

Overexpression of a Novel Vacuolar Serine Protease-Encoding Gene (spt1) to Enhance Cellulase Production in Trichoderma Reesei

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Strains, Plasmids and Culture Conditions
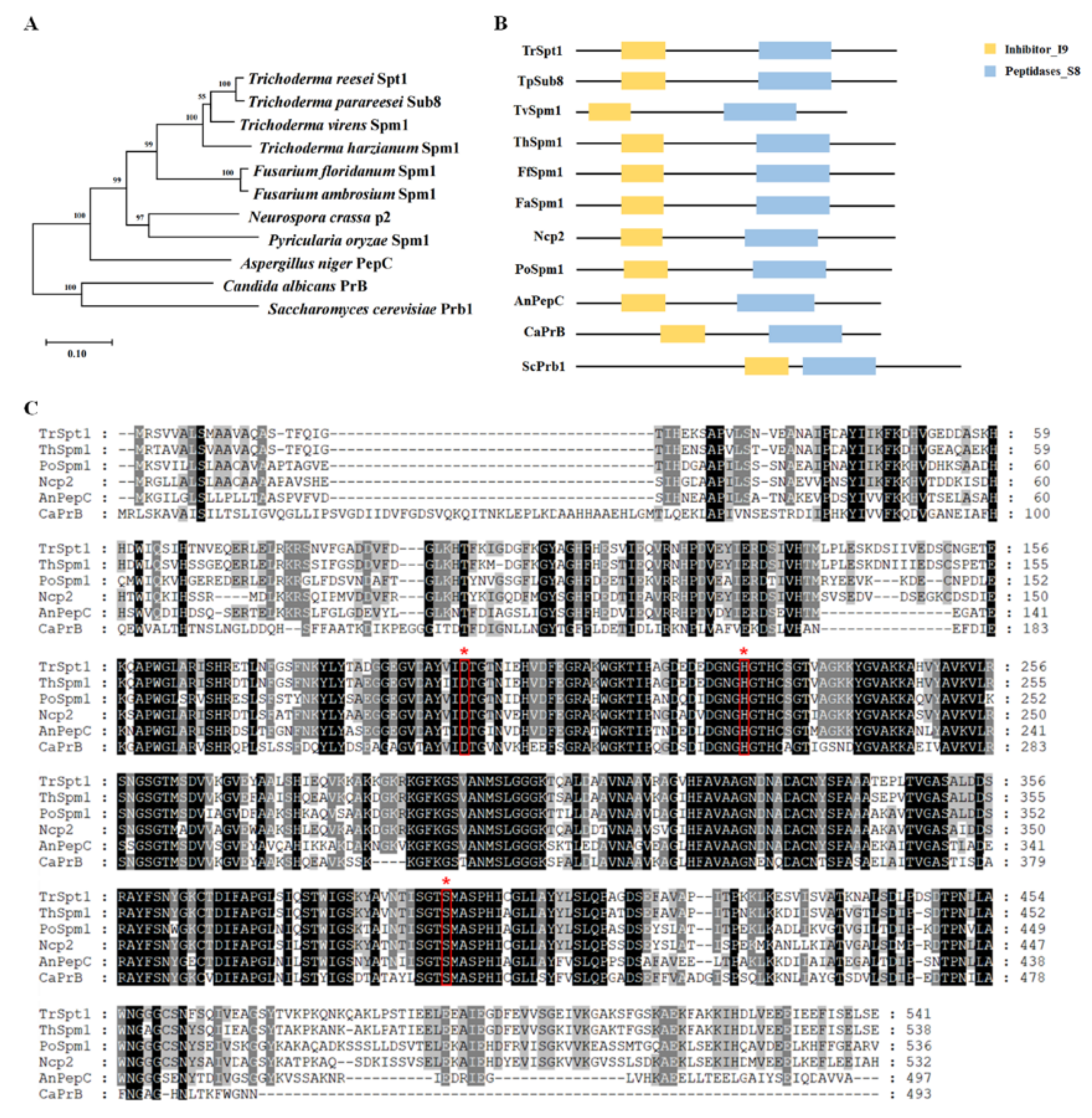
2.2. Phylogenetic Analysis and Sequence Alignments of spt1 and Its Homologs
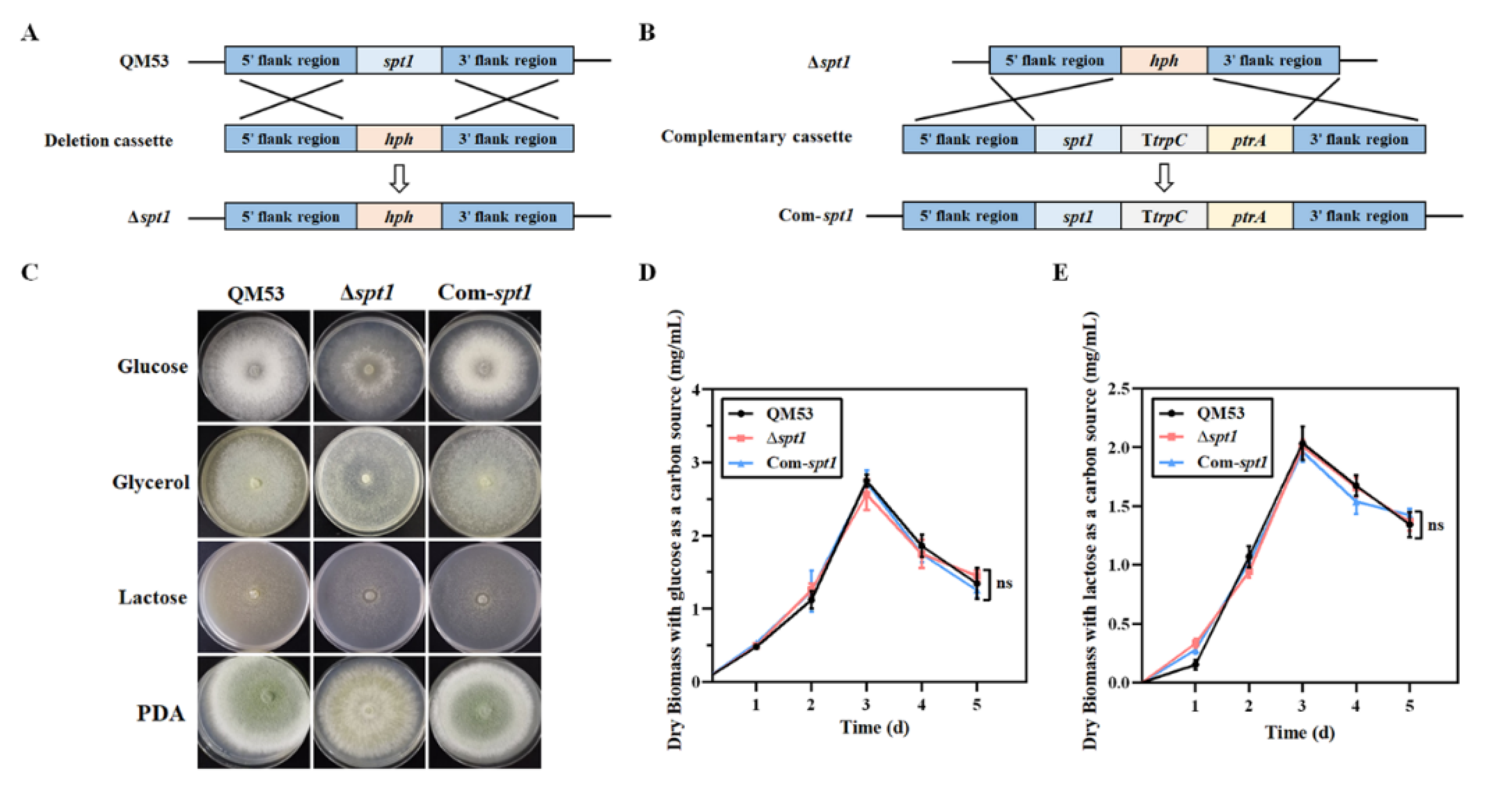
2.3. Construction of the spt1 Deletion Strain
2.4. Complementation of the spt1 Gene in the Δspt1 Strain
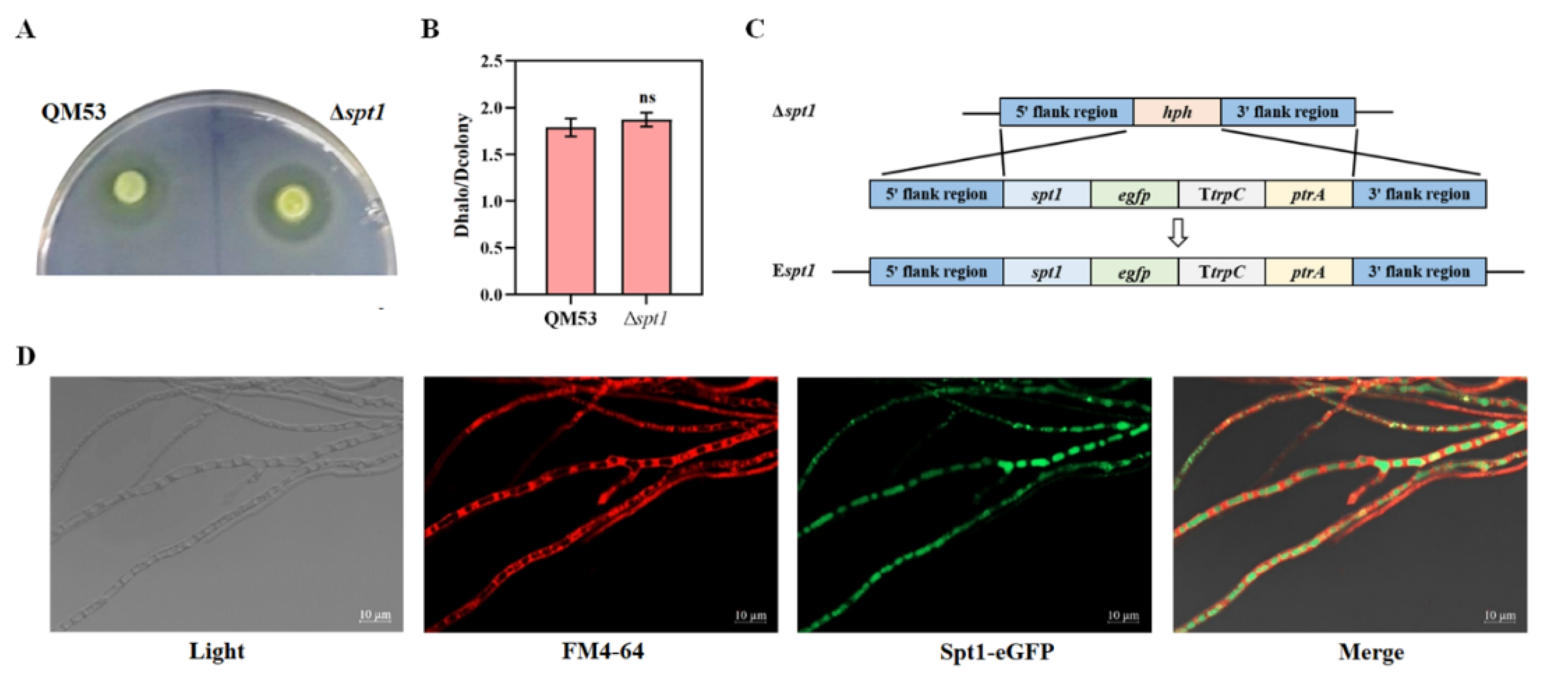
2.5. Construction of the eGFP-Labeled spt1 Strain
2.6. Confocal Fluorescence Microscope
2.7. Overexpression of the spt1 Gene in T. reesei QP4
2.8. Enzyme Activity Assay
3. Results
3.1. Sequence and Phylogenetic Analysis of T. reesei spt1 and Its Homologs in Fungi
3.2. Construction of the spt1 Deletion Strain Δspt1 and the spt1 Complementation Strain Com-spt1
3.3. Subcellular Localization of spt1 in T. reesei
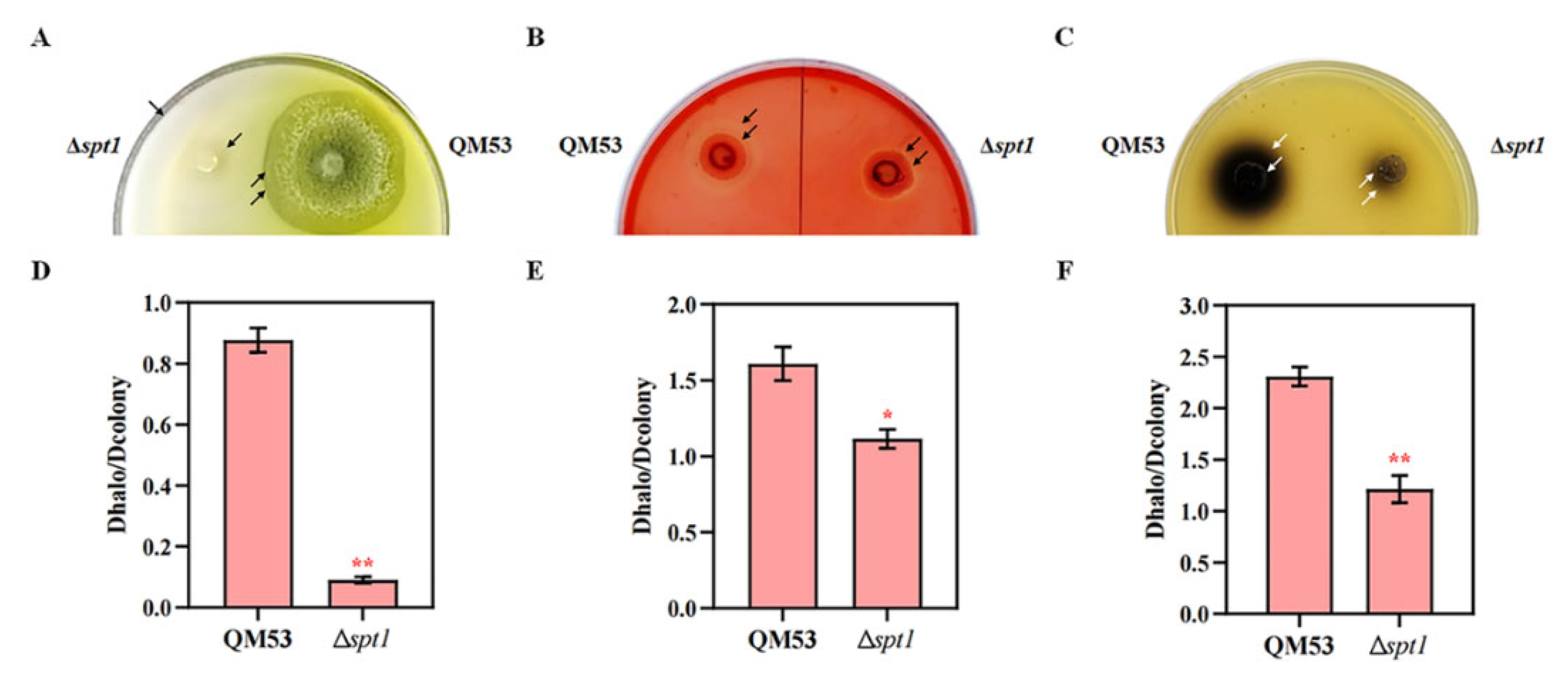
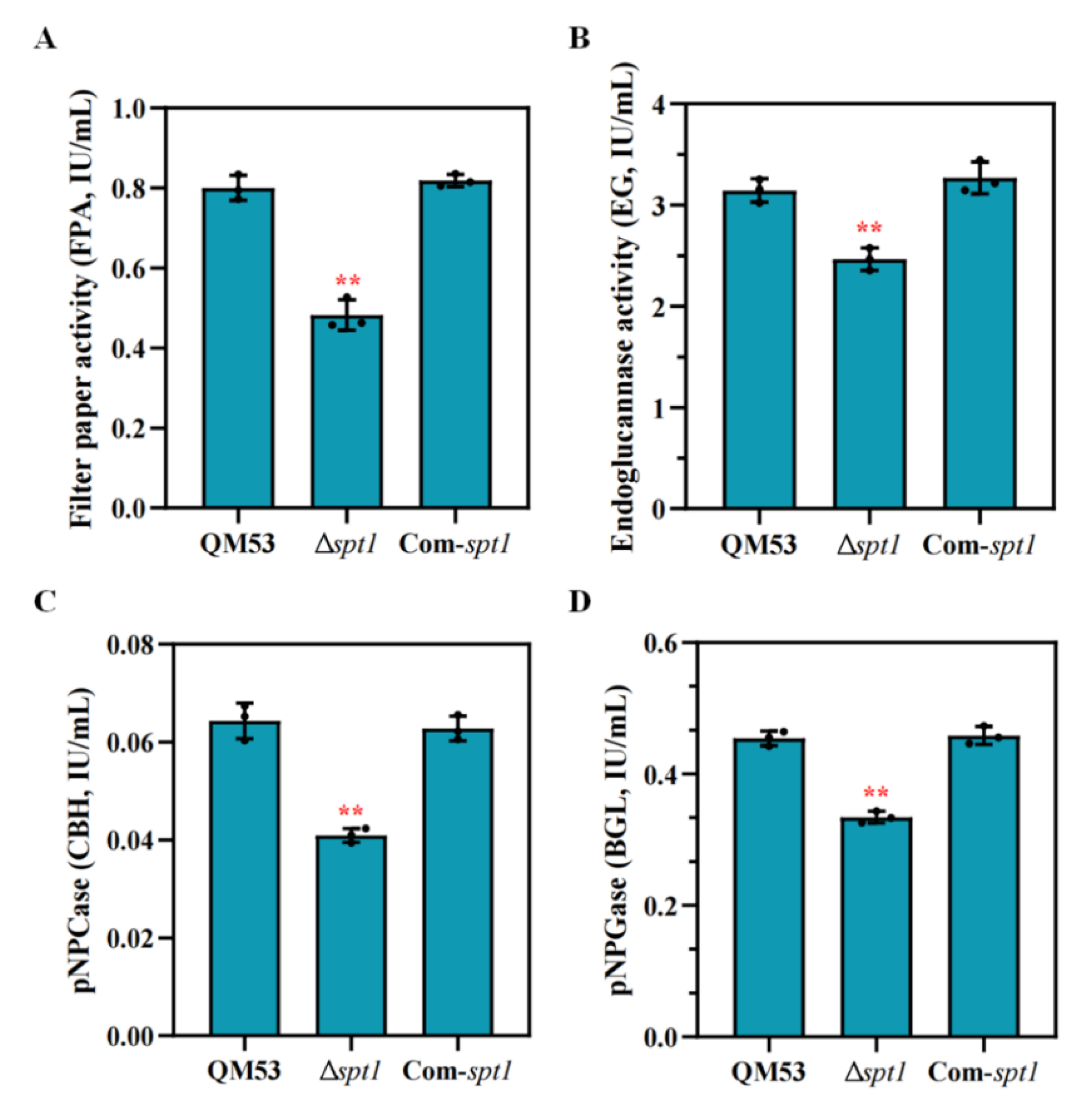
3.4. Detection of the Cellulase Secretion Capacity of Δspt1
3.5. Deletion of spt1 Resulting in Decreased Cellulase Production in T. reesei
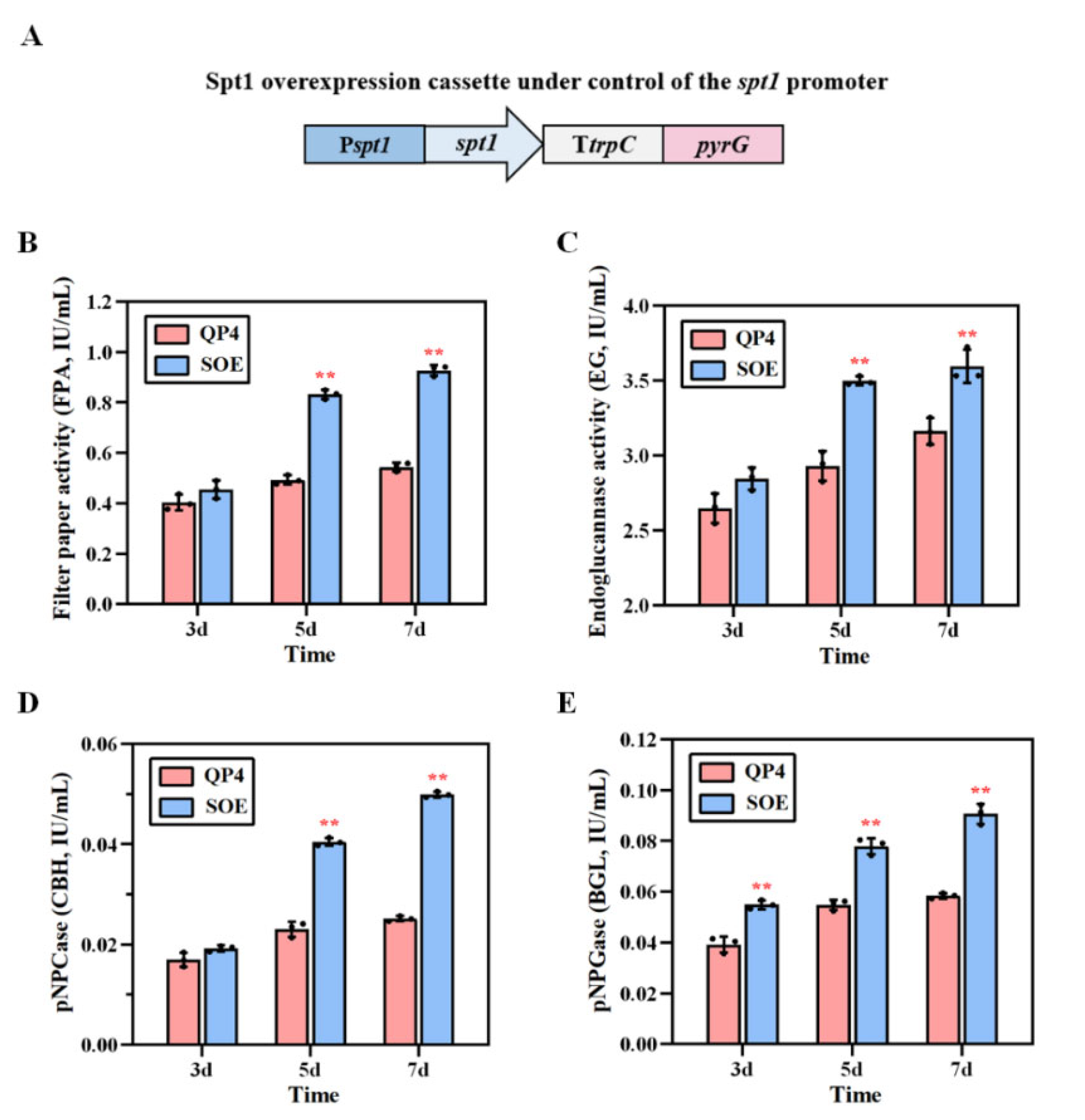
3.6. Overexpression of spt1, with Multiple Copies Improving Cellulase Production
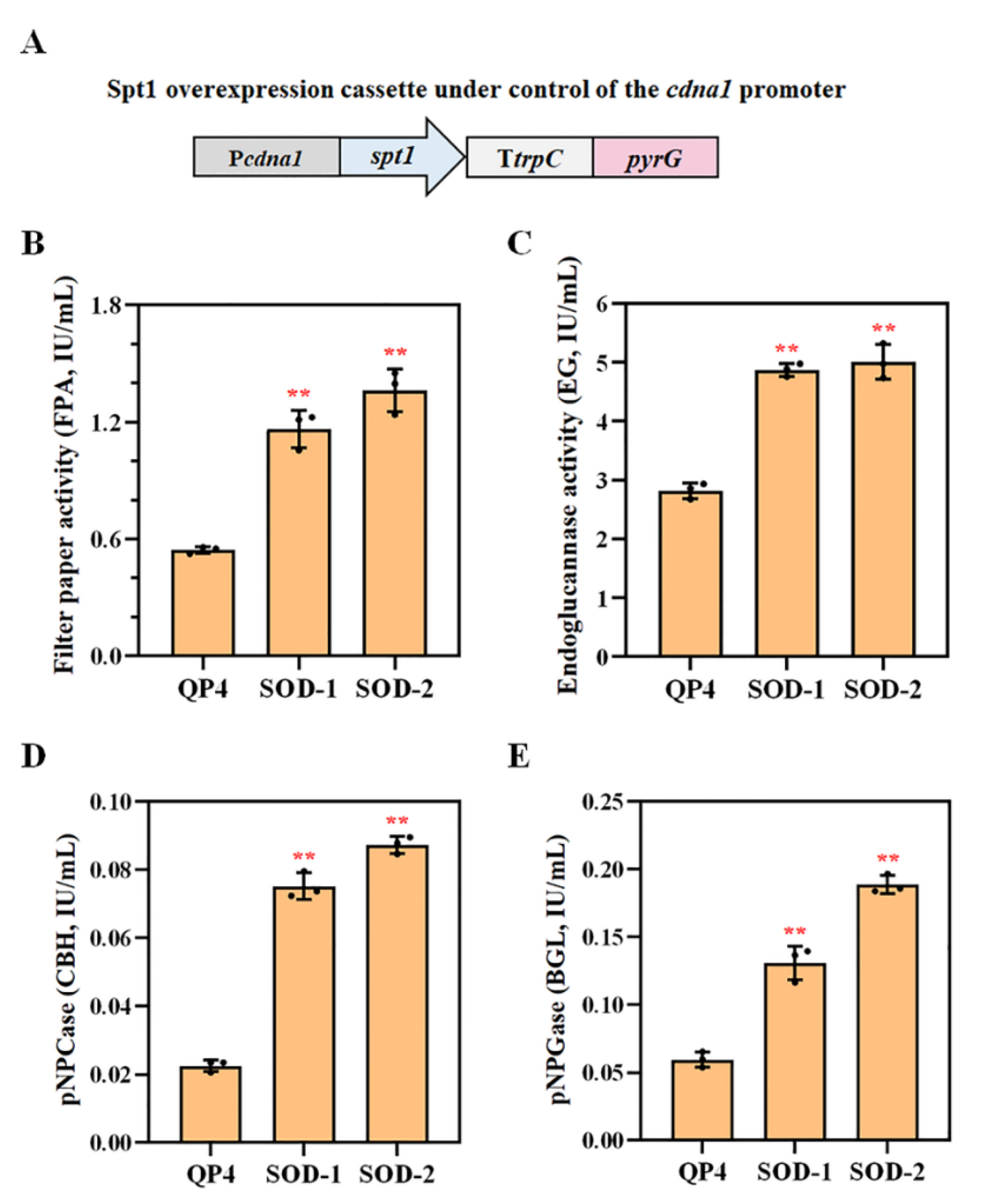
3.7. Overexpression of spt1 with the Strong Promoter Pcdna1 Enhancing the Cellulase Production
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liu, G.; Qu, Y. Engineering of filamentous fungi for efficient conversion of lignocellulose: Tools, recent advances and prospects. Biotechnol. Adv. 2018, 37, 519–529. [Google Scholar] [CrossRef]
- Limayem, A.; Ricke, S.C. Lignocellulosic biomass for bioethanol production: Current perspectives, potential issues and future prospects. Prog. Energy Combus. Sci. 2012, 38, 449–467. [Google Scholar] [CrossRef]
- Balan, V. Current challenges in commercially producing biofuels from lignocellulosic biomass. ISRN Biotechnol. 2014, 31, 463074. [Google Scholar] [CrossRef] [Green Version]
- Bilal, M.; Iqbal, H.M.N. Recent Advancements in the Life Cycle Analysis of Lignocellulosic Biomass. Curr. Sustain. Renew. Energy Rep. 2020, 7, 100–107. [Google Scholar] [CrossRef]
- Garvey, M.; Klose, H.; Fischer, R.; Lambertz, C.; Commandeur, U. Cellulases for biomass degradation: Comparing recombinant cellulase expression platforms. Trends Biotechnol. 2013, 31, 581–593. [Google Scholar] [CrossRef]
- Bischof, R.H.; Ramoni, J.; Seiboth, B. Cellulases and beyond: The first 70 years of the enzyme producer Trichoderma reesei. Microb. Cell Fact. 2016, 15, 106. [Google Scholar] [CrossRef] [Green Version]
- Kubicek, C.P.; Mikus, M.; Schuster, A.; Schmoll, M.; Seiboth, B. Metabolic engineering strategies for the improvement of cellulase production by Hypocrea jecorina. Biotechnol. Biofuels. 2009, 2, 19. [Google Scholar] [CrossRef] [Green Version]
- Martinez, D.; Berka, R.M.; Henrissat, B.; Saloheimo, M.; Arvas, M.; Baker, S.E.; Chapman, J.; Chertkov, O.; Coutinho, P.M.; Cullen, D.; et al. Genome sequencing and analysis of the biomass-degrading fungus Trichoderma reesei (syn. Hypocrea jecorina). Nat. Biotechnol. 2008, 26, 553–560. [Google Scholar] [CrossRef] [Green Version]
- Arai, T.; Ichinose, S.; Shibata, N.; Kakeshita, H.; Kodama, H.; Igarashi, K.; Takimura, Y. Inducer-free cellulase production system based on the constitutive expression of mutated XYR1 and ACE3 in the industrial fungus Trichoderma reesei. Sci. Rep. 2022, 12, 19445. [Google Scholar] [CrossRef]
- Adnan, M.; Ma, X.; Olsson, S.; Wang, J.; Liu, G. Promoter regulation and genetic engineering strategies for enhanced cellulase expression in Trichoderma reesei. Microbiol. Res. 2022, 259, 127011. [Google Scholar] [CrossRef]
- Li, C.; Pang, A.P.; Yang, H.; Lv, R.; Zhou, Z.; Wu, F.G.; Lin, F. Tracking localization and secretion of cellulase spatiotemporally and directly in living Trichoderma reesei. Biotechnol. Biofuels Bioprod. 2019, 12, 200. [Google Scholar] [CrossRef]
- Yan, S.; Xu, Y.; Yu, X.W. From induction to secretion: A complicated route for cellulase production in Trichoderma reesei. Bioresour. Bioprocess. 2021, 8, 107. [Google Scholar] [CrossRef]
- Shen, L.; Gao, J.; Wang, Y.; Li, X.; Liu, H.; Zhong, Y. Engineering the endoplasmic reticulum secretory pathway in Trichoderma reesei for improved cellulase production. Enzyme Microb. Technol. 2021, 152, 109923. [Google Scholar] [CrossRef]
- Gao, F.; Hao, Z.; Sun, X.; Qin, L.; Zhao, T.; Liu, W.; Luo, H.; Yao, B.; Su, X. A versatile system for fast screening and isolation of Trichoderma reesei cellulase hyperproducers based on DsRed and fluorescence-assisted cell sorting. Biotechnol. Biofuels. 2018, 11, 261. [Google Scholar] [CrossRef]
- Yao, C.; Yan, M.; Li, K.; Gao, W.; Li, X.; Zhang, J.; Liu, H.; Zhong, Y. The ERAD Pathway Participates in Fungal Growth and Cellulase Secretion in Trichoderma reesei. J. Fungi 2023, 9, 74. [Google Scholar] [CrossRef]
- Wu, Y.; Sun, X.; Xue, X.; Luo, H.; Yao, B.; Xie, X.; Su, X. Overexpressing key component genes of the secretion pathway for enhanced secretion of an Aspergillus niger glucose oxidase in Trichoderma reesei. Enzyme Microb. Technol. 2017, 106, 83–87. [Google Scholar] [CrossRef]
- Tang, H.; Song, M.; He, Y.; Wang, J.; Wang, S.; Shen, Y.; Hou, J.; Bao, X. Engineering vesicle trafficking improves the extracellular activity and surface display efficiency of cellulases in Saccharomyces cerevisiae. Biotechnol. Biofuels. 2017, 10, 53. [Google Scholar] [CrossRef] [Green Version]
- Li, S.C.; Kane, P.M. The yeast lysosome-like vacuole: Endpoint and crossroads. Biochim. Biophys. Acta. 2009, 1793, 650–663. [Google Scholar] [CrossRef] [Green Version]
- Pei, X.; Fan, F.; Lin, L.; Chen, Y.; Sun, W.; Zhang, S.; Tian, C. Involvement of the adaptor protein 3 complex in lignocellulase secretion in Neurospora crassa revealed by comparative genomic screening. Biotechnol. Biofuels. 2015, 8, 124. [Google Scholar] [CrossRef] [Green Version]
- Yoon, J.; Kikuma, T.; Maruyama, J.; Kitamoto, K. Enhanced production of bovine chymosin by autophagy deficiency in the filamentous fungus Aspergillus oryzae. PloS ONE 2013, 8, e62512. [Google Scholar] [CrossRef] [Green Version]
- Saitoh, H.; Fujisawa, S.; Ito, A.; Mitsuoka, C.; Berberich, T.; Tosa, Y.; Asakura, M.; Takano, Y.; Terauchi, R. SPM1 encoding a vacuole-localized protease is required for infection-related autophagy of the rice blast fungus Magnaporthe oryzae. FEMS Microbiol. Lett. 2009, 300, 115–121. [Google Scholar] [CrossRef] [Green Version]
- Shi, L.; Li, R.; Liao, S.; Bai, L.; Lu, Q.; Chen, B. Prb1, a subtilisin-like protease, is required for virulence and phenotypical traits in the chestnut blight fungus. FEMS Microbiol. Lett. 2014, 359, 26–33. [Google Scholar] [CrossRef] [Green Version]
- Häkkinen, M.; Valkonen, M.J.; Westerholm-Parvinen, A.; Aro, N.; Arvas, M.; Vitikainen, M.; Penttilä, M.; Saloheimo, M.; Pakula, T.M. Screening of candidate regulators for cellulase and hemicellulase production in Trichoderma reesei and identification of a factor essential for cellulase production. Biotechnol. Biofuels. 2014, 7, 14. [Google Scholar] [CrossRef] [Green Version]
- Steiger, M.G.; Vitikainen, M.; Uskonen, P.; Brunner, K.; Adam, G.; Pakula, T.; Penttilä, M.; Saloheimo, M.; Mach, R.L.; Mach-Aigner, A.R. Transformation system for Hypocrea jecorina (Trichoderma reesei) that favors homologous integration and employs reusable bidirectionally selectable markers. Appl. Environ. Microbiol. 2011, 77, 114–121. [Google Scholar] [CrossRef] [Green Version]
- Qian, Y.; Zhong, L.; Hou, Y.; Qu, Y.; Zhong, Y. Characterization and Strain Improvement of a Hypercellulytic Variant, Trichoderma reesei SN1, by Genetic Engineering for Optimized Cellulase Production in Biomass Conversion Improvement. Front. Microbiol. 2016, 7, 1349. [Google Scholar] [CrossRef] [Green Version]
- Punt, P.J.; van den Hondel, C.A. Transformation of filamentous fungi based on hygromycin B and phleomycin resistance markers. Methods Enzymol. 1992, 216, 447–457. [Google Scholar] [CrossRef]
- Krappmann, S.; Bayram, O.; Braus, G.H. Deletion and allelic exchange of the Aspergillus fumigatus veA locus via a novel recyclable marker module. Eukaryot. Cell. 2005, 4, 1298–1307. [Google Scholar] [CrossRef] [Green Version]
- Pöggeler, S.; Masloff, S.; Hoff, B.; Mayrhofer, S.; Kück, U. Versatile EGFP reporter plasmids for cellular localization of recombinant gene products in filamentous fungi. Curr. Genet. 2003, 43, 54–61. [Google Scholar] [CrossRef]
- Hartl, L.; Seiboth, B. Sequential gene deletions in Hypocrea jecorina using a single blaster cassette. Curr. Genet. 2005, 48, 204–211. [Google Scholar] [CrossRef]
- Penttilä, M.; Nevalainen, H.; Rättö, M.; Salminen, E.; Knowles, J. A versatile transformation system for the cellulolytic filamentous fungus Trichoderma reesei. Gene 1987, 61, 155–164. [Google Scholar] [CrossRef]
- Qian, Y.; Zhong, L.; Gao, J.; Sun, N.; Wang, Y.; Sun, G.; Qu, Y.; Zhong, Y. Production of highly efficient cellulase mixtures by genetically exploiting the potentials of Trichoderma reesei endogenous cellulases for hydrolysis of corncob residues. Microb. Cell Fact. 2017, 16, 207. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.H.; Hamari, Z.; Han, K.H.; Seo, J.A.; Reyes-Domínguez, Y.; Scazzocchio, C. Double-joint PCR: A PCR-based molecular tool for gene manipulations in filamentous fungi. Fungal Genet. Biol. 2004, 41, 973–981. [Google Scholar] [CrossRef]
- Ghose, T.K. Measurement of cellulase activities. Pure Appl. Chem. 1987, 59, 257–268. [Google Scholar] [CrossRef]
- Sun, Y.; Qian, Y.; Zhang, J.; Yao, C.; Wang, Y.; Liu, H.; Zhong, Y. Development of a novel expression platform for heterologous protein production via deleting the p53-like regulator Vib1 in Trichoderma reesei. Enzyme Microb. Technol. 2022, 155, 109993. [Google Scholar] [CrossRef]
- Nakari-Setälä, T.; Penttilä, M. Production of Trichoderma reesei cellulases on glucose-containing media. Appl. Environ. Microbiol. 1995, 61, 3650–3655. [Google Scholar] [CrossRef] [Green Version]
- Druzhinina, I.S.; Kubicek, C.P. Genetic engineering of Trichoderma reesei cellulases and their production. Microb. Biotechnol. 2017, 10, 1485–1499. [Google Scholar] [CrossRef]
- Fang, H.; Li, C.; Zhao, J.; Zhao, C. Biotechnological advances and trends in engineering Trichoderma reesei towards cellulase hyperproducer. Biotechnol. Bioprocess Eng. 2021, 26, 517–528. [Google Scholar] [CrossRef]
- Yoon, J.; Aishan, T.; Maruyama, J.; Kitamoto, K. Enhanced production and secretion of heterologous proteins by the filamentous fungus Aspergillus oryzae via disruption of vacuolar protein sorting receptor gene Aovps10. Appl. Environ. Microbiol. 2010, 76, 5718–5727. [Google Scholar] [CrossRef] [Green Version]
- Idiris, A.; Tohda, H.; Sasaki, M.; Okada, K.; Kumagai, H.; Giga-Hama, Y.; Takegawa, K. Enhanced protein secretion from multiprotease-deficient fission yeast by modification of its vacuolar protein sorting pathway. Appl. Microbiol. Biotechnol. 2010, 85, 667–677. [Google Scholar] [CrossRef]
- Marsalek, L.; Gruber, C.; Altmann, F.; Aleschko, M.; Mattanovich, D.; Gasser, B.; Puxbaum, V. Disruption of genes involved in CORVET complex leads to enhanced secretion of heterologous carboxylesterase only in protease deficient Pichia pastoris. Biotechnol. J. 2017, 12, 584. [Google Scholar] [CrossRef]
- Takeshige, K.; Baba, M.; Tsuboi, S.; Noda, T.; Ohsumi, Y. Autophagy in yeast demonstrated with proteinase-deficient mutants and conditions for its induction. J. Cell Biol. 1992, 119, 301–311. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Wang, H.; Zhang, C.; Wang, J.; Chen, A.; Chen, Y.; Ma, Z. System-wide characterization of subtilases reveals that subtilisin-like protease FgPrb1 of Fusarium graminearum regulates fungal development and virulence. Fungal Genet. Biol. 2020, 144, 103449. [Google Scholar] [CrossRef]
- Fu, H.; Chung, K.R.; Liu, X.; Li, H. Aaprb1, a subtilsin-like protease, required for autophagy and virulence of the tangerine pathotype of Alternaria alternata. Microbiol. Res. 2020, 240, 126537. [Google Scholar] [CrossRef]
- Yao, G.; Li, Z.; Wu, R.; Qin, Y.; Liu, G.; Qu, Y. Penicillium oxalicum PoFlbC regulates fungal asexual development and is important for cellulase gene expression. Fungal Genet. Biol. 2016, 86, 91–102. [Google Scholar] [CrossRef]
- Lei, Y.; Liu, G.; Li, Z.; Gao, L.; Qin, Y.; Qu, Y. Functional characterization of protein kinase CK2 regulatory subunits regulating Penicillium oxalicum asexual development and hydrolytic enzyme production. Fungal Genet. Biol. 2014, 66, 44–53. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yao, C.; Sun, N.; Gao, W.; Sun, Y.; Zhang, J.; Liu, H.; Zhong, Y. Overexpression of a Novel Vacuolar Serine Protease-Encoding Gene (spt1) to Enhance Cellulase Production in Trichoderma Reesei. Fermentation 2023, 9, 191. https://doi.org/10.3390/fermentation9020191
Yao C, Sun N, Gao W, Sun Y, Zhang J, Liu H, Zhong Y. Overexpression of a Novel Vacuolar Serine Protease-Encoding Gene (spt1) to Enhance Cellulase Production in Trichoderma Reesei. Fermentation. 2023; 9(2):191. https://doi.org/10.3390/fermentation9020191
Chicago/Turabian StyleYao, Cheng, Ningning Sun, Weihao Gao, Yu Sun, Jiaxin Zhang, Hong Liu, and Yaohua Zhong. 2023. "Overexpression of a Novel Vacuolar Serine Protease-Encoding Gene (spt1) to Enhance Cellulase Production in Trichoderma Reesei" Fermentation 9, no. 2: 191. https://doi.org/10.3390/fermentation9020191
APA StyleYao, C., Sun, N., Gao, W., Sun, Y., Zhang, J., Liu, H., & Zhong, Y. (2023). Overexpression of a Novel Vacuolar Serine Protease-Encoding Gene (spt1) to Enhance Cellulase Production in Trichoderma Reesei. Fermentation, 9(2), 191. https://doi.org/10.3390/fermentation9020191