

Illumina Sequencing and Metabolomic Analysis Explored the Effects of the Mixed Silage of Rice Straw and Chinese Cabbage Waste on Fecal Microorganisms and Metabolites in Hu Sheep


 , , and
, , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Diet, Experimental Animals, and Feeding
2.2. Sample Collection and Measurement
2.2.1. Growth Performance
2.2.2. Fecal Sample Collection
2.2.3. 16S rRNA Microbial Community Analysis
2.2.4. Untargeted Metabolomics Based on Liquid Chromatography-Mass Spectrometry and Data Processing
2.3. Data Analysis
3. Results
3.1. Growth Performance
3.2. Fecal Microbiota Structure
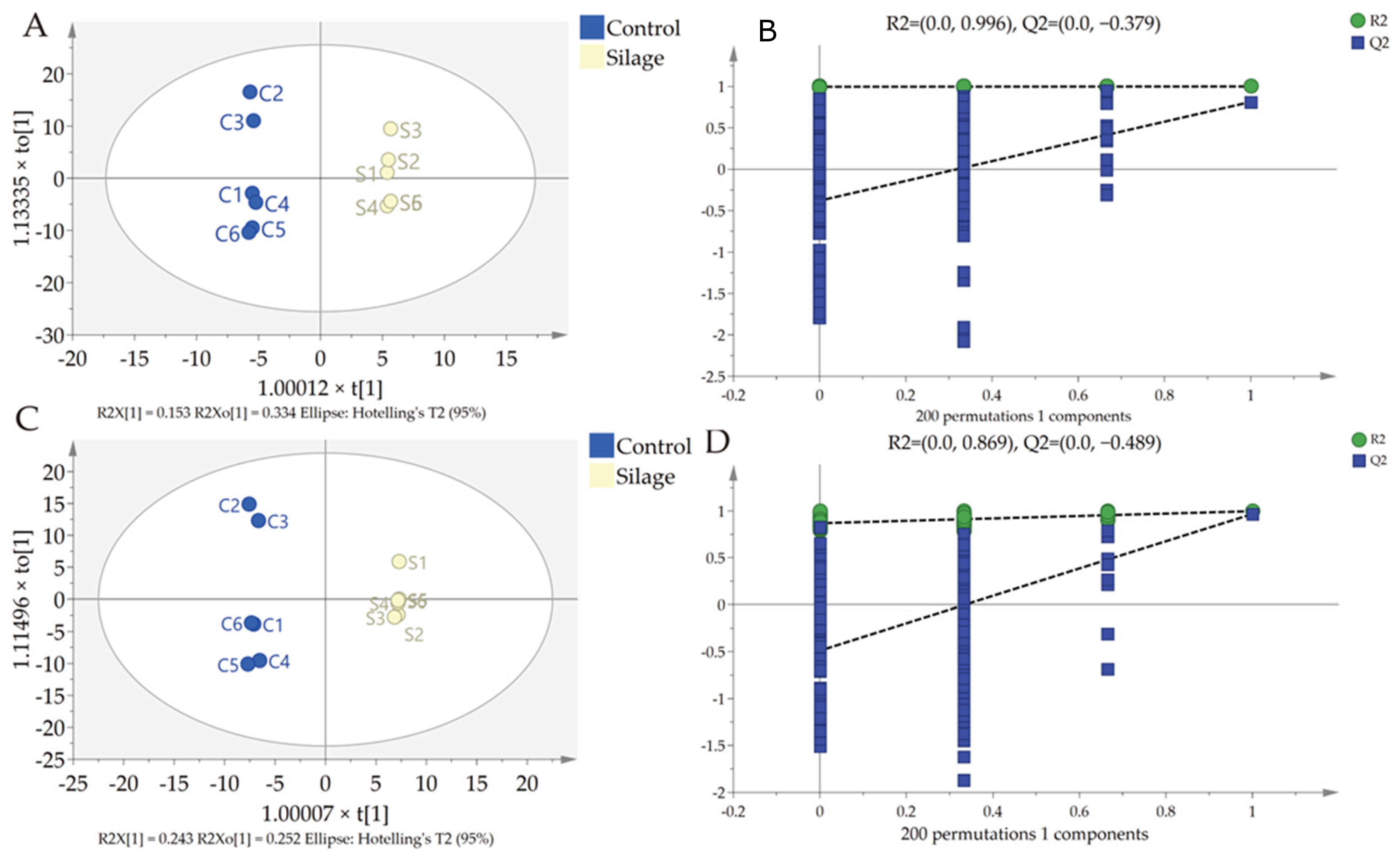
3.3. Fecal Metabolomics
3.4. KEGG Enrichment Analysis
3.5. Analysis of Differential Metabolites and Fecal Microbial Correlations
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alexandratos, N.; Bruinsma, J. World Agriculture towards 2030/2050: The 2012 Revision; Food and Agriculture Organization: Rome, Italy, 2012. [Google Scholar]
- Wang, C.; Zheng, M.; Wu, S.; Zou, X.; Chen, X.; Ge, L.; Zhang, Q. Effects of gallic acid on fermentation parameters, protein fraction, and bacterial community of whole plant soybean silage. Front. Microbiol. 2021, 12, 662966. [Google Scholar] [CrossRef]
- Wilkinson, J.; Rinne, M. Highlights of progress in silage conservation and future perspectives. Grass Forage Sci. 2018, 73, 40–52. [Google Scholar] [CrossRef]
- Wanapat, M.; Sundstøl, F.; Garmo, T. A comparison of alkali treatment methods to improve the nutritive value of straw. I. Digestibility and metabolizability. Anim. Feed Sci. Technol. 1985, 12, 295–309. [Google Scholar] [CrossRef]
- Weinberg, Z.; Chen, Y. Effects of storage period on the composition of whole crop wheat and corn silages. Anim. Feed Sci. Technol. 2013, 185, 196–200. [Google Scholar] [CrossRef]
- Ren, H.; Xu, N.; Li, J.; Li, Z.; Zhao, T.; Pei, F.; Yao, X.; Sun, Y. Effects of different mixed ratio of maize straw and cabbage wastes on silage quality. J. Biobased Mater. Bioenergy 2015, 9, 88–94. [Google Scholar] [CrossRef]
- Partovi, E.; Rouzbehan, Y.; Fazaeli, H.; Rezaei, J. Broccoli byproduct-wheat straw silage as a feed resource for fattening lambs. Transl. Anim. Sci. 2020, 4, txaa078. [Google Scholar] [CrossRef]
- Lu, H.; Yan, H.; Almeida, V.; Adeola, O.; Ajuwon, K. Effects of dietary resistant starch content on nutrient and energy digestibility and fecal metabolomic profile in growing pigs. J. Anim. Sci. 2016, 94, 364–368. [Google Scholar] [CrossRef]
- Kim, J.-H.; Hong, S.W.; Park, B.-Y.; Yoo, J.G.; Oh, M.-H. Characterisation of the bacterial community in the gastrointestinal tracts of elk (Cervus canadensis). Antonie Van Leeuwenhoek 2019, 112, 225–235. [Google Scholar] [CrossRef]
- Al-Lahham, S.a.H.; Roelofsen, H.; Priebe, M.; Weening, D.; Dijkstra, M.; Hoek, A.; Rezaee, F.; Venema, K.; Vonk, R.J. Regulation of adipokine production in human adipose tissue by propionic acid. Eur. J. Clin. Investig. 2010, 40, 401–407. [Google Scholar] [CrossRef]
- Ussar, S.; Fujisaka, S.; Kahn, C.R. Interactions between host genetics and gut microbiome in diabetes and metabolic syndrome. Mol. Metab. 2016, 5, 795–803. [Google Scholar] [CrossRef]
- Jia, W.; Li, H.; Zhao, L.; Nicholson, J.K. Gut microbiota: A potential new territory for drug targeting. Nat. Rev. Drug Discov. 2008, 7, 123–129. [Google Scholar] [CrossRef]
- Ley, R.E.; Hamady, M.; Lozupone, C.; Turnbaugh, P.J.; Ramey, R.R.; Bircher, J.S.; Schlegel, M.L.; Tucker, T.A.; Schrenzel, M.D.; Knight, R. Evolution of mammals and their gut microbes. Science 2008, 320, 1647–1651. [Google Scholar] [CrossRef]
- Johnson, A.J.; Vangay, P.; Al-Ghalith, G.A.; Hillmann, B.M.; Ward, T.L.; Shields-Cutler, R.R.; Kim, A.D.; Shmagel, A.K.; Syed, A.N.; Personalized Microbiome Class, S.; et al. Daily Sampling Reveals Personalized Diet-Microbiome Associations in Humans. Cell Host Microbe 2019, 25, 789–802.e785. [Google Scholar] [CrossRef]
- Li, C.; Chen, N.; Zhang, X.; Shahzad, K.; Qi, R.; Zhang, Z.; Lu, Z.; Lu, Y.; Yu, X.; Zafar, M.H. Mixed silage with Chinese cabbage waste enhances antioxidant ability by increasing ascorbate and aldarate metabolism through rumen Prevotellaceae UCG-004 in Hu sheep. Front. Microbiol. 2022, 13, 978940. [Google Scholar] [CrossRef]
- Nicholson, J.K.; Lindon, J.C. Metabonomics. Nature 2008, 455, 1054–1056. [Google Scholar] [CrossRef]
- Zhang, Z.; Shahzad, K.; Shen, S.; Dai, R.; Lu, Y.; Lu, Z.; Li, C.; Chen, Y.; Qi, R.; Gao, P. Altering Dietary Soluble Protein Levels With Decreasing Crude Protein May Be a Potential Strategy to Improve Nitrogen Efficiency in Hu Sheep Based on Rumen Microbiome and Metabolomics. Front. Nutr. 2021, 8, 815358. [Google Scholar] [CrossRef]
- Yi, H.; Yang, G.; Xiong, Y.; Wu, Q.; Xiao, H.; Wen, X.; Yang, X.; Wang, L.; Jiang, Z. Integrated metabolomic and proteomics profiling reveals the promotion of Lactobacillus reuteri LR1 on amino acid metabolism in the gut–liver axis of weaned pigs. Food Funct. 2019, 10, 7387–7396. [Google Scholar] [CrossRef]
- Plaizier, J.C.; Li, S.; Danscher, A.M.; Derakshani, H.; Andersen, P.H.; Khafipour, E. Changes in microbiota in rumen digesta and feces due to a grain-based subacute ruminal acidosis (SARA) challenge. Microb. Ecol. 2017, 74, 485–495. [Google Scholar] [CrossRef]
- Warner, R.D.; Jacob, R.H.; Rosenvold, K.; Rochfort, S.; Trenerry, C.; Plozza, T.; McDonagh, M.B. Altered post-mortem metabolism identified in very fast chilled lamb M. longissimus thoracis et lumborum using metabolomic analysis. Meat Sci. 2015, 108, 155–164. [Google Scholar] [CrossRef]
- National Research Council (NRC). Nutrient Requirements of Small Ruminants, Sheep, Goats, Cervids, and New World Camelids; National Academy Press: Washington, DC, USA, 2007. [Google Scholar]
- Magoč, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef] [PubMed]
- Lozupone, C.; Knight, R. UniFrac: A new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 2005, 71, 8228–8235. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.Z.; Yang, H.L.; Ma, R.L.; Zhang, C.X.; Lin, W.Y. Effect of dietary administration of Psychrobacter sp. on the growth, feed utilization, digestive enzymes and immune responses of grouper Epinephelus coioides. Aquacult. Nutr. 2011, 17, e733–e740. [Google Scholar] [CrossRef]
- Tanca, A.; Fraumene, C.; Manghina, V.; Palomba, A.; Abbondio, M.; Deligios, M.; Pagnozzi, D.; Addis, M.F.; Uzzau, S. Diversity and functions of the sheep faecal microbiota: A multi-omic characterization. Microb. Biotechnol. 2017, 10, 541–554. [Google Scholar] [CrossRef] [PubMed]
- Shabana, I.; Albakri, N.; Bouqellah, N. Metagenomic investigation of faecal microbiota in sheep and goats of the same ages. J. Taibah Univ. Sci. 2021, 15, 1–9. [Google Scholar] [CrossRef]
- Spence, C.; Wells, W.G.; Smith, C.J. Characterization of the primary starch utilization operon in the obligate anaerobe Bacteroides fragilis: Regulation by carbon source and oxygen. J. Bacteriol. 2006, 188, 4663–4672. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Fan, H.; Han, Y.; Zhao, J.; Zhou, Z. Characterization of the microbial communities along the gastrointestinal tract of sheep by 454 pyrosequencing analysis. Asian Australas. J. Anim. Sci. 2017, 30, 100. [Google Scholar] [CrossRef]
- Waters, J.L.; Ley, R.E. The human gut bacteria Christensenellaceae are widespread, heritable, and associated with health. BMC Biol. 2019, 17, 83. [Google Scholar] [CrossRef]
- Asma, Z.; Sylvie, C.; Laurent, C.; Jérôme, M.; Christophe, K.; Olivier, B.; Annabelle, T.-M.; Francis, E. Microbial ecology of the rumen evaluated by 454 GS FLX pyrosequencing is affected by starch and oil supplementation of diets. FEMS Microbiol. Ecol. 2013, 83, 504–514. [Google Scholar]
- Huang, C.; Ge, F.; Yao, X.; Guo, X.; Bao, P.; Ma, X.; Wu, X.; Chu, M.; Yan, P.; Liang, C. Microbiome and metabolomics reveal the effects of different feeding systems on the growth and ruminal development of yaks. Front. Microbiol. 2021, 12, 682989. [Google Scholar] [CrossRef]
- Li, Y.; Hu, X.; Yang, S.; Zhou, J.; Qi, L.; Sun, X.; Fan, M.; Xu, S.; Cha, M.; Zhang, M. Comparison between the fecal bacterial microbiota of healthy and diarrheic captive musk deer. Front. Microbiol. 2018, 9, 300. [Google Scholar] [CrossRef]
- Zhang, S.; Song, W.; Yu, M.; Lin, X. Comparative genomics analysis of five Psychrobacter strains isolated from world-wide habitats reveal high intra-genus variations. Extremophiles 2017, 21, 581–589. [Google Scholar] [CrossRef]
- Amato, P.; Christner, B.C. Energy metabolism response to low-temperature and frozen conditions in Psychrobacter cryohalolentis. Appl. Environ. Microbiol. 2009, 75, 711–718. [Google Scholar] [CrossRef]
- Lin, X.; Cui, S.; Xu, G.; Wang, S.; Du, N.; Shen, J. Cloning and heterologous expression of two cold-active lipases from the Antarctic bacterium Psychrobacter sp. G. Polar Res. 2010, 29, 421–429. [Google Scholar]
- Van Gylswyk, N. Succiniclasticum ruminis gen. nov., sp. nov., a ruminal bacterium converting succinate to propionate as the sole energy-yielding mechanism. Int. J. Syst. Evol. Microbiol. 1995, 45, 297–300. [Google Scholar] [CrossRef] [PubMed]
- Hammes, W.P.; Hertel, C. The genera lactobacillus and carnobacterium. Prokaryotes 2006, 4, 320–403. [Google Scholar]
- Zeineldin, M.; Barakat, R.; Elolimy, A.; Salem, A.Z.; Elghandour, M.M.; Monroy, J.C. Synergetic action between the rumen microbiota and bovine health. Microb. Pathog. 2018, 124, 106–115. [Google Scholar] [CrossRef]
- Murros, K.E.; Huynh, V.A.; Takala, T.M.; Saris, P.E. Desulfovibrio Bacteria Are Associated with Parkinson’s Disease. Front. Cell. Infect. Microbiol. 2021, 11, 378. [Google Scholar] [CrossRef]
- Xu, Y.; Shao, M.; Fang, X.; Tang, W.; Zhou, C.; Hu, X.; Zhang, X.; Su, K.-P. Antipsychotic-induced gastrointestinal hypomotility and the alteration in gut microbiota in patients with schizophrenia. Brain Behav. Immun. 2022, 99, 119–129. [Google Scholar] [CrossRef]
- Vinayavekhin, N.; Homan, E.A.; Saghatelian, A. Exploring disease through metabolomics. ACS Chem. Biol. 2010, 5, 91–103. [Google Scholar] [CrossRef] [PubMed]
- Mariz, L.; Amaral, P.; Valadares Filho, S.; Santos, S.; Detmann, E.; Marcondes, M.; Pereira, J.; Silva Júnior, J.; Prados, L.; Faciola, A. Dietary protein reduction on microbial protein, amino acid digestibility, and body retention in beef cattle: 2. Amino acid intestinal absorption and their efficiency for whole-body deposition. J. Anim. Sci. 2018, 96, 670–683. [Google Scholar] [CrossRef] [PubMed]
- Michonneau, P.; Fleurat-Lessard, P.; Cantereau, A.; Crépin, A.; Roblin, G.; Berjeaud, J.M. Implication of actin in the uptake of sucrose and valine in the tap root and leaf of sugar beet. Physiol. Plant. 2021, 172, 218–232. [Google Scholar] [CrossRef] [PubMed]
- Rose, A.J. Amino acid nutrition and metabolism in health and disease. Nutrients 2019, 11, 2623. [Google Scholar] [CrossRef] [PubMed]
- Shigesada, K.; Tatibana, M. Enzymatic synthesis of acetylglutamate by mammalian liver preparations and its stimulation by arginine. Biochem. Biophys. Res. Commun. 1971, 44, 1117–1124. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Xue, Y.; Seddik, H.-E.; Yin, Y.; Hu, F.; Mao, S. Dynamic changes of plasma metabolome in response to severe feed restriction in pregnant ewes. Metabolites 2019, 9, 112. [Google Scholar] [CrossRef] [PubMed]
- Daneshmand, A.; Kermanshahi, H.; Mesgaran, M.D.; King, A.J.; Ibrahim, S.A.; Klasing, K.C. Combination of purine and pyrimidine nucleosides influences growth performance, gut morphology, digestive enzymes, serum biochemical indices and immune functions in broiler chickens. Anim. Feed Sci. Technol. 2017, 228, 186–193. [Google Scholar] [CrossRef]
- Jung, B.; Batal, A. Effect of dietary nucleotide supplementation on performance and development of the gastrointestinal tract of broilers. Brit. Poult. Sci. 2012, 53, 98–105. [Google Scholar] [CrossRef]
- Superchi, P.; Saleri, R.; Borghetti, P.; De Angelis, E.; Ferrari, L.; Cavalli, V.; Amicucci, P.; Ossiprandi, M.; Sabbioni, A. Effects of dietary nucleotide supplementation on growth performance and hormonal and immune responses of piglets. Animal 2012, 6, 902–908. [Google Scholar] [CrossRef]
- Ma, Y.; Yang, X.; Hua, G.; Deng, X.; Xia, T.; Li, X.; Feng, D.; Deng, X. Contribution of gut microbiomes and their metabolomes to the performance of Dorper and Tan sheep. Front. Microbiol. 2022, 13, 1047744. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Items | Treatment 1 | |
|---|---|---|
| Control | Silage | |
| Ingredients (% of DM) | ||
| Peanut seedling | 30 | - |
| Corn husk | 15 | - |
| Sorghum shell | 5 | - |
| Mixed Silage | 0 | 50 |
| Corn | 34 | 34 |
| Soybean meal | 7 | 5.5 |
| Bran | 7.5 | 8 |
| Corn gluten meal | - | 1 |
| NaHCO3 | 0.5 | 0.5 |
| Premix contained 2 | 0.5 | 0.5 |
| Salt | 0.5 | 0.5 |
| Total | 100 | 100 |
| Nutrient composition (% of DM) | ||
| Digestive energy/DE (MJ/kg) 3 | 13.52 | 14.73 |
| Metabolizable energy/ME (MJ/kg) 4 | 18.93 | 20.62 |
| Crude protein, CP | 15.08 | 15.11 |
| Ash | 4.36 | 12.33 |
| Neutral Detergent Fiber, NDF | 47.64 | 48.23 |
| Acid Detergent Fiber, ADF | 23.71 | 27.17 |
| Ca | 0.48 | 0.45 |
| P | 0.38 | 0.39 |
| Metabolites | Group 1 | VIP 2 | p | FC 3 | Trend | Mode | |
|---|---|---|---|---|---|---|---|
| Control | Silage | ||||||
| Amino acids, peptides, and analogues | |||||||
| DL-Alanine | 0.079 | 0.242 | 1.403 | 0.018 | 3.071 | Up | ESI+ |
| Glycine | 0.003 | 0.011 | 1.637 | 0.001 | 3.633 | Up | ESI+ |
| LEVODOPA | 0.004 | 0.008 | 1.430 | 0.013 | 1.950 | Up | ESI+ |
| N,N-Dimethylglycine | 0.012 | 0.003 | 1.277 | 0.042 | 0.268 | Down | ESI+ |
| Ala-Ile | 0.065 | 0.027 | 1.119 | 0.045 | 0.407 | Down | ESI− |
| Arginine | 0.053 | 0.004 | 1.299 | 0.006 | 0.079 | Down | ESI− |
| D-ASPARTATE | 0.644 | 0.093 | 1.375 | 0.002 | 0.145 | Down | ESI− |
| gamma-Glutamylleucine | 0.083 | 0.027 | 1.407 | 0.001 | 0.328 | Down | ESI− |
| L-Histidine | 0.055 | 0.007 | 1.147 | 0.046 | 0.136 | Down | ESI− |
| L-Phenylalanine | 1.630 | 0.474 | 1.165 | 0.040 | 0.291 | Down | ESI− |
| L-Valine | 0.077 | 0.018 | 1.197 | 0.022 | 0.231 | Down | ESI− |
| N-Acetylglutamic acid (NAG) | 0.641 | 0.230 | 1.220 | 0.012 | 0.359 | Down | ESI− |
| N-Isobutyrylglycine | 1.345 | 0.017 | 1.206 | 0.015 | 0.012 | Down | ESI− |
| N-Tigloylglycine | 0.861 | 0.003 | 1.199 | 0.025 | 0.004 | Down | ESI− |
| Benzoic acids and derivatives | |||||||
| 2-Hydroxyhippuric acid | 0.544 | 0.007 | 1.199 | 0.028 | 0.013 | Down | ESI− |
| 5-Methoxysalicylic acid | 0.018 | 0.005 | 1.135 | 0.049 | 0.277 | Down | ESI− |
| Butylparaben | 0.231 | 0.063 | 1.144 | 0.019 | 0.275 | Down | ESI− |
| Bile acids, alcohols, and derivatives | |||||||
| Cholic Acid | 0.015 | 0.009 | 1.419 | 0.011 | 0.578 | Down | ESI+ |
| Glycochenodeoxycholate | 0.014 | 0.001 | 1.274 | 0.045 | 0.040 | Down | ESI+ |
| GLYCOCHOLATE | 0.005 | 0.002 | 1.411 | 0.011 | 0.441 | Down | ESI+ |
| Carbohydrates and carbohydrate conjugates | |||||||
| N-Acetylneuraminic acid | 0.018 | 0.133 | 1.487 | 0.006 | 7.483 | Up | ESI+ |
| D-SACCHARIC ACID | 0.166 | 0.053 | 1.425 | 0.001 | 0.318 | Down | ESI− |
| N-Acetylmannosamine | 8.252 | 2.333 | 1.232 | 0.010 | 0.283 | Down | ESI− |
| N-Acetylmuramic Acid | 0.636 | 0.141 | 1.425 | 0.001 | 0.222 | Down | ESI− |
| Carbonyl compounds | |||||||
| Acetophenone | 0.243 | 0.035 | 1.627 | 0.001 | 0.145 | Down | ESI+ |
| 4-Hydroxybenzaldehyde | 0.844 | 0.276 | 1.424 | 0.001 | 0.327 | Down | ESI− |
| Eicosanoids | |||||||
| Prostaglandin B1 | 0.007 | 0.079 | 1.272 | 0.007 | 11.976 | Up | ESI− |
| Resolvin E1 | 0.011 | 0.054 | 1.376 | 0.002 | 4.752 | Up | ESI− |
| Fatty acids and conjugates | |||||||
| 3,3-Dimethylglutaric acid | 0.642 | 0.110 | 1.577 | <0.001 | 0.172 | Down | ESI− |
| 3-Methylglutaric acid | 0.050 | 0.224 | 1.154 | 0.032 | 4.455 | Up | ESI− |
| Arachidic acid | 0.322 | 1.498 | 1.126 | 0.016 | 4.655 | Up | ESI− |
| Lauric acid | 0.061 | 0.027 | 1.224 | 0.006 | 0.454 | Down | ESI− |
| Isoflav-2-enes | |||||||
| Daidzein | 0.176 | 0.050 | 1.147 | 0.024 | 0.283 | Down | ESI− |
| Genistein | 2.530 | 0.442 | 1.376 | 0.002 | 0.175 | Down | ESI− |
| Pyrimidine 2′-deoxyribonucleosides | |||||||
| 2′-Deoxyuridine | 0.002 | 0.006 | 1.564 | 0.003 | 3.150 | Up | ESI+ |
| Thymidine | 0.018 | 0.051 | 1.330 | 0.030 | 2.752 | Up | ESI+ |
| Tetrahydrofuran lignans | |||||||
| Enterolactone | 0.018 | 0.110 | 1.118 | 0.029 | 6.082 | Up | ESI− |
| matairesinol | 0.163 | 0.015 | 1.202 | 0.010 | 0.091 | Down | ESI− |
| Others | |||||||
| Benzothiazole | 1.248 | 1.073 | 1.295 | 0.033 | 0.860 | Down | ESI+ |
| Metaxalone | 0.003 | 0.001 | 1.319 | 0.029 | 0.219 | Down | ESI+ |
| 9-Fluorenone | 5.195 | 0.812 | 1.172 | 0.017 | 0.156 | Down | ESI− |
| Abietic acid | 0.005 | 0.016 | 1.135 | 0.034 | 3.514 | Up | ESI− |
| Biotin | 0.038 | 0.082 | 1.157 | 0.047 | 2.143 | Up | ESI− |
| Bis(4-hydroxyphenyl)methane | 0.028 | 0.074 | 1.168 | 0.022 | 2.634 | Up | ESI− |
| cirsimaritin | 0.033 | 0.002 | 1.508 | <0.001 | 0.074 | Down | ESI− |
| delta7-Dafachronic acid | 0.090 | 0.047 | 1.104 | 0.048 | 0.525 | Down | ESI− |
| Ecgonine | 0.381 | 0.062 | 1.137 | 0.050 | 0.162 | Down | ESI− |
| Piceatannol | 0.010 | 0.059 | 1.393 | 0.001 | 6.047 | Up | ESI− |
| Pseudouridine | 1.798 | 0.457 | 1.160 | 0.033 | 0.254 | Down | ESI− |
| Resveratrol | 0.071 | 0.172 | 1.129 | 0.031 | 2.420 | Up | ESI− |
| santin | 0.019 | 0.085 | 1.286 | 0.009 | 4.525 | Up | ESI− |
| URIDINE | 1.502 | 0.455 | 1.120 | 0.043 | 0.303 | Down | ESI− |
| 3-Hydroxyphenylacetic acid | 0.108 | 0.021 | 1.519 | <0.001 | 0.199 | Down | ESI− |
| adrenosterone | 0.044 | 0.001 | 1.329 | 0.002 | 0.025 | Down | ESI− |
| 3-Indoxyl sulphate | 2.911 | 0.004 | 1.160 | 0.042 | 0.001 | Down | ESI− |
| Epinephrine | 0.190 | 0.000 | 1.203 | 0.024 | 0.002 | Down | ESI− |
| Isobutyric acid | 0.085 | 0.019 | 1.208 | 0.020 | 0.220 | Down | ESI− |
| Bisphenol A | 0.003 | 0.045 | 1.409 | 0.001 | 16.963 | Up | ESI− |
| Glabranine | 1.708 | 0.069 | 1.608 | <0.001 | 0.040 | Down | ESI− |
| Stearamide | 0.022 | 0.010 | 1.365 | 0.016 | 0.448 | Down | ESI+ |
| Chrysin | 0.027 | 0.001 | 1.553 | <0.001 | 0.045 | Down | ESI− |
| Ferulic acid | 0.005 | 0.012 | 1.375 | 0.023 | 2.257 | Up | ESI+ |
| afzelechin | 0.020 | 0.116 | 1.388 | 0.002 | 5.671 | Up | ESI− |
| 3-(2-Hydroxyethyl)indole | 0.073 | 0.001 | 1.171 | 0.035 | 0.019 | Down | ESI− |
| D-(+)-Tryptophan | 0.459 | 0.165 | 1.162 | 0.041 | 0.360 | Down | ESI− |
| Equol | 0.072 | 0.003 | 1.478 | <0.001 | 0.043 | Down | ESI− |
| Dihydrojasmonic Acid | 0.306 | 0.128 | 1.269 | 0.011 | 0.419 | Down | ESI− |
| 13-HPODE | 2.260 | 4.175 | 1.091 | 0.034 | 1.847 | Up | ESI− |
| Vanillin | 0.025 | 0.005 | 1.362 | 0.003 | 0.192 | Down | ESI− |
| Formononetin | 0.069 | 0.003 | 1.522 | <0.001 | 0.045 | Down | ESI− |
| 2-Hydroxyphenylacetic acid | 0.060 | 0.004 | 1.318 | 0.003 | 0.060 | Down | ESI− |
| 2′-Deoxyinosine | 0.029 | 0.065 | 1.360 | 0.021 | 2.221 | Up | ESI+ |
| Uric acid | 0.671 | 0.083 | 1.165 | 0.038 | 0.124 | Down | ESI− |
| 4-Pyridoxic acid | 0.060 | 0.026 | 1.294 | 0.042 | 0.426 | Down | ESI+ |
| Cytosine | 0.045 | 0.099 | 1.520 | 0.004 | 2.218 | Up | ESI+ |
| Acetylcholine | 0.151 | 0.030 | 1.295 | 0.043 | 0.200 | Down | ESI+ |
| 4-Methyl-2-oxovaleric Acid | 1.376 | 0.415 | 1.162 | 0.013 | 0.302 | Down | ESI− |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, C.; Lu, Z.; Qi, R.; Zhang, Z.; Lu, Y.; Zafar, M.H.; Yang, K.; Wang, M. Illumina Sequencing and Metabolomic Analysis Explored the Effects of the Mixed Silage of Rice Straw and Chinese Cabbage Waste on Fecal Microorganisms and Metabolites in Hu Sheep. Fermentation 2024, 10, 233. https://doi.org/10.3390/fermentation10050233
Li C, Lu Z, Qi R, Zhang Z, Lu Y, Zafar MH, Yang K, Wang M. Illumina Sequencing and Metabolomic Analysis Explored the Effects of the Mixed Silage of Rice Straw and Chinese Cabbage Waste on Fecal Microorganisms and Metabolites in Hu Sheep. Fermentation. 2024; 10(5):233. https://doi.org/10.3390/fermentation10050233
Chicago/Turabian StyleLi, Chuang, Zhiqi Lu, Ruxin Qi, Zhenbin Zhang, Yue Lu, Muhammad Hammad Zafar, Kailun Yang, and Mengzhi Wang. 2024. "Illumina Sequencing and Metabolomic Analysis Explored the Effects of the Mixed Silage of Rice Straw and Chinese Cabbage Waste on Fecal Microorganisms and Metabolites in Hu Sheep" Fermentation 10, no. 5: 233. https://doi.org/10.3390/fermentation10050233
APA StyleLi, C., Lu, Z., Qi, R., Zhang, Z., Lu, Y., Zafar, M. H., Yang, K., & Wang, M. (2024). Illumina Sequencing and Metabolomic Analysis Explored the Effects of the Mixed Silage of Rice Straw and Chinese Cabbage Waste on Fecal Microorganisms and Metabolites in Hu Sheep. Fermentation, 10(5), 233. https://doi.org/10.3390/fermentation10050233