
Analysis of Bacterial Diversity in Fermented Grains of Baijiu Based on Culturomics and Amplicon Sequencing
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Amplificon Sequencing Sample Pretreatment
2.3. DNA Extraction and PCR Amplification
2.4. Illumina MiSeq Sequencing and Data Processing
2.5. Bioinformation Analysis
2.6. Enrichment Culture of Samples
2.7. Pure Cultures Isolation
2.8. Preliminary Identification of Bacteria
2.9. Preservation of Bacteria
3. Results and Discussion
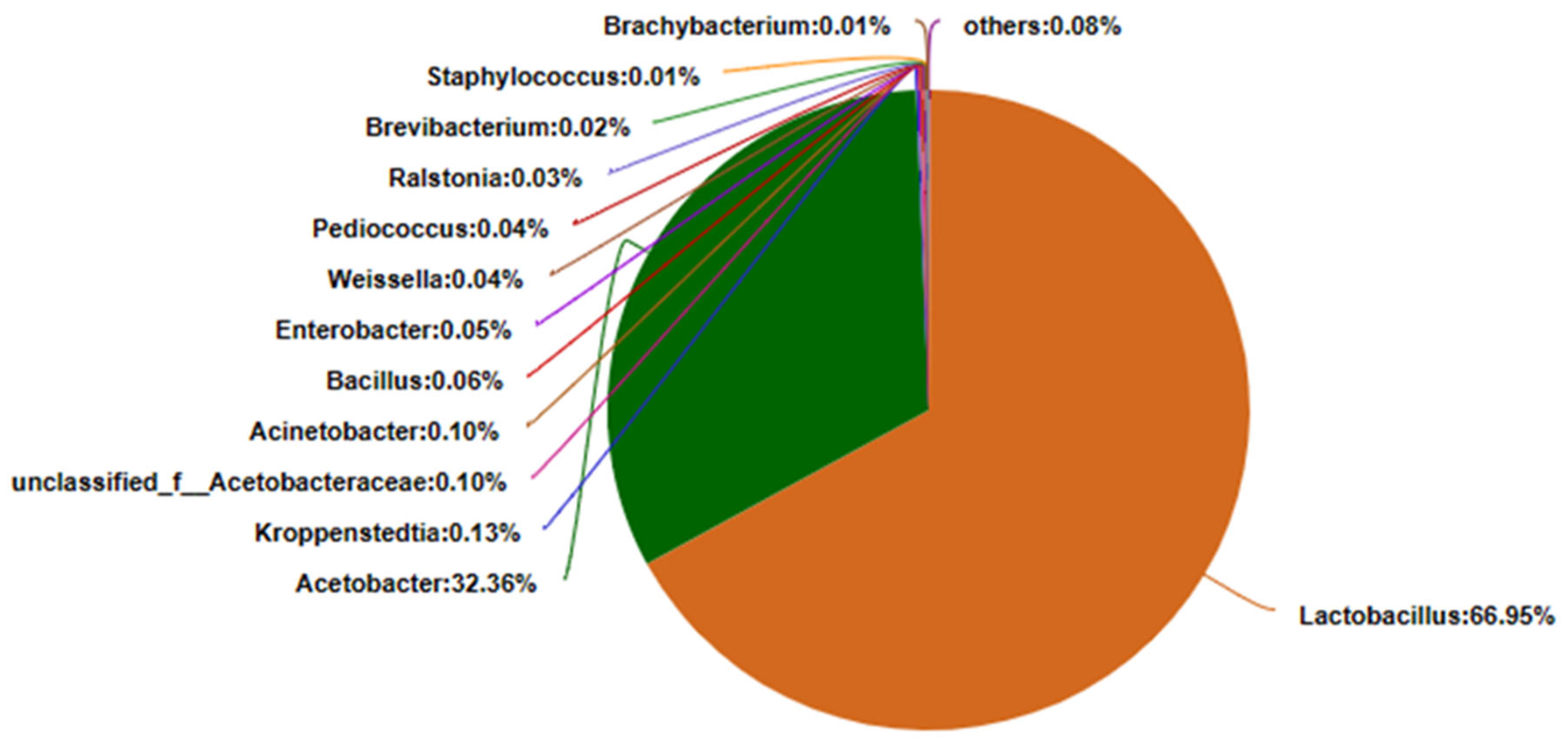
3.1. Amplicon Sequencing of Bacteria
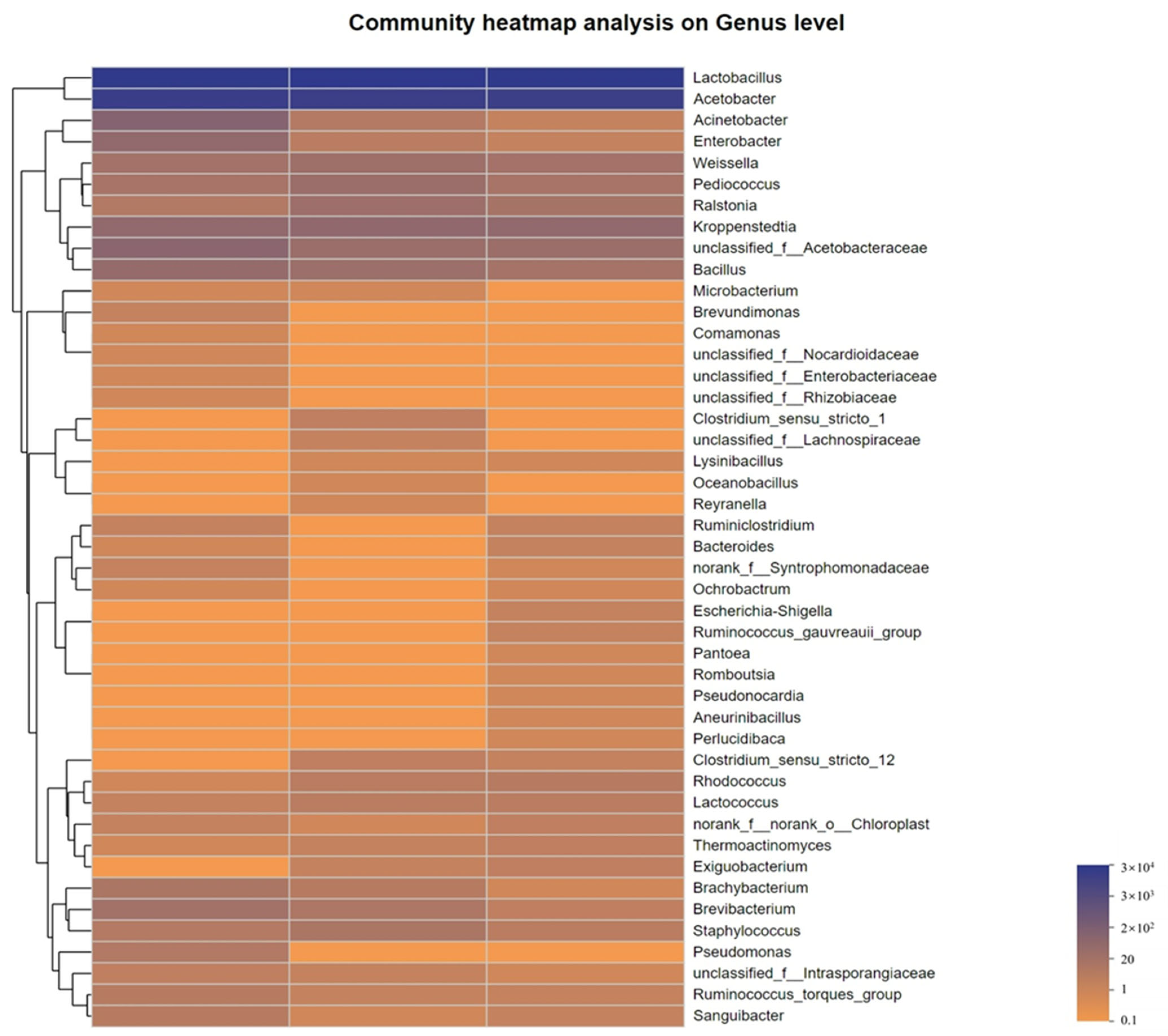
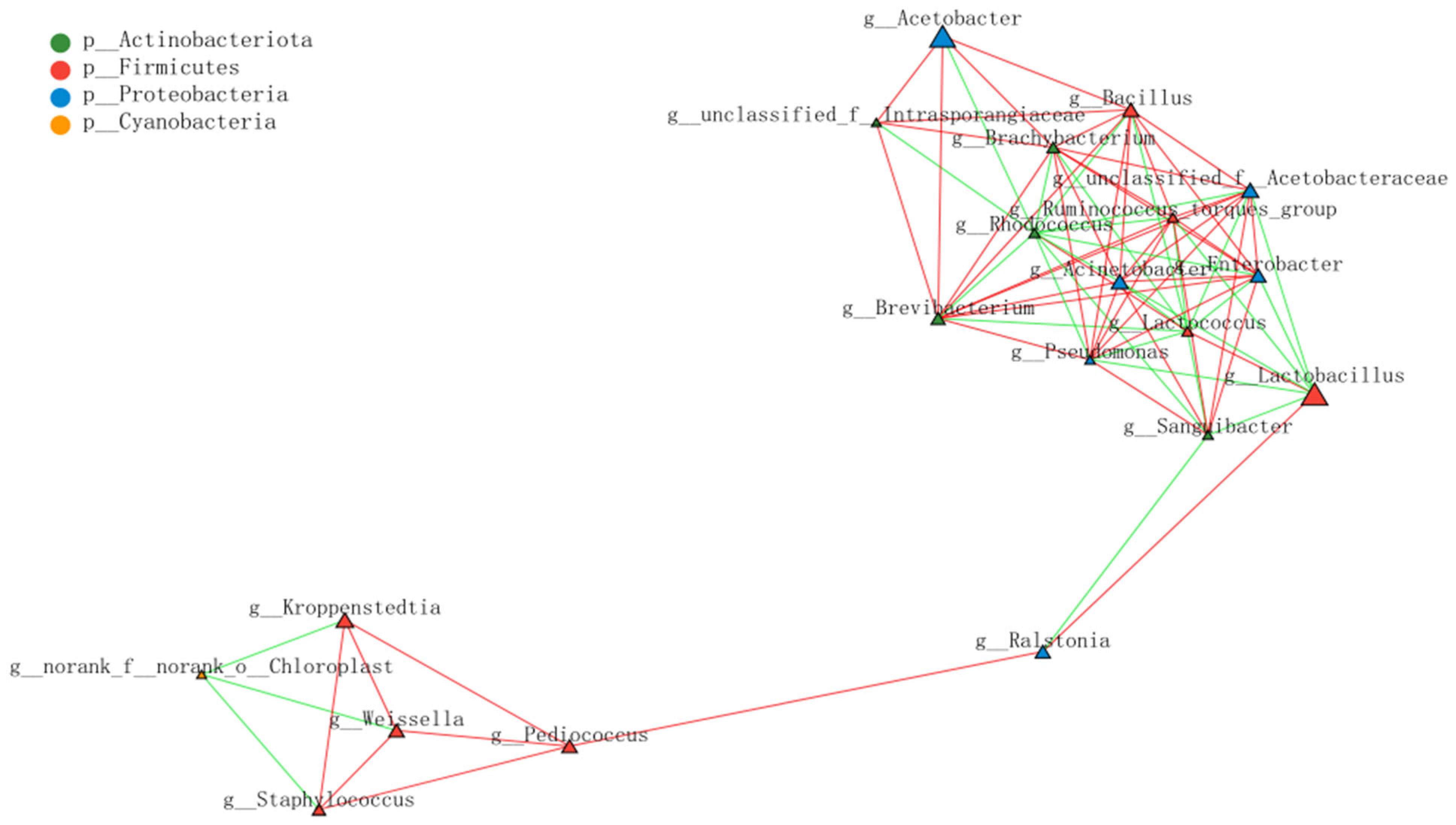
3.2. Analysis of Bacterial Community Structure
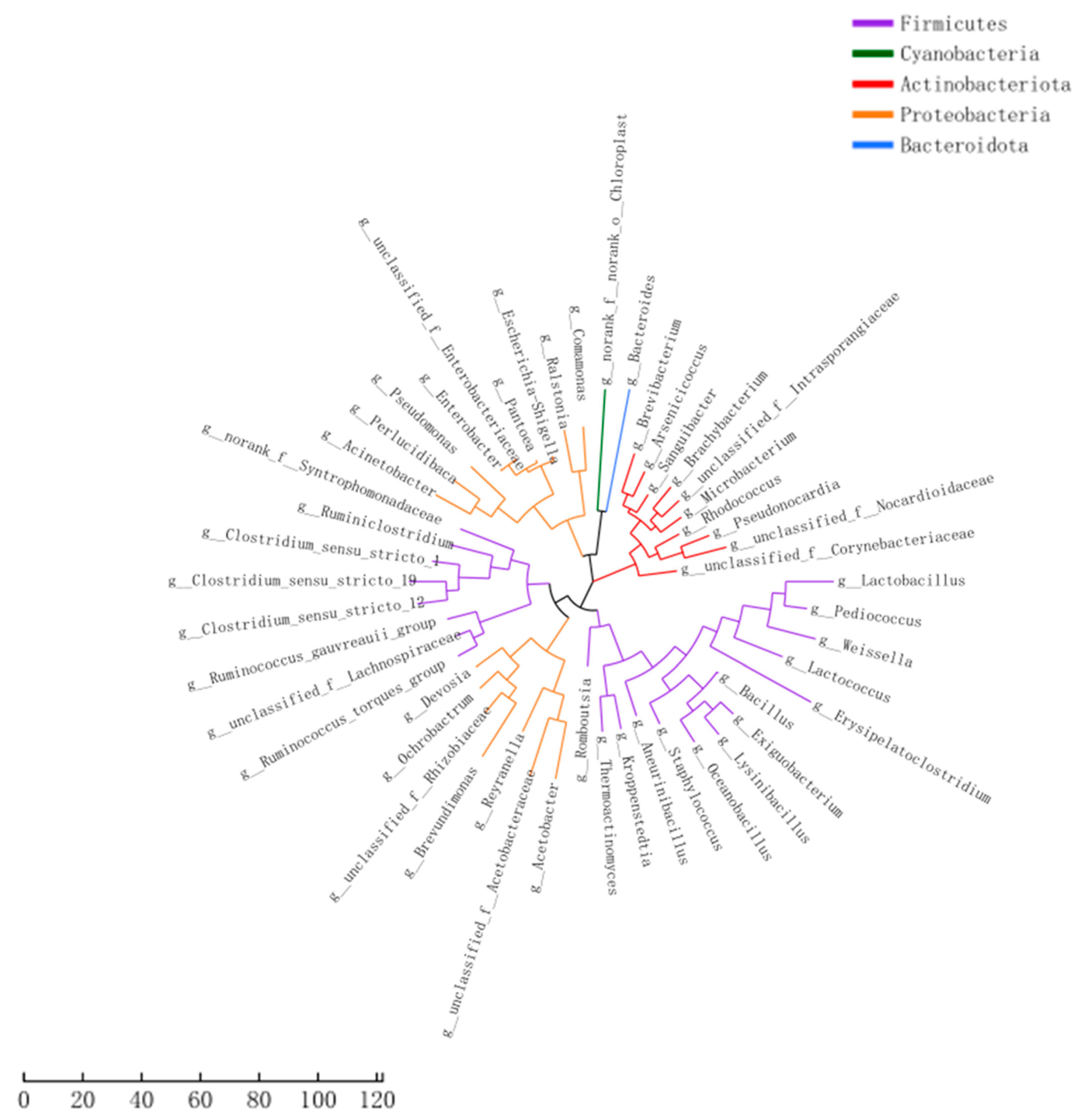
3.3. Construction of Phylogenetic Evolutionary Trees
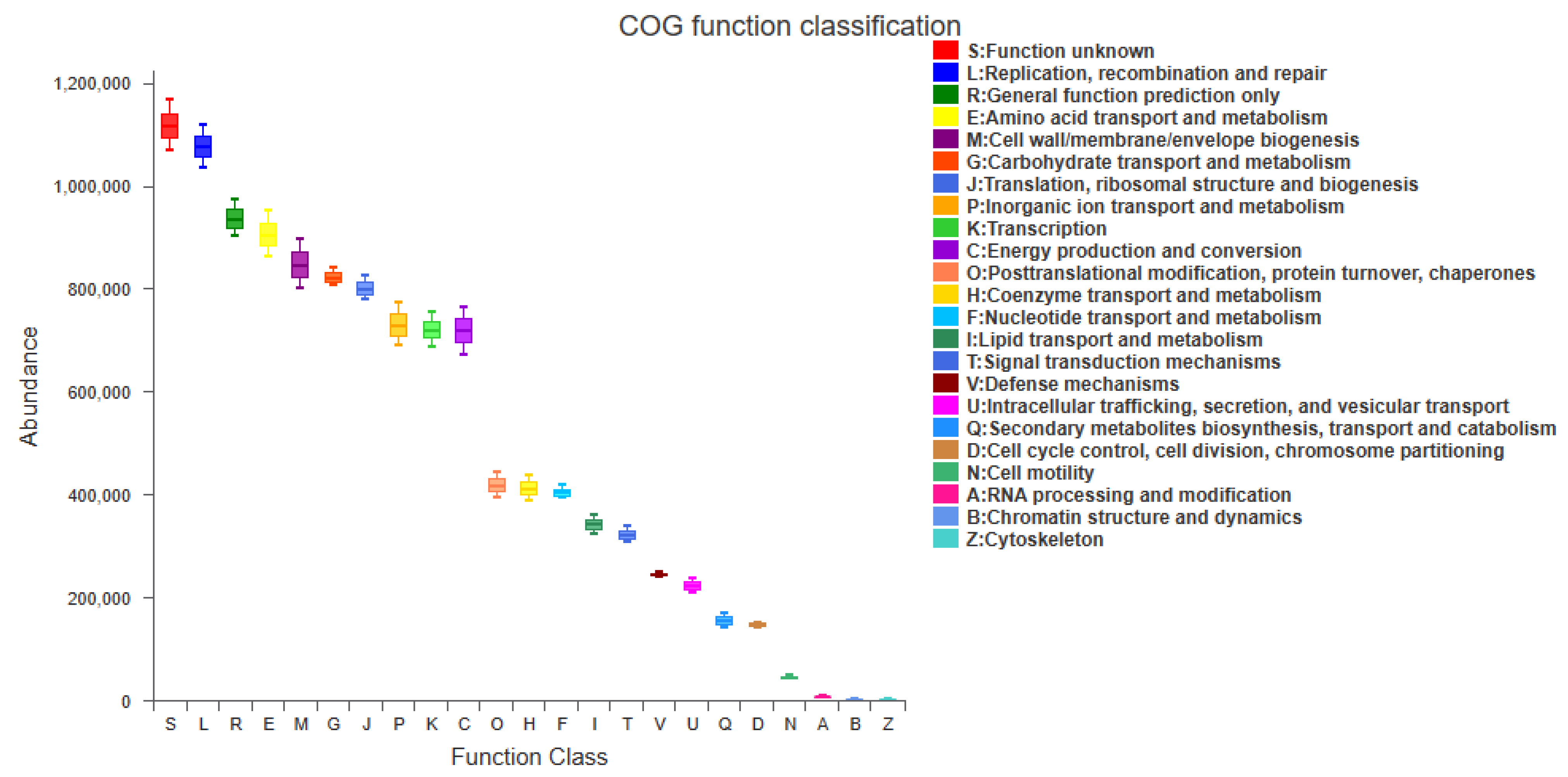
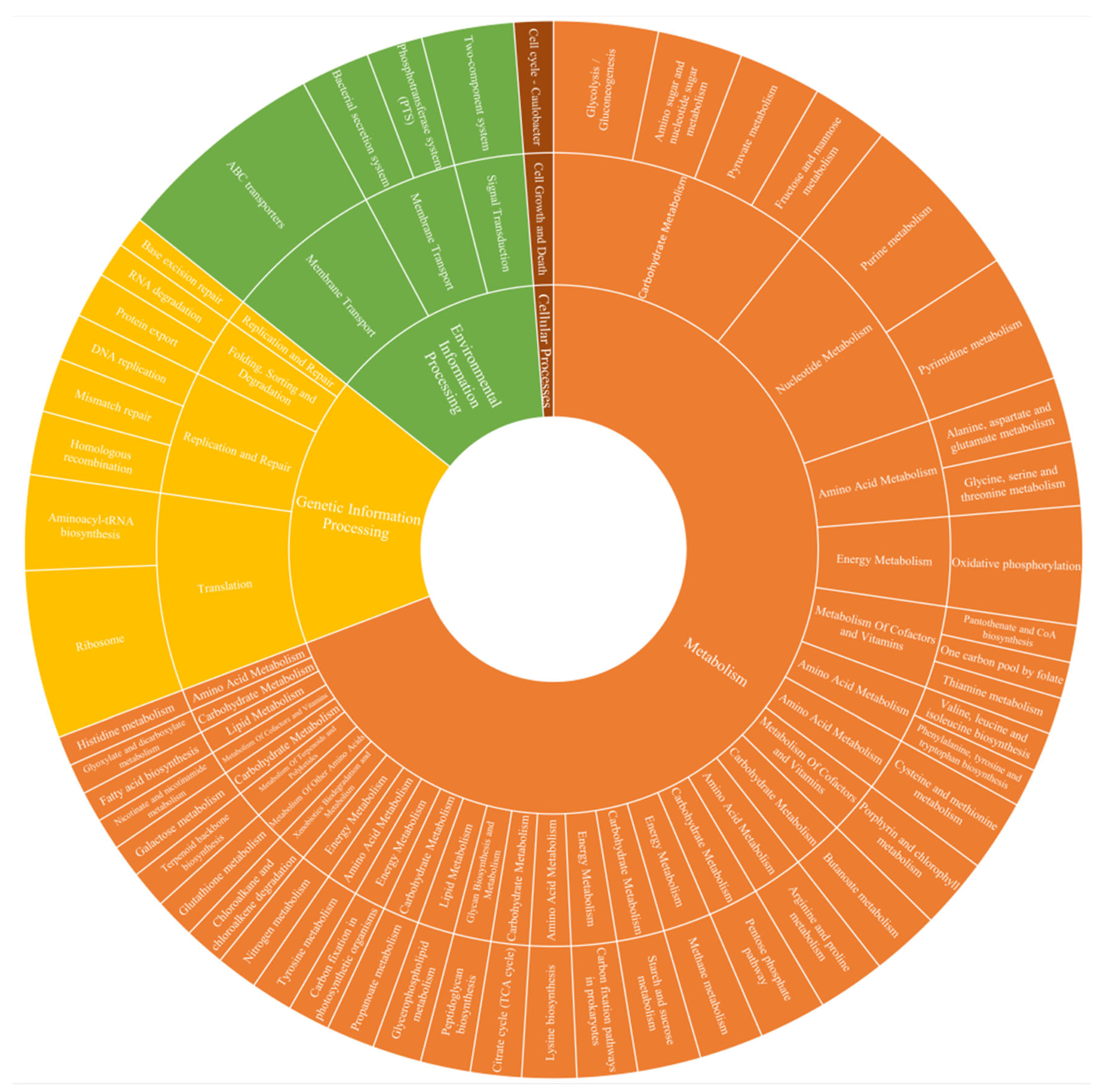
3.4. Functional Predictive Analysis
3.5. Analysis of Culturomics Results of Bacteria
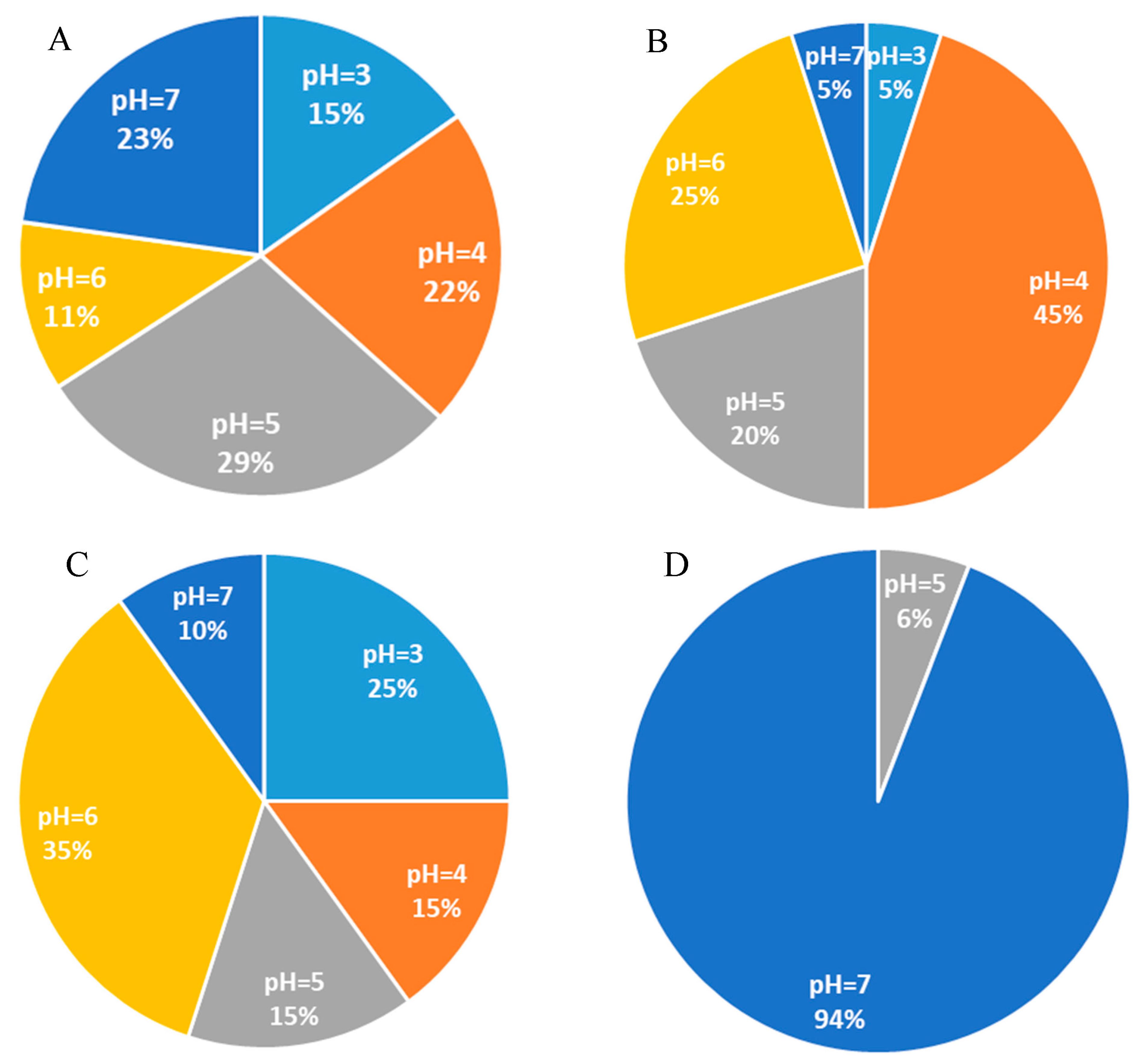
3.6. Correlation Analysis between Dominant Bacteria and Culture Conditions
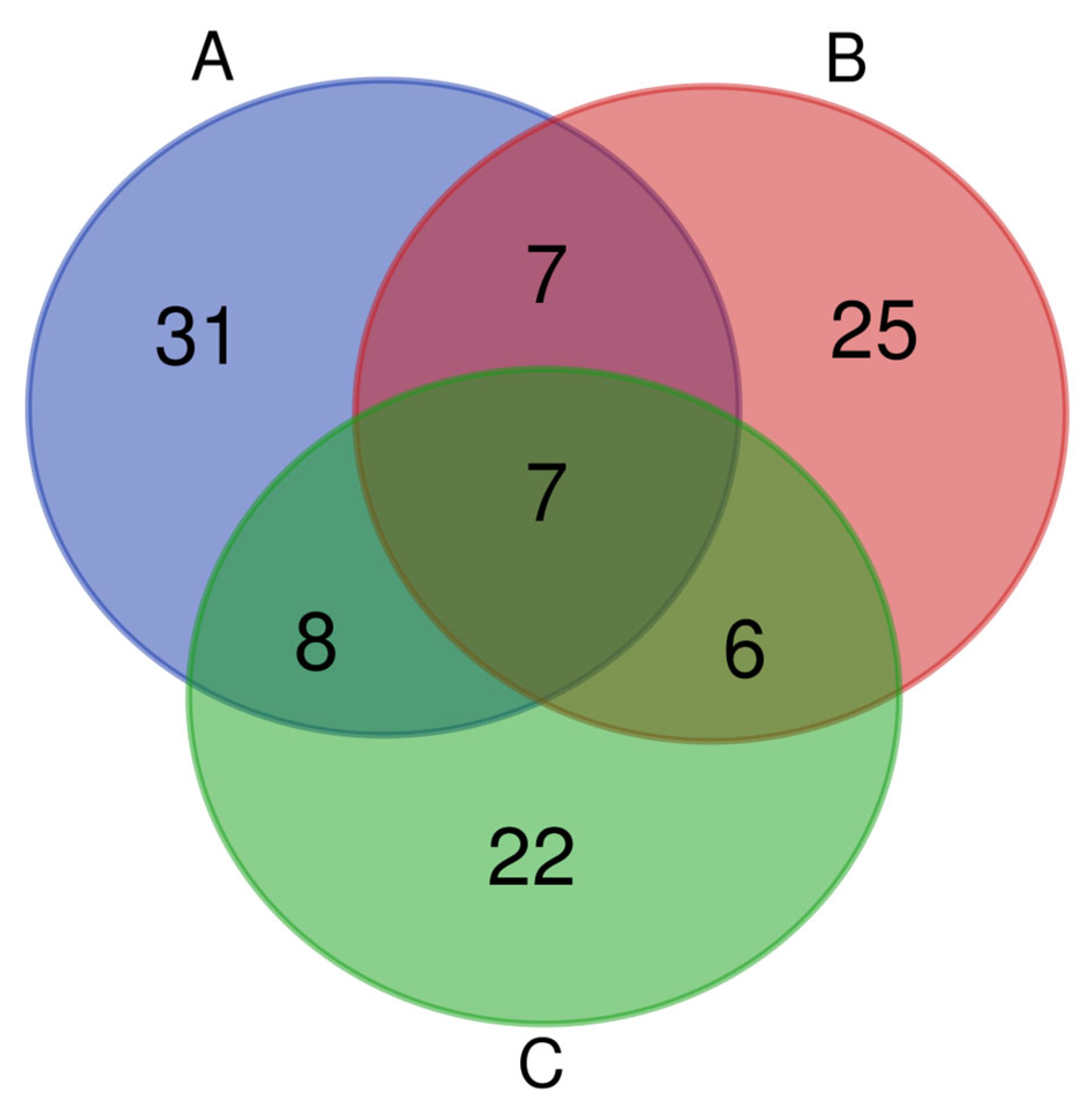
3.7. Comparison of Bacterial Differences in Fermented Grains Cultured by Culturomic, Amplicon Sequencing and Traditional Culture Method at the Genus Level
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tu, W.; Cao, X.; Cheng, J.; Li, L.; Zhang, T.; Wu, Q.; Xiang, P.; Shen, C.; Li, Q. Chinese Baijiu: The Perfect Works of Microorganisms. Front. Microbiol. 2022, 13, 919044. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.M.; Ren, C.; Nie, Y.; Xu, Y. Cultivation strategy for unculturable microbiota in pit mud involved in strong-flavor Baijiu fermentation. Food Ferment. Ind. 2020, 46, 9–16. [Google Scholar]
- Xia, Y.; Luo, H.; Wu, Z.; Zhang, W. Microbial diversity in jiuqu and its fermentation features: Saccharification, alcohol fermentation and flavors generation. Appl. Microbiol. Biotechnol. 2023, 107, 25–41. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Shang, Z.; Chen, J.; Shen, Y.; Li, Z.; Huang, D.; Luo, H. Differences in structure, volatile metabolites, and functions of microbial communities in Nongxiangxing daqu from different production areas. LWT 2022, 166, 113784. [Google Scholar] [CrossRef]
- Hu, X.; Tian, R.; Wang, K.; Cao, Z.; Yan, P.; Li, F.; Li, X.; Li, S.; He, P. The prokaryotic community, physicochemical properties and flavors dynamics and their correlations in fermented grains for Chinese strong-flavor Baijiu production. Food Res. Int. 2021, 148, 110626. [Google Scholar] [CrossRef]
- Jiao, W.; Xie, F.; Gao, L.; Du, L.; Wei, Y.; Zhou, J.; He, G. Identification of core microbiota in the fermented grains of a Chinese strong-flavor liquor from Sichuan. LWT 2022, 158, 113140. [Google Scholar] [CrossRef]
- Zhang, S.; Miu, L.H.; Zhang, M.C.; Liu, P.L.; Liao, W.F. Isolation and identification of lactic acid bacteria and growth characteristic of Lactobacillus buchneri in fermented grains of strong-sauce-flavor Baijiu. China Brew. 2020, 39, 46–50. [Google Scholar]
- Liu, F.; Qiu, Y.; Zhou, X.; Chen, X.; Li, Z.; Chen, J. The correlation between organic acid producing bacteria and organic acids biosynthesis in fermented grains of Yanghe strong-aroma spirit. Food Ferment. Ind. 2018, 44, 22–29. [Google Scholar]
- Liu, M.K.; Tang, Y.M.; Xiong, H.; Liu, Y.; Jiang, P.; Ren, D.Q.; Tian, X.H.; Yao, W.C. Characterization of the diversity and activity of cellulose-degrading bacteria in Zaopei used for Chinese Baijiu production. Food Ferment. Ind. 2018, 44, 35–41. [Google Scholar]
- Dou, X.; Han, P.; Liu, L.; Zhang, Y.; He, J.; Zhuo, X.; Wu, Y.Y.; Bai, F.Y.; Yang, J.G. Study on isolation and identification and population succession law of bacterial in fermented grains during the brewing of Luzhou-flavour Liquor. Sci. Technol. Food Ind. 2017, 38, 169–174. [Google Scholar]
- Li, H.; Huang, J.; Liu, X.; Zhou, R.; Ding, X.; Xiang, Q.; Zhang, L.; Wu, C. Characterization of Interphase Microbial Community in Luzhou-Flavored Liquor Manufacturing Pits of Various Ages by Polyphasic Detection Methods. J. Microbiol. Biotechnol. 2017, 27, 130–140. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; She, M.; Liu, K.; Zhang, Z.; Shuang, Q. Evaluation of the Bacterial Diversity of Inner Mongolian Acidic Gruel Using Illumina MiSeq and PCR-DGGE. Curr. Microbiol. 2020, 77, 434–442. [Google Scholar] [CrossRef]
- Zhao, H.W.; Yan, P.M. Analysis of Microbial Diversity of Yeast in Pickled Chinese Cabbage by PCR-DGGE Method. China Condiment 2020, 45, 51–54. [Google Scholar]
- Ling, Y.; Li, W.; Tong, T.; Li, Z.; Li, Q.; Bai, Z.; Wang, G.; Chen, J.; Wang, Y. Assessing the Microbial Communities in Four Different Daqus by Using PCR-DGGE, PLFA, and Biolog Analyses. Pol. J. Microbiol. 2020, 69, 27–37. [Google Scholar] [CrossRef] [Green Version]
- Sun, W.; Xiao, H.; Peng, Q.; Zhang, Q.; Li, X.; Han, Y. Analysis of bacterial diversity of Chinese Luzhou-flavor liquor brewed in different seasons by Illumina Miseq sequencing. Ann. Microbiol. 2016, 66, 1293–1301. [Google Scholar] [CrossRef]
- Du, R.B. Quantitation analysis and metabolic profiling of lactic acid bacteria in sesame-flavor liquor fermentation. Master’s Thesis, Jiangnan University, Wuxi, China, 2019; p. 56. [Google Scholar]
- Zhan, C.X. Study on the Structure Characteristics and Variation for Microbial Community in Daqu and Fermented Grains of Jiang-flavour Chinese Spirits Production. Master’s Thesis, Guizhou University, Guiyang, China, 2021; p. 87. [Google Scholar]
- Lagier, J.-C.; Armougom, F.; Million, M.; Hugon, P.; Pagnier, I.; Robert, C.; Bittar, F.; Fournous, G.; Gimenez, G.; Maraninchi, M.; et al. Microbial culturomics: Paradigm shift in the human gut microbiome study. Clin. Microbiol. Infect. 2012, 18, 1185–1193. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Sun, L.; Xing, X.; Sun, Z.; Gu, H.; Lu, X.; Li, Z.; Ren, Q. Culturing Bacteria from Fermentation Pit Muds of Baijiu with Culturomics and Amplicon-Based Metagenomic Approaches. Front. Microbiol. 2020, 11, 1223. [Google Scholar] [CrossRef]
- Huang, Z.Q.; Qiu, J.X.; Li, J.; Dp Xu, L.Q. Exploration of microbial diversity based on 16S rRNA gene sequence analysis. Acta Microbiol. Sin. 2020, 61, 1044–1063. [Google Scholar]
- Magoč, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef] [Green Version]
- Lagier, J.-C.; Hugon, P.; Khelaifia, S.; Fournier, P.-E.; La Scola, B.; Raoult, D. The rebirth of culture in microbiology through the example of culturomics to study human gut microbiota. Clin. Microbiol. Rev. 2015, 28, 237–264. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Yoon, S.-H.; Ha, S.-M.; Kwon, S.; Lim, J.; Kim, Y.; Seo, H.; Chun, J. Introducing EzBioCloud: A taxonomically united database of 16S rRNA gene sequences and whole-genome assemblies. Int. J. Syst. Evol. Microbiol. 2017, 67, 1613–1617. [Google Scholar] [CrossRef] [PubMed]
- Lagier, J.-C.; Dubourg, G.; Million, M.; Cadoret, F.; Bilen, M.; Fenollar, F.; Levasseur, A.; Rolain, J.-M.; Fournier, P.-E.; Raoult, D. Culturing the human microbiota and culturomics. Nat. Rev. Microbiol. 2018, 16, 540–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goris, J.; Konstantinidis, K.T.; Klappenbach, J.A.; Coenye, T.; Vandamme, P.; Tiedje, J.M. DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int. J. Syst. Evol. Microbiol. 2007, 57, 81–91. [Google Scholar] [CrossRef] [Green Version]
- Jiao, J.-Y.; Liu, L.; Hua, Z.-S.; Fang, B.-Z.; Zhou, E.-M.; Salam, N.; Hedlund, B.P.; Li, W.-J. Microbial dark matter coming to light: Challenges and opportunities. Natl. Sci. Rev. 2020, 8, nwaa280. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.S.; Roberts, N.; Singleton, F.L.; Attwell, R.W.; Grimes, D.J.; Colwell, R.R. Survival and viability of nonculturable Escherichia coli and Vibriocholerae in the estuarine and marine environment. Microb. Ecol. 1982, 8, 313–323. [Google Scholar] [CrossRef]
- Lewis, W.H.; Tahon, G.; Geesink, P.; Sousa, D.Z.; Ettema, T.J.G. Innovations to culturing the uncultured microbial majority. Nature Rev. Microbiology 2020, 19, 225–240. [Google Scholar] [CrossRef]
- Ren, Q.; Chen, H.; Sun, Z.; Guo, L.; Dai, F.; Xu, J.; Zhang, W. Umezawaea beigongshangensis sp. nov., Isolated from the Mash of Baijiu. Curr. Microbiol. 2021, 78, 4127–4131. [Google Scholar] [CrossRef]
- Sun, Z.; Dai, F.; Yan, Y.; Guo, L.; Gu, H.; Xu, J.; Ren, Q. Pseudoxanthomonas beigongshangi sp. nov. a novel bacteria with predicted nitrite and nitrate reduce ability isolated from pit mud of Baijiu. Antonie Leeuwenhoek 2021, 114, 1307–1314. [Google Scholar] [CrossRef]
- Sun, Z.; Guo, L.; Yan, Y.; Zhang, X.; Wang, J.; Liu, B.; Xu, J.; Ren, Q. Sporosarcina beigongshangi sp. nov., isolated from pit mud of Baijiu. Arch. Microbiol. 2021, 204, 10. [Google Scholar] [CrossRef]
- Zhu, X.; Zhou, Y.; Wang, Y.; Wu, T.; Li, X.; Li, D.; Tao, Y. Production of high-concentration n-caproic acid from lactate through fermentation using a newly isolated Ruminococcaceae bacterium CPB6. Biotechnol. Biofuels 2017, 10, 102. [Google Scholar] [CrossRef] [Green Version]
- Tao, Y.; Hu, X.; Zhu, X.; Jin, H.; Xu, Z.; Tang, Q.; Li, X. Production of Butyrate from Lactate by a Newly Isolated Clostridium sp. BPY5. Appl. Biochem. Biotechnol. 2016, 179, 361–374. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number | Medium Name (Abbreviation) | Complementary Ingredient | Type of Medium |
|---|---|---|---|
| 1 | MRS Agar (MRS) | 200U/plate Catalase solution | Selective culture medium |
| 2 | Actinomycetes Culture Medium (ACM) | -- | Selective culture medium |
| 3 | Tryptone Soy Agar (TSA) | Vitamin K | Non-selective culture medium |
| 4 | R2A Agar (R2A) | 5% sterile defibrillated sheep blood | Non-selective culture medium |
| 5 | ISP Medium 2 (ISP2) | -- | Selective culture medium |
| 6 | ISP Medium 3 (ISP3) | -- | Selective culture medium |
| 7 | In-situ Simulation Medium (ISM) | Meat extracts, Peptone, NaCl, Glucose | In situ simulated culture medium |
| 8 | Lactobacillus Selective Agar (LSA) | 200U/plate Catalase solution | Selective culture medium |
| 9 | Nutrient Agar (NA) | -- | Non-selective culture medium |
| Sample Name | Sequence Number | Base Number | Mean Length | Minimum Sequence Length | Longest Sequence Length |
|---|---|---|---|---|---|
| Fermented grains 1 | 48430 | 20354273 | 420.282325 | 277 | 450 |
| Fermented grains 2 | 46219 | 19488183 | 421.648738 | 287 | 478 |
| Fermented grains 3 | 48407 | 20390609 | 421.232652 | 277 | 452 |
| Number | Genus Name | Specific Epithet |
|---|---|---|
| 1 | Bacillus | B. aerius |
| B. aerophilus | ||
| B. altitudinis | ||
| B. anthracis | ||
| B. subtilis | ||
| B. pumilus | ||
| B. oleronius | ||
| B. megaterium | ||
| B. licheniformis | ||
| B. safensis | ||
| B. cereus | ||
| B. kochii | ||
| B. aryabhattai | ||
| B. simplex | ||
| B. toyonensis | ||
| B. methylotrophicus | ||
| B. idriensis | ||
| B. siamensis | ||
| B. amyloliquefaciens | ||
| B. velezensis | ||
| B. australimaris | ||
| B. infantis | ||
| B. marisflavi | ||
| B. zhangzhouensis | ||
| B. invictae | ||
| B. thuringiensis | ||
| B. stratosphericus | ||
| 2 | Microbacterium | M. paraoxydans |
| M. paraoxydans | ||
| M. testaceum | ||
| M.trichothecenolyticum | ||
| M. resistens | ||
| M. esteraromaticum | ||
| M. ginsengisoli | ||
| 3 | Acetobacter | A. oryzoeni |
| A. pasteurianus | ||
| 4 | Brevundimonas | B. diminuta |
| B. aurantiaca | ||
| B. faecalis | ||
| B. albigilva | ||
| B. vancanneytii | ||
| 5 | Pseudomonas | P. fluorescens |
| P. oryzihabitans | ||
| P. hibiscicola | ||
| 6 | Ralstonia | R. pickettii |
| 7 | Enterobacter | E. hormaechei |
| E. cloacae | ||
| 8 | Rhizobium | R. radiobacter |
| 9 | Variovorax | V. paradoxus |
| 10 | Stenotrophomonas | S. maltophilia |
| 11 | Paracoccus | P. yeei |
| P. chinensis | ||
| P. salipaludis | ||
| 12 | Providencia | P. rettgeri |
| 13 | Micrococcus | M. flavus |
| M. luteus | ||
| 14 | Brevibacterium | B. frigoritolerans |
| B. casei | ||
| B. aureum | ||
| 15 | Moraxella | M. osloensis |
| 16 | Sphingomonas | S. aquatilis |
| S. melonis | ||
| S. carotinifaciens | ||
| 17 | Cupriavidus | C. metallidurans |
| 18 | Ochrobactrum | O. anthropi |
| 19 | Staphylococcus | S. hominis |
| S. gallinarum | ||
| S. haemolyticus | ||
| S. capitis | ||
| 20 | Nocardioides | N. exalbidus |
| 21 | Lysinibacillus | L. sphaericus |
| L. massiliensis | ||
| L. boronitolerans | ||
| 22 | Gordonia | G. rubripertincta |
| 23 | Streptomyces | S. griseoruber |
| S. scabrisporus | ||
| 24 | Acinetobacter | A. lwoffii |
| 25 | Kocuria | K. rosea |
| 26 | Brachybacterium | B. paraconglomeratum |
| 27 | Micromonospora | M. aurantiaca |
| M. echinospora | ||
| 28 | Klebsiella | K. aerogenes |
| 29 | Methylobacterium | M. extorquens |
| M. hispanicum | ||
| M. aquaticum | ||
| 30 | Rhodococcus | R. yunnanensis |
| 31 | Tsukamurella | T. tyrosinosolvens |
| 32 | Alcaligenes | A. faecalis |
| 33 | Enterococcus | E. gallinarum |
| 34 | Paenibacillus | P. barengoltzii |
| P. alvei | ||
| 35 | Neobacillus | N. mesonae |
| 36 | Serratia | S. marcescens |
| 37 | Rummeliibacillus | R. suwonensis |
| R. stabekisii | ||
| R. pycnus | ||
| 38 | Novosphingobium | N. panipatense |
| 39 | Pseudochrobactrum | P. asaccharolyticum |
| 40 | Sphingobacterium | S. daejeonense |
| 41 | Bosea | B. eneae |
| 42 | Chryseobacterium | C. taklimakanense |
| C. hominis | ||
| 43 | Paraburkholderia | P. fungorum |
| 44 | Agrobacterium | A. deltaense |
| 45 | Pigmentiphaga | P. kullae |
| 46 | Brevibacillus | B. choshinensis |
| B. nitrificans |
| Strain Number | Genus Name | Closest Type Species | BLAST Similarity | EZ Similarity |
|---|---|---|---|---|
| H9 | Sphingomonas | S. aquatilis | 100 | 98.34 |
| H18 | Microbacterium | M. proteolyticum | 99.93 | 98.86 |
| H14 | Microbacterium | M. proteolyticum | 100 | 98.93 |
| H56 | Curvibacter | C.lanceolatus | 98.39 | 98.19 |
| HC15 | Microvirga | M. indica | 99.41 | 98.38 |
| HD21 | Xanthobacter | X. flavus | 96.95 | 96.89 |
| HC47 | Xanthobacter | X. flavus | 96.96 | 96.85 |
| HD30 | Alcanivorax | A. pacificus | 96.45 | 96.10 |
| HC49 | Aneurinibacillus | A. aneurinilyticus | 98.74 | 98.34 |
| HD31 | Cohnella | C. nanjingensis | 98.04 | 97.99 |
| HD23B | Alcanivorax | A. pacificus | 96.73 | 96.58 |
| H9-1 | Lysinibacillus | L. macroides | 98.33 | 97.39 |
| HD59 | Altererythrobacter | A. terrae | 98.9 | 99.49 |
| HS3 | Microbacterium | M. pseudoresistens | 98.24 | 98.17 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, J.; Hao, S.; Ren, Q. Analysis of Bacterial Diversity in Fermented Grains of Baijiu Based on Culturomics and Amplicon Sequencing. Fermentation 2023, 9, 260. https://doi.org/10.3390/fermentation9030260
Wang J, Hao S, Ren Q. Analysis of Bacterial Diversity in Fermented Grains of Baijiu Based on Culturomics and Amplicon Sequencing. Fermentation. 2023; 9(3):260. https://doi.org/10.3390/fermentation9030260
Chicago/Turabian StyleWang, Jiaxuan, Shuyue Hao, and Qing Ren. 2023. "Analysis of Bacterial Diversity in Fermented Grains of Baijiu Based on Culturomics and Amplicon Sequencing" Fermentation 9, no. 3: 260. https://doi.org/10.3390/fermentation9030260
APA StyleWang, J., Hao, S., & Ren, Q. (2023). Analysis of Bacterial Diversity in Fermented Grains of Baijiu Based on Culturomics and Amplicon Sequencing. Fermentation, 9(3), 260. https://doi.org/10.3390/fermentation9030260