, I.; Zimmerman, S.; Twik, M.; Fishilevich, S.; Stein, T.I.; Nudel, R.; Lieder, I.; Mazor, Y.; et al. The GeneCards Suite: From Gene Data Mining to Disease Genome Sequence Analyses. Curr. Protoc. Bioinform. 2016, 54, 1–30. [Google Scholar] [CrossRef]
- Di Tommaso, P.; Chatzou, M.; Floden, E.W.; Barja, P.P.; Palumbo, E.; Notredame, C. Nextflow enables reproducible computational workflows. Nat. Biotechnol. 2017, 35, 316–319. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Ning, B.; Shi, T. Single-Cell RNA-Seq Technologies and Related Computational Data Analysis. Front. Genet. 2019, 10, 317. [Google Scholar] [CrossRef] [PubMed]
- Lahnemann, D.; Koster, J.; Szczurek, E.; McCarthy, D.J.; Hicks, S.C.; Robinson, M.D.; Vallejos, C.A.; Campbell, K.R.; Beerenwinkel, N.; Mahfouz, A.; et al. Eleven grand challenges in single-cell data science. Genome Biol. 2020, 21, 31. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, M.; Huber, W.; Pages, H.; Aboyoun, P.; Carlson, M.; Gentleman, R.; Morgan, M.T.; Carey, V.J. Software for computing and annotating genomic ranges. PLoS Comput. Biol. 2013, 9, e1003118. [Google Scholar] [CrossRef]
- Xie, Z.; Bailey, A.; Kuleshov, M.V.; Clarke, D.J.B.; Evangelista, J.E.; Jenkins, S.L.; Lachmann, A.; Wojciechowicz, M.L.; Kropiwnicki, E.; Jagodnik, K.M.; et al. Gene Set Knowledge Discovery with Enrichr. Curr. Protoc. 2021, 1, e90. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis. In Use R! 2nd ed.; Springer International Publishing: Cham, Switzerland, 2016. [Google Scholar] [CrossRef]
- Howe, E.A.; Sinha, R.; Schlauch, D.; Quackenbush, J. RNA-Seq analysis in MeV. Bioinformatics 2011, 27, 3209–3210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Chang, W.; Cheng, J.; Allaire, J.; Xie, Y.; McPherson, J. Shiny: Web application framework for R. In R Package Version; R Foundation for Statistical Computing: Vienna, Austria, 2017; Volume 1. [Google Scholar]
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef] [Green Version]
- Durinck, S.; Spellman, P.T.; Birney, E.; Huber, W. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat. Protoc. 2009, 4, 1184–1191. [Google Scholar] [CrossRef] [Green Version]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3—New capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ilieva, M.; Dao, J.; Miller, H.E.; Madsen, J.H.; Bishop, A.J.R.; Kauppinen, S.; Uchida, S. Systematic Analysis of Long Non-Coding RNA Genes in Nonalcoholic Fatty Liver Disease. Non-Coding RNA 2022, 8, 56. https://doi.org/10.3390/ncrna8040056
Ilieva M, Dao J, Miller HE, Madsen JH, Bishop AJR, Kauppinen S, Uchida S. Systematic Analysis of Long Non-Coding RNA Genes in Nonalcoholic Fatty Liver Disease. Non-Coding RNA. 2022; 8(4):56. https://doi.org/10.3390/ncrna8040056
Chicago/Turabian StyleIlieva, Mirolyuba, James Dao, Henry E. Miller, Jens Hedelund Madsen, Alexander J. R. Bishop, Sakari Kauppinen, and Shizuka Uchida. 2022. "Systematic Analysis of Long Non-Coding RNA Genes in Nonalcoholic Fatty Liver Disease" Non-Coding RNA 8, no. 4: 56. https://doi.org/10.3390/ncrna8040056
APA StyleIlieva, M., Dao, J., Miller, H. E., Madsen, J. H., Bishop, A. J. R., Kauppinen, S., & Uchida, S. (2022). Systematic Analysis of Long Non-Coding RNA Genes in Nonalcoholic Fatty Liver Disease. Non-Coding RNA, 8(4), 56. https://doi.org/10.3390/ncrna8040056