New Insights into H2S Adsorption on Graphene and Graphene-Like Structures: A Comparative DFT Study

 ,
,  ,
,  ,
,  ,
,  and
and 
Abstract
:
1. Introduction
2. Computational Method
3. Results and Discussion
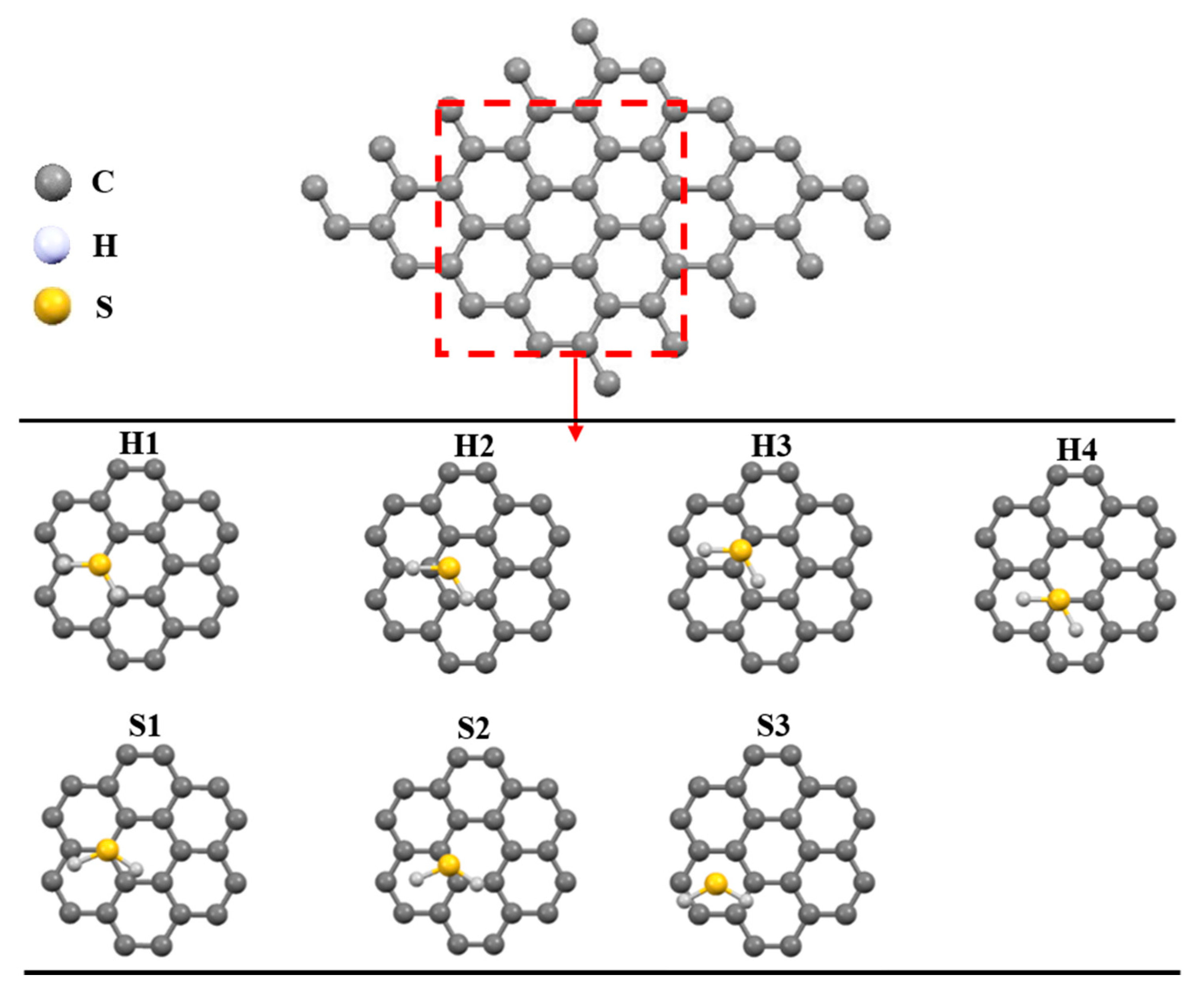
3.1. Geometrical Design and Optimization
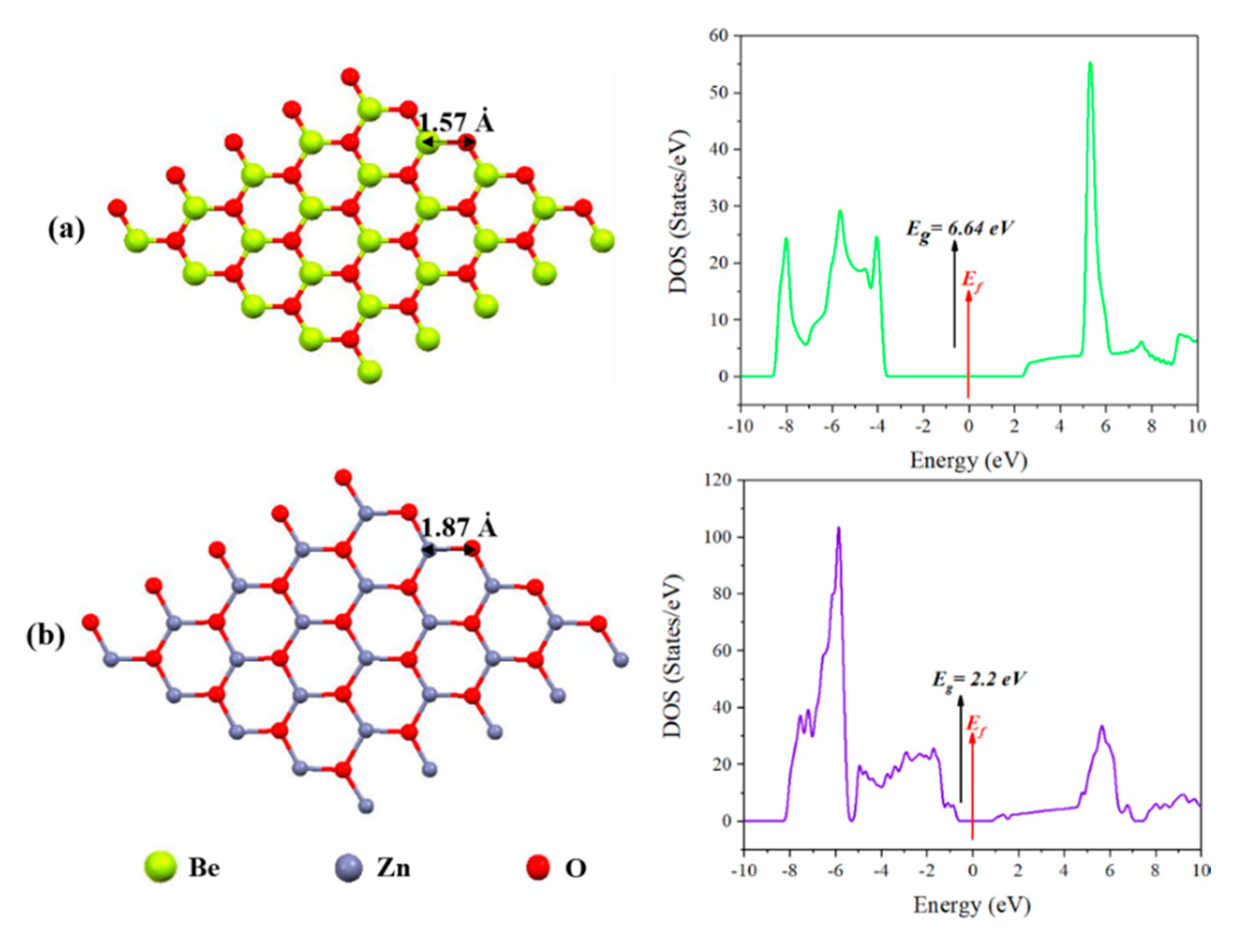
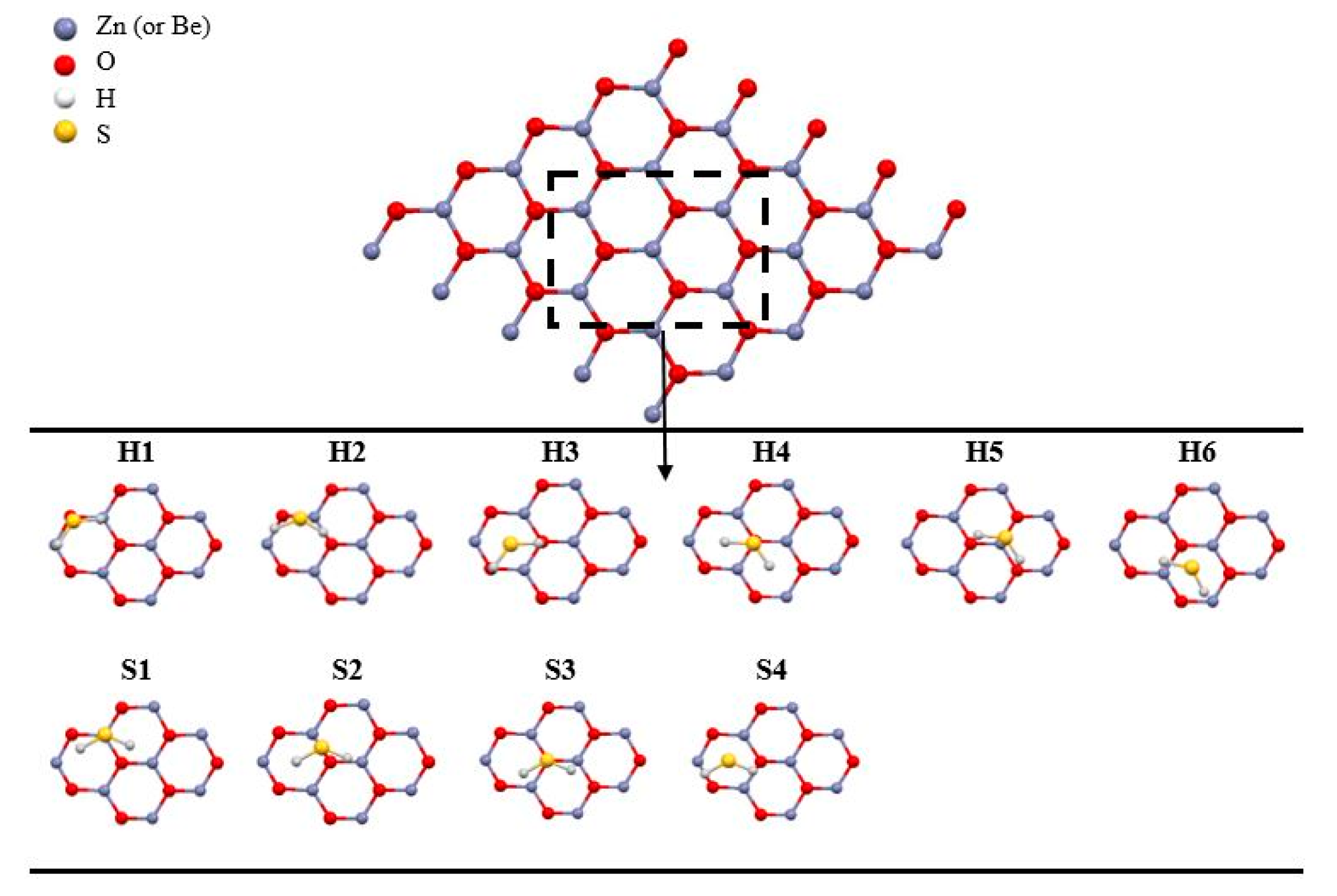
3.2. Adsorption of H2S on Graphene-Like Metal Oxide Surfaces
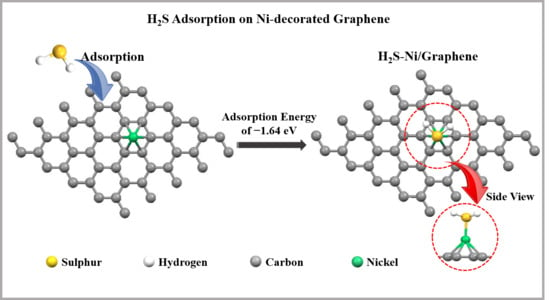
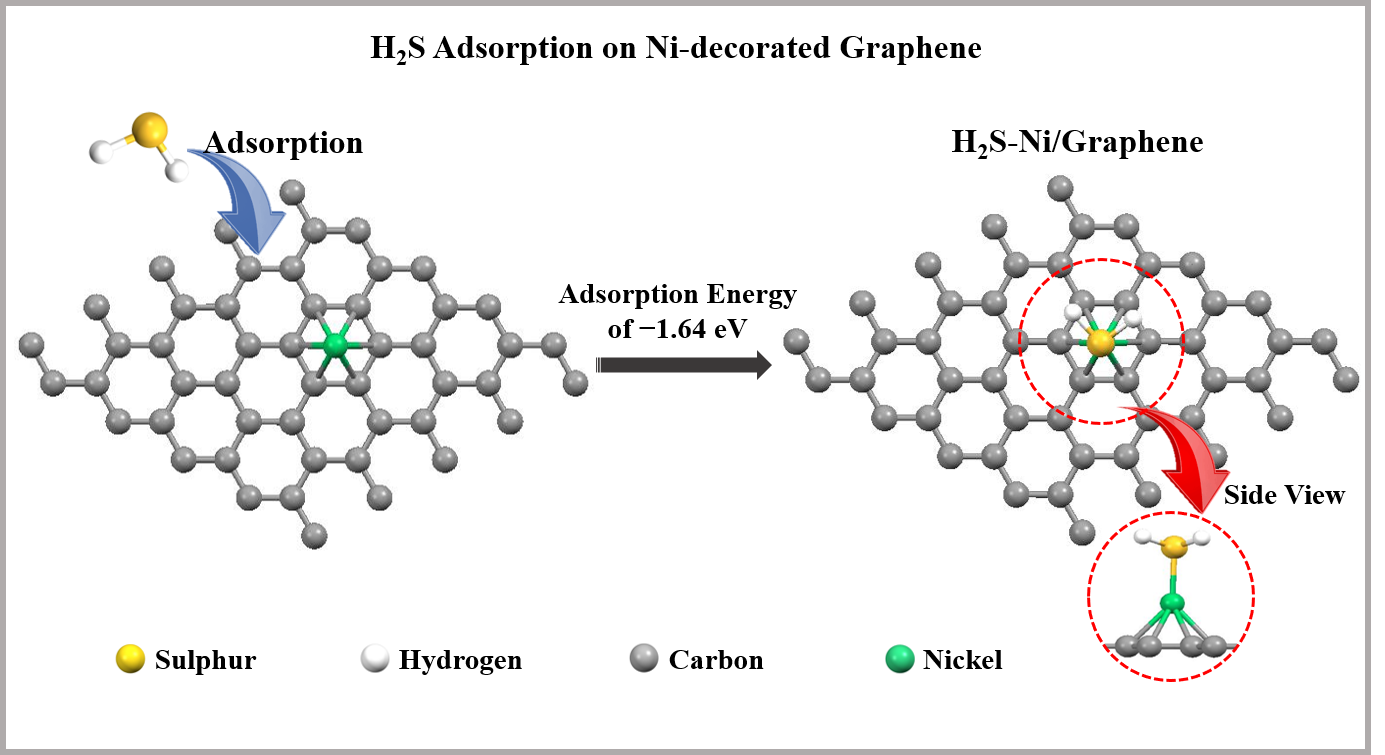
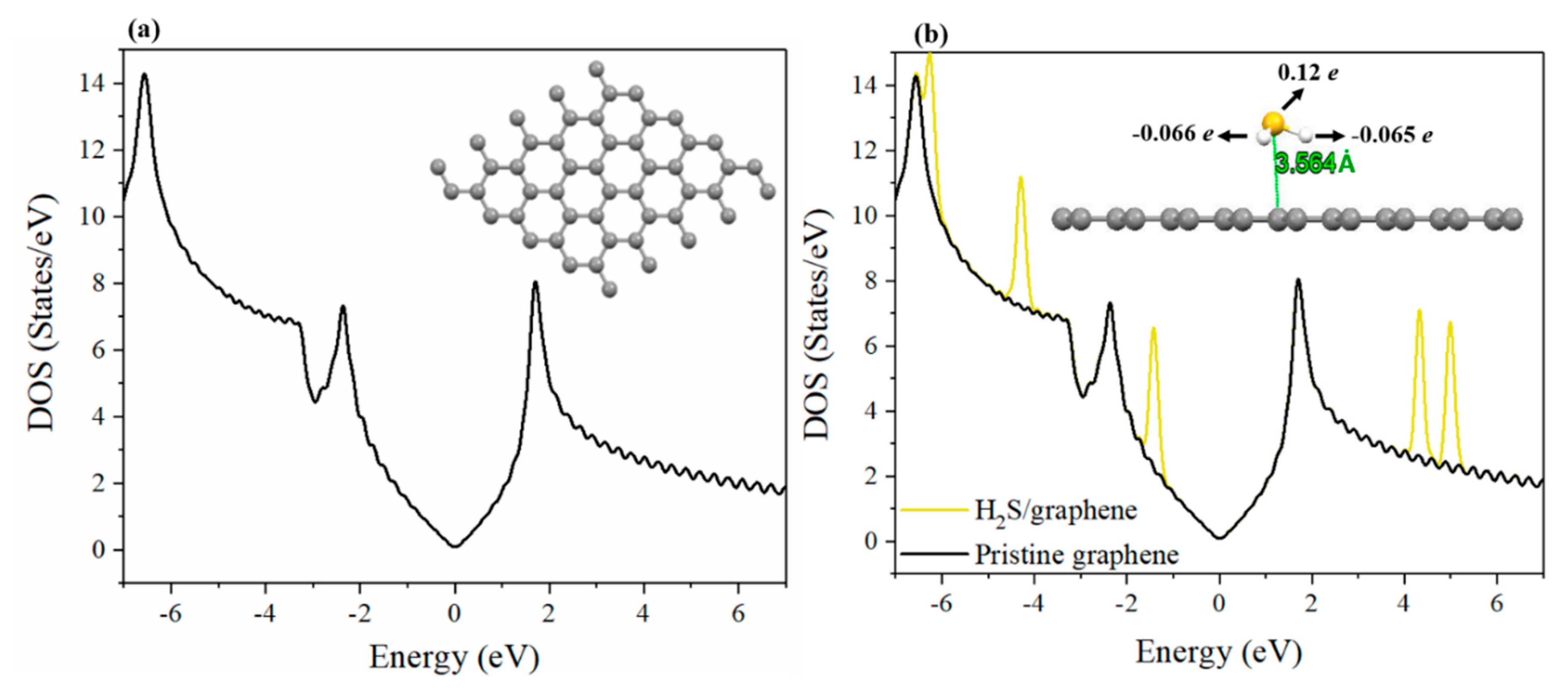
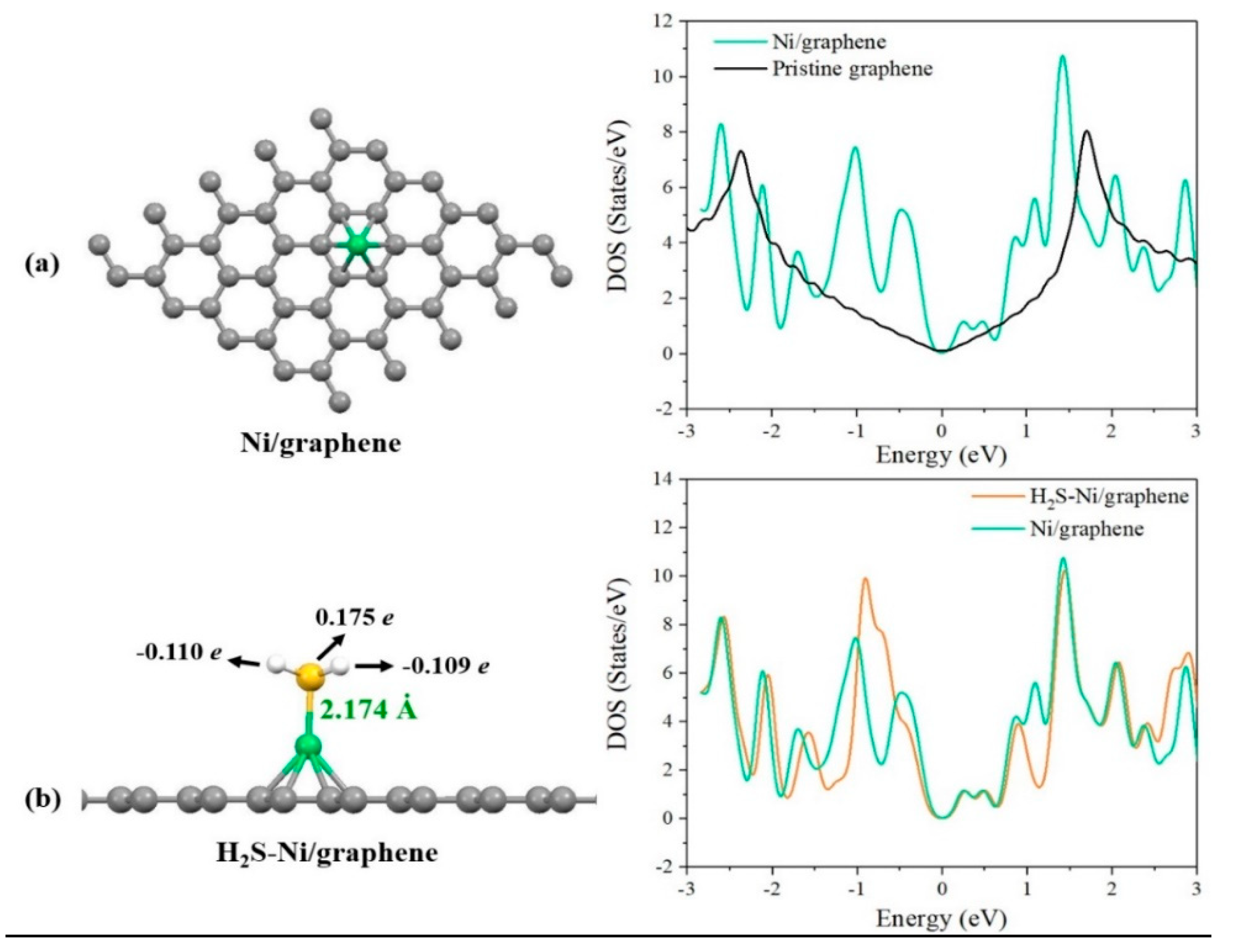
3.3. Adsorption of H2S on Pristine and Ni-Decorated Graphene Nanosheets
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Lins, V.F.C.; Guimarães, E.M. Failure of a heat exchanger generated by an excess of SO2 and H2S in the Sulfur Recovery Unit of a petroleum refinery. J. Loss Prev. Process Ind. 2007, 20, 91–97. [Google Scholar] [CrossRef]
- Mochizuki, Y.; Tsubouchi, N. Removal of Hydrogen Sulfide in Simulated Coke Oven Gas with Low-Grade Iron Ore. Energy Fuels 2017, 31, 8087–8094. [Google Scholar] [CrossRef]
- Bak, C.-U.; Lim, C.-J.; Lee, J.-G.; Kim, Y.-D.; Kim, W.-S. Removal of sulfur compounds and siloxanes by physical and chemical sorption. Sep. Purif. Technol. 2019, 209, 542–549. [Google Scholar] [CrossRef]
- Jiang, X.; Wu, J.; Jin, Z.; Yang, S.; Shen, L. Enhancing the removal of H2S from biogas through refluxing of outlet gas in biological bubble-column. Bioresour. Technol. 2020, 299, 122621. [Google Scholar] [CrossRef]
- Barbera, N.; Montana, A.; Indorato, F.; Arbouche, N.; Romano, G. Evaluation of the Role of Toxicological Data in Discriminating Between H2S Femoral Blood Concentration Secondary to Lethal poisoning and Endogenous H2S Putrefactive Production. J. Forensic Sci. 2017, 62, 392–394. [Google Scholar] [CrossRef]
- Shefa, U.; Kim, M.-S.; Jeong, N.Y.; Jung, J. Antioxidant and Cell-Signaling Functions of Hydrogen Sulfide in the Central Nervous System. Oxidative Med. Cell. Longev. 2018, 2018, 17. [Google Scholar] [CrossRef] [PubMed]
- Jia, X.; Zhang, H.; Zhang, Z.; An, L. First-principles investigation of vacancy-defected graphene and Mn-doped graphene towards adsorption of H2S. Superlattices Microstruct. 2019, 134, 106235. [Google Scholar] [CrossRef]
- Sudalma, S.; Purwanto, P.; Santoso, L.W. The Effect of SO2 and NO2 from Transportation and Stationary Emissions Sources to SO42− and NO3− in Rain Water in Semarang. Procedia Environ. Sci. 2015, 23, 247–252. [Google Scholar] [CrossRef] [Green Version]
- Santiago, R.; Lemus, J.; Outomuro, A.X.; Bedia, J.; Palomar, J. Assessment of ionic liquids as H2S physical absorbents by thermodynamic and kinetic analysis based on process simulation. Sep. Purif. Technol. 2020, 233, 116050. [Google Scholar] [CrossRef]
- Atlaskin, A.A.; Kryuchkov, S.S.; Yanbikov, N.R.; Smorodin, K.A.; Petukhov, A.N.; Trubyanov, M.M.; Vorotyntsev, V.M.; Vorotyntsev, I.V. Comprehensive experimental study of acid gases removal process by membrane-assisted gas absorption using imidazolium ionic liquids solutions absorbent. Sep. Purif. Technol. 2020, 239, 116578. [Google Scholar] [CrossRef]
- Ge, K.; Wu, Y.; Wang, T.; Wu, J. Humidity swing adsorption of H2S by fibrous polymeric ionic liquids (PILs). Sep. Purif. Technol. 2019, 217, 1–7. [Google Scholar] [CrossRef]
- Zhang, H.-P.; Luo, X.-G.; Song, H.-T.; Lin, X.-Y.; Lu, X.; Tang, Y. DFT study of adsorption and dissociation behavior of H2S on Fe-doped graphene. Appl. Surf. Sci. 2014, 317, 511–516. [Google Scholar] [CrossRef]
- Alfonso, D.R. First-principles studies of H2S adsorption and dissociation on metal surfaces. Surf. Sci. 2008, 602, 2758–2768. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Fu, Z.; Yu, Y. Adsorption and decomposition of H2S on the Ni(111) and Ni(211) surfaces: A first-principles density functional study. Appl. Surf. Sci. 2019, 473, 657–667. [Google Scholar] [CrossRef]
- Sigot, L.; Ducom, G.; Germain, P. Adsorption of hydrogen sulfide (H2S) on zeolite (Z): Retention mechanism. Chem. Eng. J. 2016, 287, 47–53. [Google Scholar] [CrossRef]
- Berhe Gebreegziabher, T.; Wang, S.; Nam, H. Adsorption of H2S, NH3 and TMA from indoor air using porous corncob activated carbon: Isotherm and kinetics study. J. Environ. Chem. Eng. 2019, 7, 103234. [Google Scholar] [CrossRef]
- Sitthikhankaew, R.; Predapitakkun, S.; Kiattikomol, R.; Pumhiran, S.; Assabumrungrat, S.; Laosiripojana, N. Comparative Study of Hydrogen Sulfide Adsorption by using Alkaline Impregnated Activated Carbons for Hot Fuel Gas Purification. Energy Procedia 2011, 9, 15–24. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Cui, H.; Chen, D.; Dong, X.; Tang, J. Electronic structure and H2S adsorption property of Pt3 cluster decorated (8, 0) SWCNT. Appl. Surf. Sci. 2018, 428, 82–88. [Google Scholar] [CrossRef]
- Srivastava, R.; Suman, H.; Shrivastava, S.; Srivastava, A. DFT analysis of pristine and functionalized zigzag CNT: A case of H2S sensing. Chem. Phys. Lett. 2019, 731, 136575. [Google Scholar] [CrossRef]
- Yousefian, Z.; Ghasemy, E.; Askarieh, M.; Rashidi, A. Theoretical studies on B, N, P, S, and Si doped fullerenes toward H2S sensing and adsorption. Phys. E Low-Dimens. Syst. Nanostruct. 2019, 114, 113626. [Google Scholar] [CrossRef]
- Faye, O.; Raj, A.; Mittal, V.; Beye, A.C. H2S adsorption on graphene in the presence of sulfur: A density functional theory study. Comput. Mater. Sci. 2016, 117, 110–119. [Google Scholar] [CrossRef]
- Rodriguez, J.A.; Maiti, A. Adsorption and Decomposition of H2S on MgO(100), NiMgO(100), and ZnO(0001) Surfaces: A First-Principles Density Functional Study. J. Phys. Chem. B 2000, 104, 3630–3638. [Google Scholar] [CrossRef]
- Ranea, V.A.; Dammig Quiña, P.L.; Yalet, N.M. General adsorption model for H2S, H2Se, H2Te, NH3, PH3, AsH3 and SbH3 on the V2O5(001) surface including the van der Waals interaction. Chem. Phys. Lett. 2019, 720, 58–63. [Google Scholar] [CrossRef]
- Bo, Z.; Guo, X.; Wei, X.; Yang, H.; Yan, J.; Cen, K. Density functional theory calculations of NO2 and H2S adsorption on the group 10 transition metal (Ni, Pd and Pt) decorated graphene. Phys. E Low-Dimens. Syst. Nanostruct. 2019, 109, 156–163. [Google Scholar] [CrossRef]
- Faye, O.; Eduok, U.; Szpunar, J.; Samoura, A.; Beye, A. H2S adsorption and dissociation on NH-decorated graphene: A first principles study. Surf. Sci. 2018, 668, 100–106. [Google Scholar] [CrossRef]
- Ganji, M.D.; Sharifi, N.; Ardjmand, M.; Ahangari, M.G. Pt-decorated graphene as superior media for H2S adsorption: A first-principles study. Appl. Surf. Sci. 2012, 261, 697–704. [Google Scholar] [CrossRef]
- Cortés-Arriagada, D.; Villegas-Escobar, N.; Ortega, D.E. Fe-doped graphene nanosheet as an adsorption platform of harmful gas molecules (CO, CO2, SO2 and H2S), and the co-adsorption in O2 environments. Appl. Surf. Sci. 2018, 427, 227–236. [Google Scholar] [CrossRef]
- Khodadadi, Z. Evaluation of H2S sensing characteristics of metals–doped graphene and metals-decorated graphene: Insights from DFT study. Phys. E Low-Dimens. Syst. Nanostruct. 2018, 99, 261–268. [Google Scholar] [CrossRef]
- Zhu, Y.; Murali, S.; Cai, W.; Li, X.; Suk, J.W.; Potts, J.R.; Ruoff, R.S. Graphene and Graphene Oxide: Synthesis, Properties, and Applications. Adv. Mater. 2010, 22, 3906–3924. [Google Scholar] [CrossRef]
- Castro Neto, A.H.; Guinea, F.; Peres, N.M.R.; Novoselov, K.S.; Geim, A.K. The electronic properties of graphene. Rev. Mod. Phys. 2009, 81, 109–162. [Google Scholar] [CrossRef] [Green Version]
- Lin, X.; Ni, J.; Fang, C. Adsorption capacity of H2O, NH3, CO, and NO2 on the pristine graphene. J. Appl. Phys. 2013, 113, 034306. [Google Scholar] [CrossRef]
- Gui, Y.; Hao, Z.; Li, X.; Tang, C.; Xu, L. Gas sensing of graphene and graphene oxide nanoplatelets to ClO2 and its decomposed species. Superlattices Microstruct. 2019, 135, 106248. [Google Scholar] [CrossRef]
- Shao, L.; Chen, G.; Ye, H.; Wu, Y.; Qiao, Z.; Zhu, Y.; Niu, H. Sulfur dioxide adsorbed on graphene and heteroatom-doped graphene: A first-principles study. Eur. Phys. J. B 2013, 86, 54. [Google Scholar] [CrossRef]
- Ovsianytskyi, O.; Nam, Y.-S.; Tsymbalenko, O.; Lan, P.-T.; Moon, M.-W.; Lee, K.-B. Highly sensitive chemiresistive H2S gas sensor based on graphene decorated with Ag nanoparticles and charged impurities. Sens. Actuators B Chem. 2018, 257, 278–285. [Google Scholar] [CrossRef]
- Song, Y.; Fang, W.; Hsu, A.L.; Kong, J. Iron (III) Chloride doping of CVD graphene. Nanotechnology 2014, 25, 395701. [Google Scholar] [CrossRef] [PubMed]
- Gaboardi, M.; Bliersbach, A.; Bertoni, G.; Aramini, M.; Vlahopoulou, G.; Pontiroli, D.; Mauron, P.; Magnani, G.; Salviati, G.; Züttel, A.; et al. Decoration of graphene with nickel nanoparticles: Study of the interaction with hydrogen. J. Mater. Chem. A 2014, 2, 1039–1046. [Google Scholar] [CrossRef]
- Hamed Mashhadzadeh, A.; Fathalian, M.; Ghorbanzadeh Ahangari, M.; Shahavi, M.H. DFT study of Ni, Cu, Cd and Ag heavy metal atom adsorption onto the surface of the zinc-oxide nanotube and zinc-oxide graphene-like structure. Mater. Chem. Phys. 2018, 220, 366–373. [Google Scholar] [CrossRef]
- Mo, Y.; Li, H.; Zhou, K.; Ma, X.; Guo, Y.; Wang, S.; Li, L. Acetone adsorption to (BeO)12, (MgO)12 and (ZnO)12 nanoparticles and their graphene composites: A density functional theory (DFT) study. Appl. Surf. Sci. 2019, 469, 962–973. [Google Scholar] [CrossRef]
- Gui, Y.; Liu, D.; Li, X.; Tang, C.; Zhou, Q. DFT-based study on H2S and SOF2 adsorption on Si-MoS2 monolayer. Results Phys. 2019, 13, 102225. [Google Scholar] [CrossRef]
- Ganji, M.; Sharifi, N.; Ahangari, M.G. Adsorption of H2S molecules on non-carbonic and decorated carbonic graphenes: A van der Waals density functional study. Comput. Mater. Sci. 2014, 92, 127–134. [Google Scholar] [CrossRef]
- Zhang, Y.-H.; Yue, L.-J.; Gong, F.-L.; Li, F.; Zhang, H.-L.; Chen, J.-L. Highly enhanced H2S gas sensing and magnetic performances of metal doped hexagonal ZnO monolayer. Vacuum 2017, 141, 109–115. [Google Scholar] [CrossRef]
- Ordejón, P.; Artacho, E.; Soler, J.M. Self-consistent order-$N$ density-functional calculations for very large systems. Phys. Rev. B 1996, 53, R10441–R10444. [Google Scholar] [CrossRef] [Green Version]
- Soler, J.M.; Artacho, E.; Gale, J.D.; García, A.; Junquera, J.; Ordejón, P.; Sánchez-Portal, D. The SIESTA method for ab initio order-N materials simulation. J. Phys. Condens. Matter 2002, 14, 2745. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; EA Kelly, R.; Otero, R.; Schöck, M.; Lægsgaard, E.; Stensgaard, I.; Kantorovich, L.N.; Besenbacher, F. Probing the Hierarchy of Thymine–Thymine Interactions in Self-Assembled Structures by Manipulation with Scanning Tunneling Microscopy. Small 2007, 3, 2011–2014. [Google Scholar] [CrossRef]
- Junquera, J.; Paz, Ó.; Sánchez-Portal, D.; Artacho, E. Numerical atomic orbitals for linear-scaling calculations. Phys. Rev. B 2001, 64, 235111. [Google Scholar] [CrossRef] [Green Version]
- Balabin, R.M. Enthalpy difference between conformations of normal alkanes: Intramolecular basis set superposition error (BSSE) in the case of n-butane and n-hexane. J. Chem. Phys. 2008, 129, 164101. [Google Scholar] [CrossRef]
- Ghorbanzadeh Ahangari, M.; Fereidoon, A.; Hamed Mashhadzadeh, A. Interlayer interaction and mechanical properties in multi-layer graphene, Boron-Nitride, Aluminum-Nitride and Gallium-Nitride graphene-like structure: A quantum-mechanical DFT study. Superlattices Microstruct. 2017. [Google Scholar] [CrossRef]
- Ansari, R.; Mirnezhad, M.; Rouhi, H. A first principles study on the mechanical properties of hexagonal zinc oxide sheets. Superlattices Microstruct. 2015, 79, 15–20. [Google Scholar] [CrossRef]
- Marana, N.L.; Casassa, S.; Longo, E.; Sambrano, J.R. Structural, Electronic, Vibrational, and Topological Analysis of Single-Walled Zinc Oxide Nanotubes. J. Phys. Chem. C 2016, 120, 6814–6823. [Google Scholar] [CrossRef] [Green Version]
- Hamed Mashhadzadeh, A.; Ghorbanzadeh Ahangari, M.; Dadrasi, A.; Fathalian, M. Theoretical studies on the mechanical and electronic properties of 2D and 3D structures of Beryllium-Oxide graphene and graphene nanobud. Appl. Surf. Sci. 2019, 476, 36–48. [Google Scholar] [CrossRef]
- Rastegar, S.F.; Ahmadi Peyghan, A.; Soleymanabadi, H. Ab initio studies of the interaction of formaldehyde with beryllium oxide nanotube. Phys. E Low-Dimens. Syst. Nanostruct. 2015, 68, 22–27. [Google Scholar] [CrossRef]
- Ahangari, M.G.; Mashhadzadeh, A.H.; Fathalian, M.; Dadrasi, A.; Rostamiyan, Y.; Mallahi, A. Effect of various defects on mechanical and electronic properties of zinc-oxide graphene-like structure: A DFT study. Vacuum 2019, 165, 26–34. [Google Scholar] [CrossRef]
- Rostamiyan, Y.; Mohammadi, V.; Hamed Mashhadzadeh, A. Mechanical, electronic and stability properties of multi-walled beryllium oxide nanotubes and nanopeapods: A density functional theory study. J. Mol. Modeling 2020, 26, 76. [Google Scholar] [CrossRef]
- Pawar, V.; Jha, P.K.; Panda, S.K.; Jha, P.A.; Singh, P. Band-Gap Engineering in ZnO Thin Films: A Combined Experimental and Theoretical Study. Phys. Rev. Appl. 2018, 9, 054001. [Google Scholar] [CrossRef]
- Arif, A.; Belahssen, O.; Gareh, S.; Benramache, S. The calculation of band gap energy in zinc oxide films. J. Semicond. 2015, 36, 013001. [Google Scholar] [CrossRef]
- Baumeier, B.; Krüger, P.; Pollmann, J. Structural, elastic, and electronic properties of SiC, BN, and BeO nanotubes. Phys. Rev. B 2007, 76, 085407. [Google Scholar] [CrossRef]
- Wu, W.; Lu, P.; Zhang, Z.; Guo, W. Electronic and Magnetic Properties and Structural Stability of BeO Sheet and Nanoribbons. ACS Appl. Mater. Interfaces 2011, 3, 4787–4795. [Google Scholar] [CrossRef]
- Derakhshandeh, M.; Anaraki-Ardakani, H. A computational study on the experimentally observed sensitivity of Ga-doped ZnO nanocluster toward CO gas. Phys. E Low-Dimens. Syst. Nanostruct. 2016, 84, 298–302. [Google Scholar] [CrossRef]
- Hamed Mashhadzadeh, A.; Fereidoon, A.; Ghorbanzadeh Ahangari, M. Combining density functional theory-finite element multi-scale method to predict mechanical properties of polypropylene/graphene nanocomposites: Experimental study. Mater. Chem. Phys. 2017, 201, 214–223. [Google Scholar] [CrossRef]
- Hamed Mashhadzadeh, A.; Ghorbanzadeh Ahangari, M.; Salmankhani, A.; Fataliyan, M. Density functional theory study of adsorption properties of non-carbon, carbon and functionalized graphene surfaces towards the zinc and lead atoms. Phys. E Low-Dimens. Syst. Nanostruct. 2018, 104, 275–285. [Google Scholar] [CrossRef]
- Ghorbanzadeh Ahangari, M.; Salmankhani, A.; Imani, A.H.; Shahab, N.; Hamed Mashhadzadeh, A. Density Functional Theory Study on the Mechanical Properties and Interlayer Interactions of Multi-layer Graphene: Carbonic, Silicon-Carbide and Silicene Graphene-like Structures. Silicon 2019, 11, 1235–1246. [Google Scholar] [CrossRef]
- Ergun, S. Structure of Graphite. Nat. Phys. Sci. 1973, 241, 65–67. [Google Scholar] [CrossRef]
- Mashhadzadeh, A.H.; Vahedi, A.M.; Ardjmand, M.; Ahangari, M.G. Investigation of heavy metal atoms adsorption onto graphene and graphdiyne surface: A density functional theory study. Superlattices Microstruct. 2016, 100, 1094–1102. [Google Scholar] [CrossRef]
- Sharifi, N.; Falamaki, C.; Ahangari, M.G. DFT study of Au adsorption on pure and Pt-decorated γ-alumina (110) surface. Appl. Surf. Sci. 2017, 416, 390–396. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
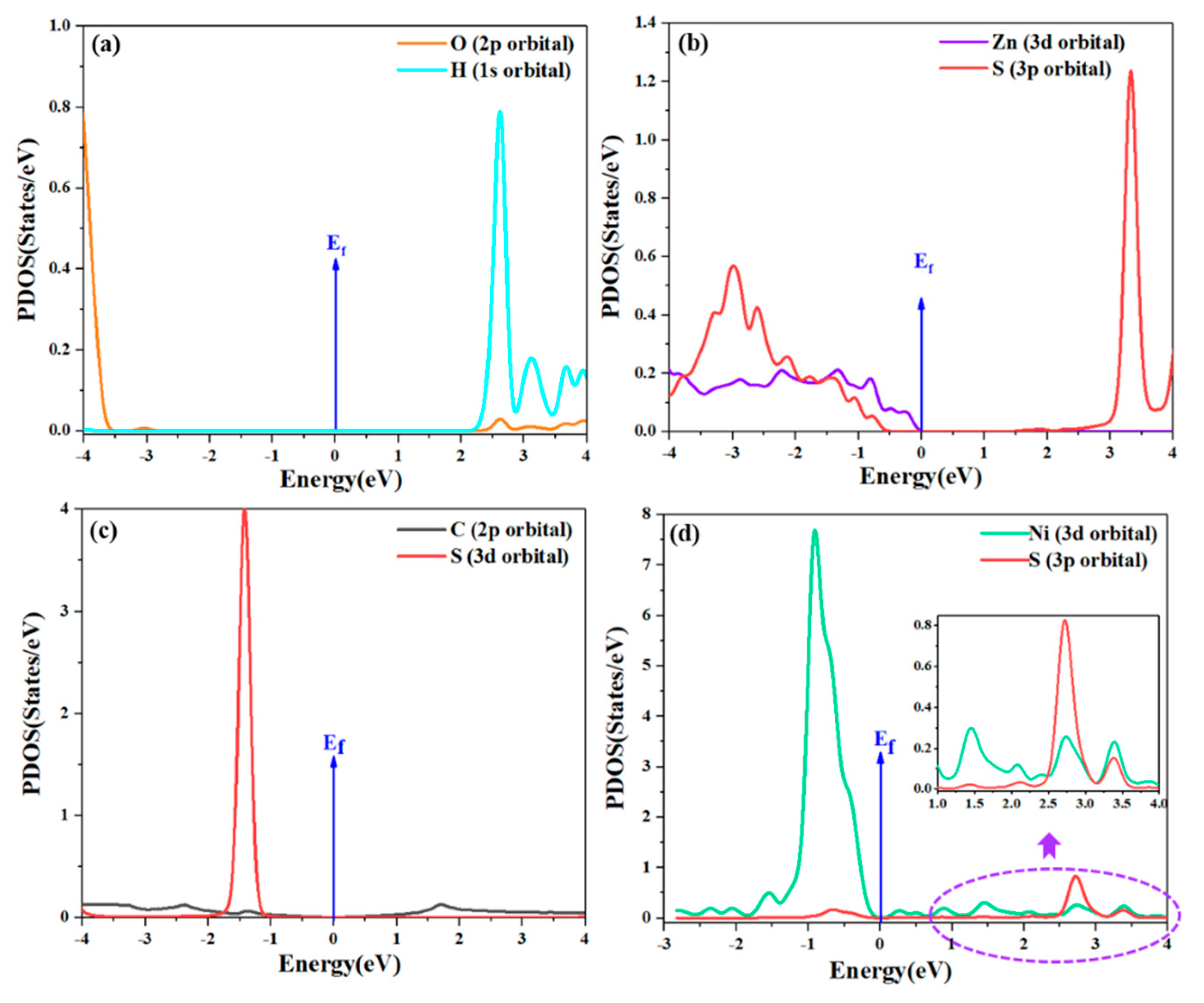{kind=link}
| Adsorption Site | Adsorption Energy (eV) | |
|---|---|---|
| on BeO Nanosheet | on ZnO Nanosheet | |
| H1 | −0.096 | −0.376 |
| H2 | −0.144 | 0.011 |
| H3 | −0.114 | −0.199 |
| H4 | −0.101 | −0.201 |
| H5 | −0.110 | −0.239 |
| H6 | −0.134 | −0.236 |
| S1 | −0.127 | −0.217 |
| S2 | −0.141 | −0.041 |
| S3 | −0.124 | −0.192 |
| S4 | −0.112 | −0.359 |
| Adsorption Site | Adsorption Energy (eV) |
|---|---|
| H1 | −0.113 |
| H2 | −0.112 |
| H3 | −0.118 |
| H4 | −0.119 |
| S1 | −0.117 |
| S2 | −0.114 |
| S3 | −0.116 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salmankhani, A.; Karami, Z.; Mashhadzadeh, A.H.; Ganjali, M.R.; Vatanpour, V.; Esmaeili, A.; Habibzadeh, S.; Saeb, M.R.; Fierro, V.; Celzard, A. New Insights into H2S Adsorption on Graphene and Graphene-Like Structures: A Comparative DFT Study. C 2020, 6, 74. https://doi.org/10.3390/c6040074
Salmankhani A, Karami Z, Mashhadzadeh AH, Ganjali MR, Vatanpour V, Esmaeili A, Habibzadeh S, Saeb MR, Fierro V, Celzard A. New Insights into H2S Adsorption on Graphene and Graphene-Like Structures: A Comparative DFT Study. C. 2020; 6(4):74. https://doi.org/10.3390/c6040074
Chicago/Turabian StyleSalmankhani, Azam, Zohre Karami, Amin Hamed Mashhadzadeh, Mohammad Reza Ganjali, Vahid Vatanpour, Amin Esmaeili, Sajjad Habibzadeh, Mohammad Reza Saeb, Vanessa Fierro, and Alain Celzard. 2020. "New Insights into H2S Adsorption on Graphene and Graphene-Like Structures: A Comparative DFT Study" C 6, no. 4: 74. https://doi.org/10.3390/c6040074
APA StyleSalmankhani, A., Karami, Z., Mashhadzadeh, A. H., Ganjali, M. R., Vatanpour, V., Esmaeili, A., Habibzadeh, S., Saeb, M. R., Fierro, V., & Celzard, A. (2020). New Insights into H2S Adsorption on Graphene and Graphene-Like Structures: A Comparative DFT Study. C, 6(4), 74. https://doi.org/10.3390/c6040074