
A Benchmark for Atlantic Salmon Conservation: Genetic Diversity and Structure in a Southern European Glacial Refuge before the Climate Changed

 ,
,  ,
,  , , and
, , and 
Abstract
:1. Introduction
2. Materials and Methods
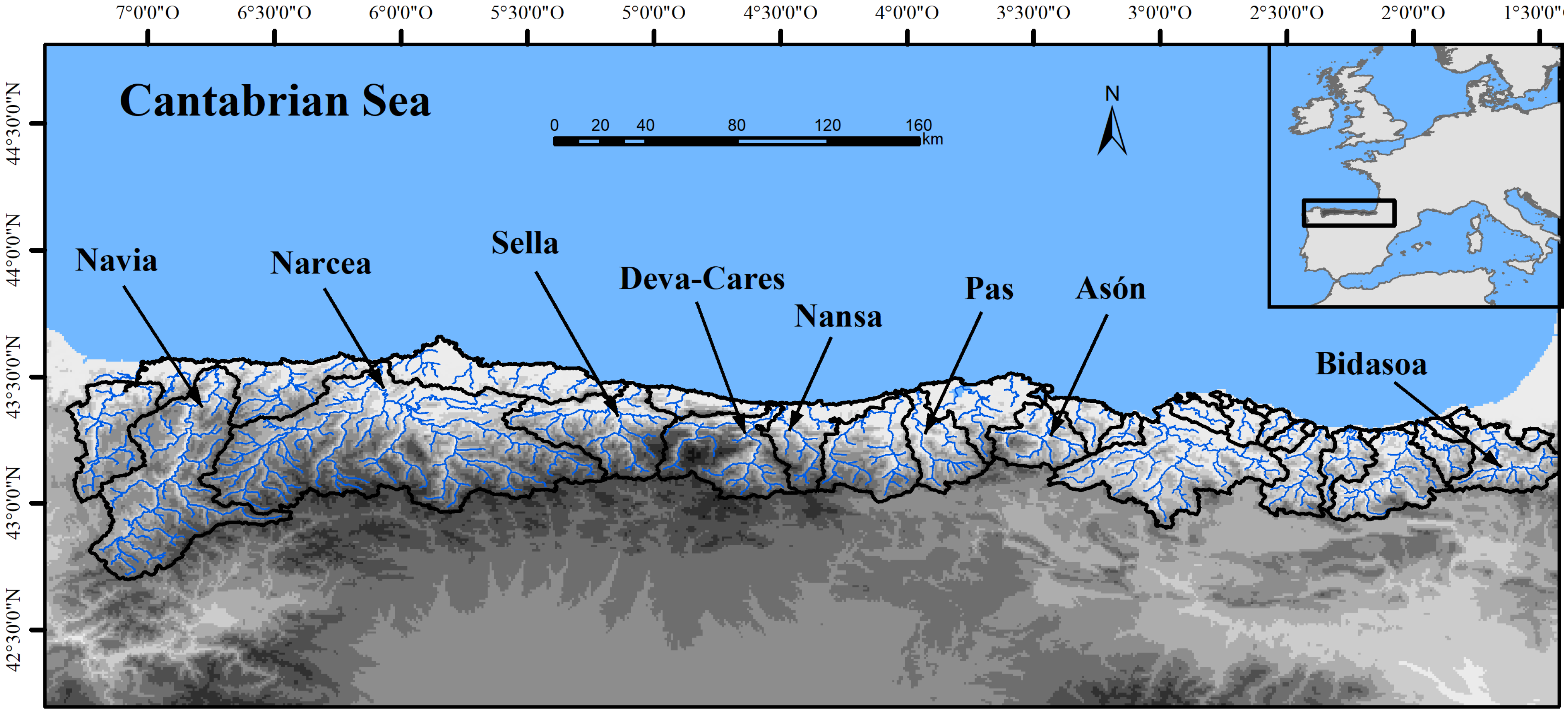
2.1. Study Area
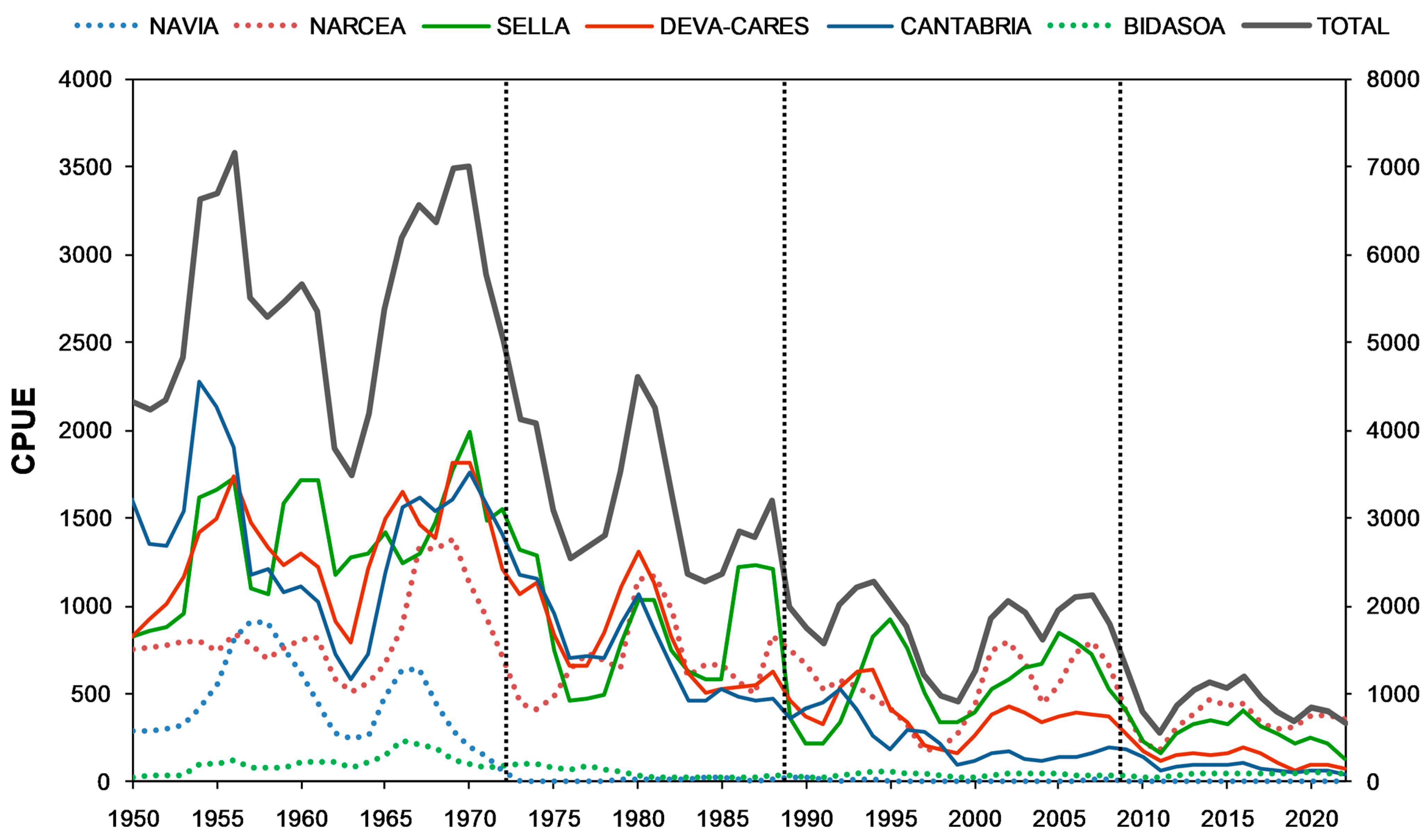
2.2. Trends in Population Abundance
2.3. DNA Extraction and Microsatellite Genotyping
2.4. Genetic Diversity
2.5. Connectivity and Population Structure
2.6. Demographic Parameters
3. Results
3.1. Long-Term Trends in Population Abundance
3.2. Genetic Diversity
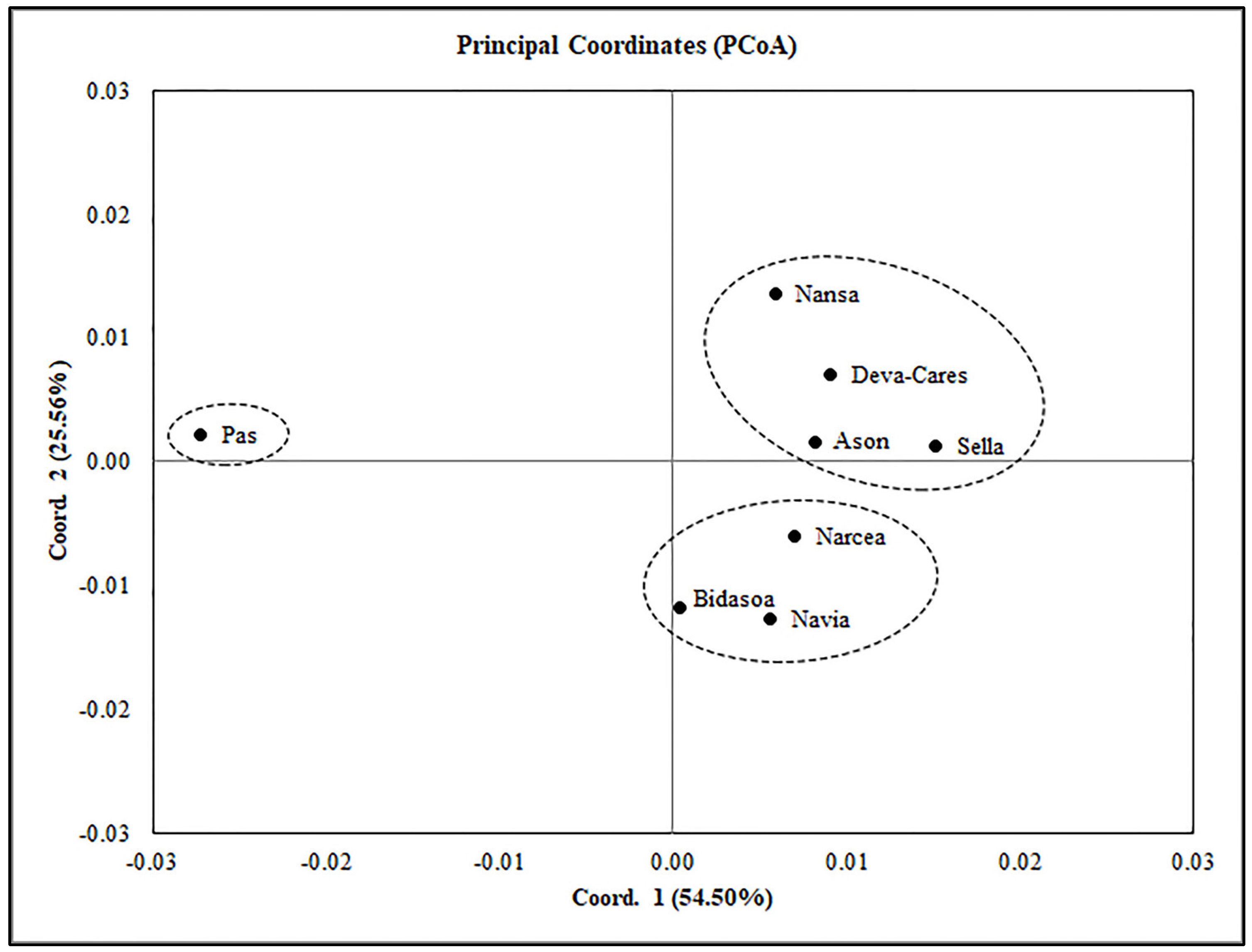
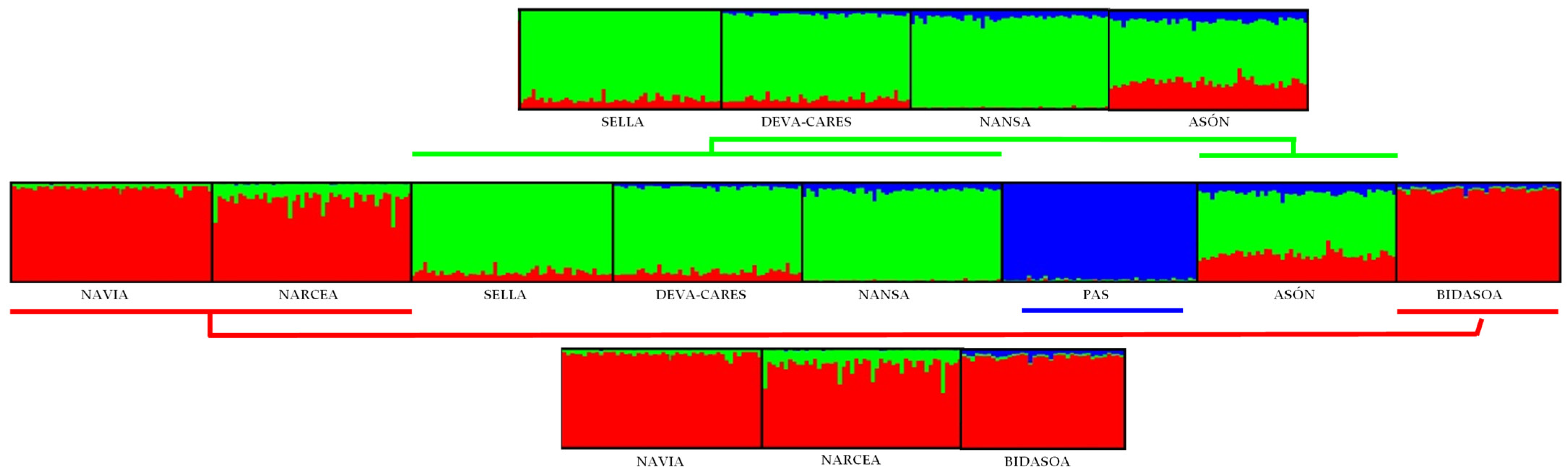
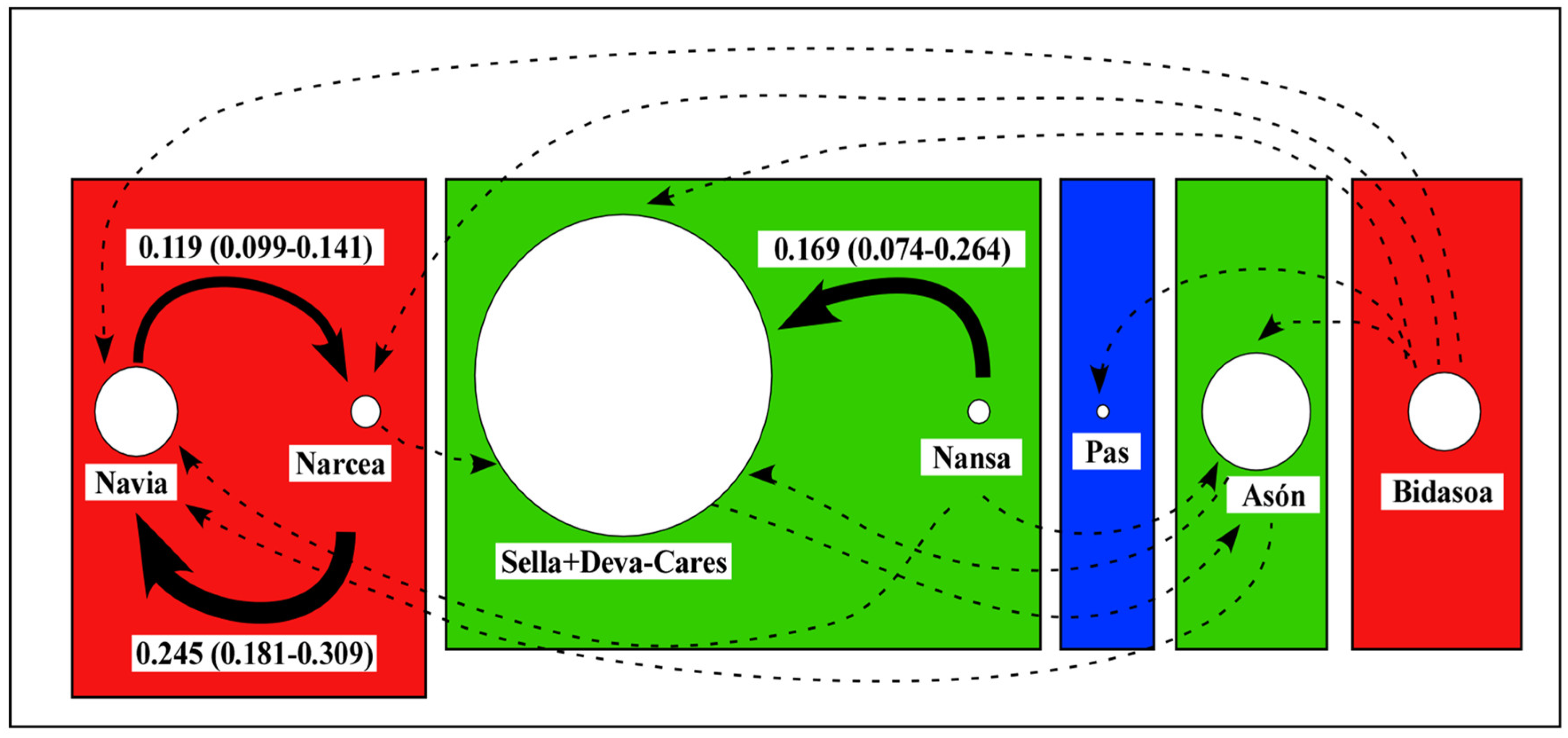
3.3. Connectivity and Population Structure
3.4. Demographic Parameters
4. Discussion
4.1. Genetic Diversity
4.2. Connectivity and Population Structure
4.3. Demographic Parameters
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schindler, D.E.; Hilborn, R.; Chasco, B.; Boatright, C.P.; Quinn, T.P.; Rogers, L.A.; Webster, M.S. Population diversity and the portfolio effect in an exploited species. Nature 2010, 465, 609–612. [Google Scholar] [CrossRef] [PubMed]
- Hoban, S.; Archer, F.I.; Bertola, L.D.; Bragg, J.G.; Breed, M.F.; Bruford, M.W.; Hunter, M.E. Global genetic diversity status and trends: Towards a suite of Essential Biodiversity Variables (EBVs) for genetic composition. Biol. Rev. 2022, 97, 1511–1538. [Google Scholar] [CrossRef]
- Luck, G.W.; Daily, G.C.; Ehrlich, P.R. Population diversity and ecosystem services. Trends Ecol. Evol. 2003, 18, 331–336. [Google Scholar] [CrossRef] [Green Version]
- Frankham, R. Genetics and extinction. Biol. Conserv. 2005, 126, 131–140. [Google Scholar] [CrossRef]
- Reusch, T.B.H.; Ehlers, A.; Hämmerli, A.; Worm, B. Ecosystem recovery after climatic extremes enhanced by genotypic diversity. Proc. Natl. Acad. Sci. USA 2005, 102, 2826–2831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dadswell, M.; Spares, A.; Reader, J.; McLean, M.; McDermott, T.; Samways, K.; Lilly, J. The decline and impending collapse of the Atlantic Salmon (Salmo salar) population in the North Atlantic Ocean: A review of possible causes. Rev. Fish. Sci. Aquac. 2021, 30, 215–258. [Google Scholar] [CrossRef]
- Thorstad, E.B.; Bliss, D.; Breau, C.; Damon-Randall, K.; Sundt-Hansen, L.E.; Hatfield, E.M.; Sutton, S.G. Atlantic salmon in a rapidly changing environment. Facing the challenges of reduced marine survival and climate change. Aquat. Conserv. Mar. Freshw. Ecosyst. 2021, 31, 2654–2665. [Google Scholar] [CrossRef]
- Nieto, A.; Ralph, G.M.; Comeros-Raynal, M.T.; Kemp, J.; García-Criado, M.; Allen, D.J.; Williams, J.T. European Red List of Marine Fishes; Publications Office of the European Union: Luxembourg, 2015. [Google Scholar]
- Perrier, C.; April, J.; Cote, G.; Bernatchez, L.; Dionne, M. Effective number of breeders in relation to census size as management tools for Atlantic salmon conservation in a context of stocked populations. Conserv. Genet. 2016, 17, 31–44. [Google Scholar] [CrossRef]
- Nicola, G.G.; Elvira, B.; Jonsson, B.; Ayllón, D.; Almodóvar, A. Local and global climatic drivers of Atlantic salmon decline in southern Europe. Fish Res. 2018, 198, 78–85. [Google Scholar] [CrossRef]
- Almodóvar, A.; Ayllón, D.; Nicola, G.G.; Jonsson, B.; Elvira, B. Climate-driven bio-physical changes in feeding and breeding environments explain the decline of southernmost European Atlantic salmon populations. Can. J. Fish. Aquat. Sci. 2019, 76, 1581–1595. [Google Scholar] [CrossRef] [Green Version]
- Almodóvar, A.; Leal, S.; Nicola, G.G.; Hórreo, J.L.; García-Vázquez, E.; Elvira, B. Long-term stocking practices threaten the original genetic diversity of the southernmost European populations of Atlantic salmon Salmo salar. Endanger. Species Res. 2020, 41, 303–317. [Google Scholar] [CrossRef]
- Gallagher, B.K.; Geargeoura, S.; Fraser, D.J. Effects of climate on salmonid productivity: A global meta-analysis across freshwater ecosystems. Glob. Chang. Biol. 2022, 28, 7250–7269. [Google Scholar] [CrossRef]
- Almodóvar, A.; Nicola, G.G.; Ayllón, D.; Trueman, C.N.; Davidson, I.; Kennedy, R.; Elvira, B. Stable isotopes suggest the location of marine feeding grounds of south European Atlantic salmon in Greenland. ICES J. Mar. Sci. 2020, 77, 593–603. [Google Scholar] [CrossRef]
- Rikardsen, A.H.; Righton, D.; Strøm, J.F.; Thorstad, E.B.; Gargan, P.; Sheehan, T.; Aarestrup, K. Redefining the oceanic distribution of Atlantic salmon. Sci. Rep. 2021, 11, 12266. [Google Scholar] [CrossRef] [PubMed]
- Hórreo, J.L.; de la Hoz, J.; Machado-Schiaffino, G.; Pola, I.G.; García-Vázquez, E. Restoration and enhancement of Atlantic salmon populations: What we have learned from North Iberian rivers. Knowl. Manag. Aquat. Ecosyst. 2011, 402, 23. [Google Scholar] [CrossRef]
- Ribeiro, A.; Morán, P.; Caballero, A. Genetic diversity and effective size of the Atlantic salmon Salmo salar L. inhabiting the River Eo (Spain) following a stock collapse. J. Fish Biol. 2008, 72, 1933–1944. [Google Scholar] [CrossRef]
- Consuegra, S.; Verspoor, E.; Knox, D.; García de Leániz, C. Asymmetric gene flow and the evolutionary maintenance of genetic diversity in small, peripheral Atlantic salmon populations. Conserv. Genet. 2005, 6, 823–842. [Google Scholar] [CrossRef]
- Ciborowski, K.; Jordan, W.C.; García de Leániz, C.; Consuegra, S. Temporal and spatial instability in neutral and adaptive (MHC) genetic variation in marginal salmon populations. Sci. Rep. 2017, 7, 42416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayllón, F.; Martínez, J.L.; García-Vázquez, E. Loss of regional population structure in Atlantic salmon, Salmo salar L. following stocking. ICES J. Mar. Sci. 2006, 63, 1269–1273. [Google Scholar] [CrossRef] [Green Version]
- Hórreo, J.L.; Machado-Schiaffino, G.; Ayllón, F.; Griffiths, A.M.; Bright, D.; Stevens, J.R.; García-Vázquez, E. Impact of climate change and human-mediated introgression on southern European Atlantic salmon populations. Glob. Chang. Biol. 2011, 17, 1778–1787. [Google Scholar] [CrossRef]
- Blanco, G.; Ramos, M.D.; Vázquez, E.; Sánchez, J.A. Assessing temporal and spatial variation in wild populations of Atlantic salmon with particular reference to Asturias (Northern Spain) rivers. J. Fish Biol. 2005, 67, 169–184. [Google Scholar] [CrossRef]
- Campos, J.L.; Posada, D.; Morán, P. Introgression and genetic structure in northern Spanish Atlantic salmon (Salmo salar L.) populations according to mtDNA data. Conserv. Genet. 2008, 9, 157–169. [Google Scholar] [CrossRef]
- Palstra, F.P.; O’Connell, M.F.; Ruzzante, D.E. Population structure and gene flow reversals in Atlantic salmon (Salmo salar) over contemporary and long-term temporal scales: Effects of population size and life history. Mol. Ecol. 2007, 16, 4504–4522. [Google Scholar] [CrossRef]
- Kaczmarczyk, D. Techniques based on the polymorphism of microsatellite DNA as tools for conservation of endangered populations. Appl. Ecol. Environ. Res. 2019, 17, 1599–1615. [Google Scholar] [CrossRef]
- Wenne, R. Microsatellites as molecular markers with applications in exploitation and conservation of aquatic animal populations. Genes 2023, 14, 808. [Google Scholar] [CrossRef]
- Gilbey, J.; Coughlan, J.; Wennevik, V.; Prodöhl, P.; Stevens, J.R.; García de Leániz, C.; Verspoor, E. A microsatellite baseline for genetic stock identification of European Atlantic salmon (Salmo salar L.). ICES J. Mar. Sci. 2018, 75, 662–674. [Google Scholar] [CrossRef] [Green Version]
- Yue, S.; Wang, C.Y. The Mann-Kendall test modified by effective sample size to detect trend in serially correlated hydrological series. Water Resour. Manag. 2004, 18, 201–218. [Google Scholar] [CrossRef]
- Patakamuri, S.K.; O’Brien, N. Modifiedmk: Modified Versions of Mann Kendall and Spearman’s Rho Trend Tests, R Package Version 1.6. 2022. Available online: https://cran.r-project.org/web/packages/modifiedmk/ (accessed on 9 May 2023).
- Webster, R. Automatic soil-boundary location from transect data. J. Int. Assoc. Math. Geol. 1973, 5, 27–37. [Google Scholar] [CrossRef]
- O’Reilly, P.T.; Hamilton, L.C.; McConnell, S.K.; Wright, J.M. Rapid analysis of genetic variation in Atlantic salmon (Salmo salar) by PCR multiplexing of dinucleotide and tetranucleotide microsatellites. Can. J. Fish. Aquat. Sci. 1996, 53, 2292–2298. [Google Scholar] [CrossRef]
- Slettan, A.; Olsaker, I.; Lie, Ø. Atlantic salmon, Salmo salar, microsatellites at the SSOSL25, SSOSL85, SSOSL311, SSOSL417 loci. Anim. Genet. 1995, 26, 281–282. [Google Scholar] [CrossRef] [PubMed]
- Slettan, A.; Olsaker, I.; Lie, Ø. Polymorphic Atlantic salmon, Salmo salar L. microsatellites at the SSOSL438, SSOSL439 and SSOSL444 loci. Anim. Genet. 1996, 27, 57–58. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, J.A.; Clabby, C.; Ramos, D.; Blanco, G.; Flavin, F.; Vázquez, E.; Powell, R. Protein and microsatellite single locus variability in Salmo salar L. (Atlantic salmon). Heredity 1996, 77, 423–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paterson, S.; Piertney, S.B.; Knox, D.; Gilbey, J.; Verspoor, E. Characterization and PCR multiplexing of novel highly variable tetranucleotide Atlantic salmon (Salmo salar L.) microsatellites. Mol. Ecol. Notes 2004, 4, 160–162. [Google Scholar] [CrossRef]
- Van Oosterhout, C.; Hutchinson, W.F.; Wills, D.P.; Shipley, P. MICRO-CHECKER: Software for identifying and correcting genotyping errors in microsatellite data. Mol. Ecol. Notes 2004, 4, 535–538. [Google Scholar] [CrossRef]
- Dabrowski, M.J.; Pilot, M.; Kruczyk, M.; Zmihorski, M.; Umer, H.M.; Gliwicz, J. Reliability assessment of null allele detection: Inconsistencies between and within different methods. Mol. Ecol. 2014, 14, 361–373. [Google Scholar] [CrossRef] [Green Version]
- Kalinowski, S.T.; Taper, M.L.; Marshall, T.C. Revising how the computer program CERVUS accommodates genotyping error increases success in paternity assignment. Mol. Ecol. 2007, 16, 1099–1106. [Google Scholar] [CrossRef]
- Kalinowski, S.T.; Taper, M.L. Maximum likelihood estimation of the frequency of null alleles at microsatellite loci. Conserv. Genet. 2006, 7, 991–995. [Google Scholar] [CrossRef]
- Rousset, F. GENEPOP’007: A complete reimplementation of the GENEPOP software for Windows and Linux. Mol. Ecol. Resour. 2008, 8, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Belkhir, K.; Borsa, P.; Chikhi, L.; Raufaste, N.; Bonhomme, F. GENETIX 4.05, logiciel sous Windows TM pour la génétique des populations. In Laboratoire Génome, Populations, Interactions, CNRS UMR 5171; Université de Montpellier II: Montpellier, France, 2004. [Google Scholar]
- Weir, B.S.; Cockerham, C.C. Estimating F-statistics for the analysis of population structure. Evolution 1984, 38, 1358–1370. [Google Scholar] [CrossRef]
- Goudet, J. FSTAT (Version 1.2): A computer program to calculate F-statistics. J. Hered. 1995, 86, 485–486. [Google Scholar] [CrossRef]
- Peakall, R.; Smouse, P.E. GENALEX 6: Genetic analysis in Excel. Population genetic software for teaching and research. Mol. Ecol. Notes 2006, 6, 288–295. [Google Scholar] [CrossRef]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [Green Version]
- Earl, D.A.; von Holdt, B.M. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Jakobsson, M.; Rosenberg, N.A. CLUMPP: A cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 2007, 23, 1801–1806. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, N.A. DISTRUCT: A program for the graphical display of population structure. Mol. Ecol. Notes 2004, 4, 137–138. [Google Scholar] [CrossRef]
- Rousset, F. Genetic differentiation and estimation of gene flow from F-statistics under isolation by distance. Genetics 1997, 145, 1219–1228. [Google Scholar] [CrossRef]
- King, T.L.; Kalinowski, S.T.; Schill, W.B.; Spidle, A.P.; Lubinski, B.A. Population structure of Atlantic salmon (Salmo salar L.): A range-wide perspective from microsatellite DNA variation. Mol. Ecol. 2001, 10, 807–821. [Google Scholar] [CrossRef] [Green Version]
- Thioulouse, J.; Chessel, D.; Dolédec, S.; Olivier, J. ADE-4: A multivariate analysis and graphical display software. Stat. Comput. 1997, 7, 75–83. [Google Scholar] [CrossRef]
- Chessel, D.; DuFour, A.B.; Thioulouse, J. The ade4 Package—I: One-table Methods. R News 2004, 4, 5–10. [Google Scholar]
- Oksanen, J.; Blanchet, F.; Guillaume, F.M.; Kindt, R.; Legendre, P.; McGlinn, D.; Wagner, H. Vegan: Community Ecology Package. R Package (Version 2.5-2). 2008. Available online: https://CRAN.R-project.org/package=vegan (accessed on 5 May 2023).
- Cornuet, J.M.; Luikart, G. Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics 1996, 144, 2001–2014. [Google Scholar] [CrossRef] [PubMed]
- Piry, S.; Luikart, G.; Cornuet, J.M. Computer note. BOTTLENECK: A computer program for detecting recent reductions in the effective size using allele frequency data. J. Hered. 1999, 90, 502–503. [Google Scholar] [CrossRef]
- Di Rienzo, A.; Peterson, A.C.; Garza, J.C.; Valdes, A.M.; Slatkin, M.; Freimer, N.B. Mutational processes of simple-sequence repeat loci in human populations. Proc. Natl. Acad. Sci. USA 1994, 91, 3166–3170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garza, J.C.; Williamson, E. Detection of reduction in population size using data from microsatellite loci. Mol. Ecol. 2001, 10, 305–318. [Google Scholar] [CrossRef]
- Williamson-Natesan, E.G. Comparison of methods for detecting bottlenecks from microsatellite loci. Conserv. Genet. 2005, 6, 551–562. [Google Scholar] [CrossRef]
- Perry, M.Z.; Kirby, R.; Reid, B.N.; Stoelting, R.; Doucet-Bëer, E.; Robinson, S.; Palsbøll, P.J. Reliability of genetic bottleneck tests for detecting recent population declines. Mol. Ecol. 2012, 21, 3403–3418. [Google Scholar] [CrossRef] [PubMed]
- Wilson, G.A.; Rannala, B. Bayesian inference of recent migration rates using multilocus genotypes. Genetics 2003, 163, 1177–1191. [Google Scholar] [CrossRef] [PubMed]
- Rannala, B. BayesAss Edition 3.0 User’s Manual; University of California: Davis, CA, USA, 2011; Available online: https://www.rannala.org/?page_id=245 (accessed on 5 May 2023).
- Beerli, P. Comparison of Bayesian and maximum likelihood inference of population genetic parameters. Bioinformatics 2006, 22, 341–345. [Google Scholar] [CrossRef] [Green Version]
- Beerli, P.; Palczewski, M. Unified framework to evaluate panmixia and migration direction among multiple sampling locations. Genetics 2010, 185, 313–326. [Google Scholar] [CrossRef] [Green Version]
- Rannala, B.; Mountain, J.L. Detecting immigration by using multilocus genotypes. Proc. Natl. Acad. Sci. USA 1997, 94, 9197–9201. [Google Scholar] [CrossRef] [Green Version]
- Piry, S.; Alapetite, A.; Cornuet, J.M.; Paetkau, D.; Baudouin, L.; Estoup, A. GENECLASS2: A software for genetic assignment and first-generation migrant detection. J. Hered. 2004, 95, 536–539. [Google Scholar] [CrossRef] [PubMed]
- Paetkau, D.; Slade, R.; Burden, M.; Estoup, A. Genetic assignment methods for the direct, real-time estimation of migration rate: A simulation-based exploration of accuracy and power. Mol. Ecol. 2004, 13, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Do, C.; Waples, R.S.; Peel, D.; Macbeth, G.M.; Tillett, B.J.; Ovenden, J.R. NeEstimator v2: Re-implementation of software for the estimation of contemporary effective population size (Ne) from genetic data. Mol. Ecol. Resour. 2014, 14, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Uchida, D.; Knight, T.W.; Ruzzante, D.E. Interaction of landscape and life history attributes on genetic diversity, neutral divergence and gene flow in a pristine community of salmonids. Mol. Ecol. 2009, 18, 4854–4869. [Google Scholar] [CrossRef] [PubMed]
- Waples, R.S.; England, P.R. Estimating contemporary effective population size on the basis of linkage disequilibrium in the face of migration. Genetics 2011, 189, 633–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilbert, K.J.; Whitlock, M.C. Evaluating methods for estimating local effective population size with and without migration. Evolution 2015, 69, 2154–2166. [Google Scholar] [CrossRef] [PubMed]
- Whiteley, A.R.; Coombs, J.A.; Donnell, M.J.O.; Nislow, K.H.; Letcher, B.H. Keeping things local: Subpopulation Nb and Ne in a stream network with partial barriers to fish migration. Evol. Appl. 2017, 10, 348–365. [Google Scholar] [CrossRef]
- Bernos, T.A.; Yates, M.C.; Fraser, D.J. Fine-scale differences in genetic and census population size ratios between two stream fishes. Conserv. Genet. 2018, 9, 265–274. [Google Scholar] [CrossRef]
- Waples, R.S.; Antao, T.; Luikart, G. Effects of overlapping generations on linkage disequilibrium estimates of effective population size. Genetics 2014, 197, 769–780. [Google Scholar] [CrossRef] [Green Version]
- Borrell, Y.J.; Bernardo, D.; Blanco, G.; Vázquez, E.; Sánchez, J.A. Spatial and temporal variation of genetic diversity and estimation of effective population sizes in Atlantic salmon (Salmo salar L.) populations from Asturias (Northern Spain) using microsatellites. Conserv. Genet. 2008, 9, 807–819. [Google Scholar] [CrossRef]
- Hórreo, J.L.; Machado-Schiaffino, G.; Griffiths, A.M.; Bright, D.; Stevens, J.R.; García-Vázquez, E. Atlantic salmon at risk: Apparent rapid declines in effective population size in southern European populations. Trans. Am. Fish. Soc. 2011, 140, 605–610. [Google Scholar] [CrossRef]
- Valiente, A.G.; Beall, E.; García-Vázquez, E. Population genetics of south European Atlantic salmon under global change. Glob. Chang. Biol. 2010, 16, 36–47. [Google Scholar] [CrossRef]
- Säisä, M.; Koljonen, M.L.; Gross, R.; Nilsson, J.; Tähtinen, J.; Koskiniemi, J.; Vasemägi, A. Population genetic structure and postglacial colonization of Atlantic salmon (Salmo salar) in the Baltic Sea area based on microsatellite DNA variation. Can. J. Fish. Aquat. Sci. 2005, 62, 1887–1904. [Google Scholar] [CrossRef] [Green Version]
- Finnegan, A.K.; Griffiths, A.M.; King, R.A.; Machado-Schiaffino, G.; Porcher, J.P.; García-Vázquez, E.; Stevens, J.R. Use of multiple markers demonstrates a cryptic western refugium and postglacial colonisation routes of Atlantic salmon (Salmo salar L.) in northwest Europe. Heredity 2013, 111, 34–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valiquette, E.; Perrier, C.; Thibault, I.; Bernatchez, L. Loss of genetic integrity in wild lake trout populations following stocking: Insights from an exhaustive study of 72 lakes from Québec, Canada. Evol. Appl. 2014, 7, 625–644. [Google Scholar] [CrossRef] [PubMed]
- Gudmundsson, L.A.; Gudjónsson, S.; Marteinsdóttir, G.; Scarnecchia, D.L.; Daníelsdóttir, A.K.; Pampoulie, C. Spatio-temporal effects of stray hatchery-reared Atlantic salmon Salmo salar on population genetic structure within a 21 km-long Icelandic river system. Conserv. Genet. 2013, 14, 1217–1231. [Google Scholar] [CrossRef]
- Griffiths, A.M.; Machado-Schiaffino, G.; Dillane, E.; Coughlan, J.; Hórreo, J.L.; Bowkett, A.E.; McGinnity, P. Genetic stock identification of Atlantic salmon (Salmo salar) populations in the southern part of the European range. BMC Genet. 2010, 11, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vähä, J.P.; Erkinaro, J.; Falkegård, M.; Orell, P.; Niemelä, E. Genetic stock identification of Atlantic salmon and its evaluation in a large population complex. Can. J. Fish. Aquat. Sci. 2016, 74, 327–338. [Google Scholar] [CrossRef] [Green Version]
- Vähä, J.P.; Erkinaro, J.; Niemelä, E.; Primmer, C.R. Temporally stable genetic structure and low migration in an Atlantic salmon population complex: Implications for conservation and management. Evol. Appl. 2008, 1, 137–154. [Google Scholar] [CrossRef]
- Fraser, D.J.; Lippe, C.; Bernatchez, L. Consequences of unequal population size, asymmetric gene flow and sex-biased dispersal on population structure in brook charr (Salvelinus fontinalis). Mol. Ecol. 2004, 13, 67–80. [Google Scholar] [CrossRef]
- Manier, M.K.; Arnold, S.J. Population genetic analysis identifies source-sink dynamics for two sympatric garter snake species (Thamnophis elegans and Thamnophis sirtalis). Mol. Ecol. 2005, 14, 3965–3976. [Google Scholar] [CrossRef]
- Hansen, M.M.; Skaala, O.; Jensen, L.F.; Bekkevold, D.; Mensberg, K.L.D. Gene flow, effective population size and selection at major histocompatibility complex genes: Brown trout in the Hardanger Fjord, Norway. Mol. Ecol. 2007, 16, 1413–1425. [Google Scholar] [CrossRef] [Green Version]
- Bradbury, I.R.; Hamilton, L.C.; Robertson, M.J.; Bourgeois, C.E.; Mansour, A.; Dempson, J.B. (2014). Landscape structure and climatic variation determine Atlantic salmon genetic connectivity in the Northwest Atlantic. Can. J. Fish. Aquat. Sci. 2014, 71, 246–258. [Google Scholar] [CrossRef]
- Sexton, J.P.; Hangartner, S.B.; Hoffman, A.A. Genetic isolation by environment or distance: Which pattern of gene flow is most common? Evolution 2013, 68, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Bowlby, H.D.; Fleming, I.A.; Gibson, A.J.F. Applying landscape genetics to evaluate threats affecting endangered Atlantic salmon populations. Conserv. Genet. 2016, 17, 823–838. [Google Scholar] [CrossRef] [Green Version]
- Lehnert, S.J.; Kess, T.; Bentzen, P.; Kent, M.P.; Lien, S.; Gilbey, J.; Clément, M.; Jeffery, N.W.; Waples, R.S.; Bradbury, I.R. Genomic signatures and correlates of widespread population declines in salmon. Nat. Commun. 2019, 10, 2996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frankham, R.; Bradshaw, C.J.A.; Brook, B.W. Genetics in conservation management: Revised recommendations for the 50/500 rules, red list criteria and population viability analyses. Biol. Conserv. 2014, 170, 56–63. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| River | Total | Locus | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Ssa197 | SSOSL85 | SSOSL311 | SSOSL417 | Sssp2210 | SsspG7 | SsaF43 | SSOSL438 | |||
| Navia | N | 48 | 48 | 48 | 48 | 48 | 48 | 48 | 48 | 47 |
| A | 7.875 | 8 | 9 | 11 | 8 | 8 | 9 | 6 | 4 | |
| AR | 7.191 | 7.706 | 8.178 | 9.756 | 7.098 | 7.458 | 8.454 | 5.387 | 3.489 | |
| Ho | 0.623 | 0.708 | 0.625 | 0.646 | 0.563 | 0.792 | 0.813 | 0.479 | 0.362 | |
| He | 0.643 | 0.808 | 0.659 | 0.717 | 0.635 | 0.806 | 0.786 | 0.412 | 0.317 | |
| FIS | 0.040 | 0.133 | 0.062 | 0.11 | 0.125 | 0.029 | −0.023 | −0.154 | −0.13 | |
| HWE | 0.081 | 0.047 | 0.268 | 0.132 | 0.112 | 0.391 | 0.448 | 0.115 | 0.209 | |
| Narcea | N | 48 | 48 | 48 | 48 | 48 | 48 | 48 | 48 | 48 |
| A | 7.875 | 10 | 7 | 9 | 9 | 9 | 10 | 5 | 4 | |
| AR | 7.515 | 9.433 | 6.454 | 8.918 | 8.701 | 8.837 | 9.116 | 4.729 | 3.929 | |
| Ho | 0.677 | 0.729 | 0.708 | 0.708 | 0.688 | 0.792 | 0.813 | 0.604 | 0.375 | |
| He | 0.687 | 0.8 | 0.661 | 0.766 | 0.707 | 0.788 | 0.799 | 0.632 | 0.34 | |
| FIS | 0.024 | 0.099 | −0.061 | 0.086 | 0.038 | 0.005 | −0.006 | 0.054 | −0.092 | |
| HWE | 0.192 | 0.095 | 0.292 | 0.156 | 0.848 | 0.515 | 0.538 | 0.342 | 0.278 | |
| Sella | N | 49 | 49 | 49 | 49 | 49 | 49 | 49 | 49 | 49 |
| A | 8.75 | 12 | 11 | 9 | 10 | 9 | 11 | 5 | 3 | |
| AR | 8.217 | 11.727 | 10.304 | 8.405 | 9.3 | 8.121 | 10.183 | 4.693 | 3 | |
| Ho | 0.722 | 0.776 | 0.776 | 0.776 | 0.837 | 0.878 | 0.694 | 0.571 | 0.469 | |
| He | 0.731 | 0.853 | 0.814 | 0.758 | 0.761 | 0.794 | 0.779 | 0.544 | 0.545 | |
| FIS | 0.023 | 0.101 | 0.057 | −0.012 | −0.09 | −0.095 | 0.119 | −0.041 | 0.149 | |
| HWE | 0.194 | 0.061 | 0.228 | 0.541 | 0.141 | 0.111 | 0.048 | 0.416 | 0.149 | |
| Deva-Cares | N | 46 | 46 | 46 | 46 | 46 | 46 | 46 | 46 | 46 |
| A | 8.5 | 10 | 8 | 12 | 12 | 9 | 9 | 4 | 4 | |
| AR | 7.811 | 9.886 | 6.988 | 10.749 | 10.51 | 8.865 | 7.792 | 3.988 | 3.706 | |
| Ho | 0.745 | 0.804 | 0.717 | 0.935 | 0.739 | 0.826 | 0.674 | 0.717 | 0.544 | |
| He | 0.697 | 0.844 | 0.688 | 0.822 | 0.729 | 0.762 | 0.642 | 0.604 | 0.488 | |
| FIS | −0.057 | 0.058 | −0.032 | −0.127 | −0.004 | −0.073 | −0.039 | −0.177 | −0.102 | |
| HWE | 0.031 | 0.213 | 0.435 | 0.029 | 0.572 | 0.195 | 0.412 | 0.063 | 0.283 | |
| Nansa | N | 48 | 44 | 47 | 48 | 47 | 46 | 48 | 48 | 47 |
| A | 7 | 10 | 11 | 11 | 6 | 6 | 5 | 4 | 3 | |
| AR | 6.702 | 9.748 | 10.347 | 10.368 | 5.984 | 5.509 | 4.724 | 4 | 2.937 | |
| Ho | 0.678 | 0.841 | 0.745 | 0.813 | 0.809 | 0.565 | 0.646 | 0.729 | 0.277 | |
| He | 0.667 | 0.866 | 0.77 | 0.855 | 0.715 | 0.599 | 0.584 | 0.673 | 0.274 | |
| FIS | −0.006 | 0.041 | 0.043 | 0.06 | −0.12 | 0.067 | −0.096 | −0.074 | 0.002 | |
| HWE | 0.472 | 0.297 | 0.31 | 0.218 | 0.105 | 0.288 | 0.238 | 0.272 | 0.638 | |
| Pas | N | 47 | 46 | 47 | 47 | 45 | 47 | 47 | 47 | 46 |
| A | 6.125 | 11 | 7 | 8 | 6 | 5 | 5 | 5 | 2 | |
| AR | 5.96 | 10.633 | 6.615 | 7.741 | 5.998 | 4.984 | 4.969 | 4.744 | 1.997 | |
| Ho | 0.583 | 0.783 | 0.617 | 0.766 | 0.733 | 0.532 | 0.553 | 0.596 | 0.087 | |
| He | 0.619 | 0.839 | 0.694 | 0.825 | 0.745 | 0.614 | 0.564 | 0.585 | 0.083 | |
| FIS | 0.068 | 0.078 | 0.121 | 0.082 | 0.027 | 0.144 | 0.03 | −0.007 | −0.034 | |
| HWE | 0.017 | 0.14 | 0.102 | 0.154 | 0.412 | 0.089 | 0.452 | 0.555 | 1 | |
| Asón | N | 48 | 48 | 48 | 48 | 48 | 48 | 48 | 48 | 48 |
| A | 7.625 | 9 | 7 | 10 | 7 | 9 | 9 | 5 | 5 | |
| AR | 7.191 | 8.378 | 6.71 | 9.116 | 6.706 | 8.417 | 8.688 | 4.928 | 4.587 | |
| Ho | 0.703 | 0.792 | 0.813 | 0.667 | 0.854 | 0.688 | 0.75 | 0.521 | 0.542 | |
| He | 0.683 | 0.774 | 0.724 | 0.736 | 0.708 | 0.725 | 0.815 | 0.469 | 0.516 | |
| FIS | −0.019 | −0.012 | −0.112 | 0.104 | −0.196 | 0.062 | 0.09 | −0.101 | −0.04 | |
| HWE | 0.289 | 0.539 | 0.12 | 0.131 | 0.013 | 0.297 | 0.131 | 0.225 | 0.441 | |
| Bidasoa | N | 40 | 38 | 37 | 40 | 35 | 40 | 40 | 40 | 36 |
| A | 8.75 | 8 | 9 | 12 | 10 | 8 | 11 | 7 | 5 | |
| AR | 8.67 | 7.989 | 8.89 | 11.845 | 10 | 7.861 | 10.971 | 6.861 | 4.944 | |
| Ho | 0.716 | 0.868 | 0.73 | 0.825 | 0.714 | 0.7 | 0.9 | 0.575 | 0.417 | |
| He | 0.728 | 0.79 | 0.721 | 0.873 | 0.709 | 0.787 | 0.875 | 0.63 | 0.437 | |
| FIS | 0.029 | −0.086 | 0.002 | 0.067 | 0.008 | 0.123 | −0.016 | 0.099 | 0.061 | |
| HWE | 0.138 | 0.194 | 0.574 | 0.173 | 0.556 | 0.087 | 0.541 | 0.172 | 0.391 | |
| River | Period | Av. AR | Av. He | Av. Ho | Av. Ne | Sources |
|---|---|---|---|---|---|---|
| Navia | 1950s | 7.19 | 0.643 | 0.623 | 667 | Present study |
| 1990s | - | - | - | - | ||
| Narcea | 1950s | 6.68 | 0.617 | 0.607 | 325 | Present study |
| 1990s | 6.32 | 0.536 | 0.487 | 114 | [75] | |
| Sella | 1950s | 8.22 | 0.731 | 0.722 | 1088 | Present study |
| 1990s | 6.65 | 0.532 | 0.487 | 107 | [75] | |
| Deva-Cares | 1950s | 7.81 | 0.697 | 0.745 | 729 | Present study |
| 1990s | 5.97 | 0.689 | 0.564 | - | [18,19,75,76] | |
| 2000s | 4.68 | 0.660 | 0.690 | - | [19] | |
| Nansa | 1950s | 6.70 | 0.667 | 0.678 | 197 | Present study |
| 1990s | 5.34 | 0.772 | 0.620 | 68 | [18,19] | |
| 2000s | 4.88 | 0.680 | 0.710 | - | [19] | |
| Pas | 1950s | 5.96 | 0.619 | 0.583 | 180 | Present study |
| 1990s | 5.15 | 0.769 | 0.650 | - | [18,19] | |
| 2000s | 4.48 | 0.670 | 0.670 | - | [19] | |
| Asón | 1950s | 7.19 | 0.683 | 0.703 | 1051 | Present study |
| 1990s | 5.20 | 0.773 | 0.650 | 42 | [18,19] | |
| 2000s | 4.99 | 0.700 | 0.740 | - | [19] | |
| Bidasoa | 1950s | 8.67 | 0.728 | 0.716 | 689 | Present study |
| 2000s | - | 0.850 | 0.750 | - | [77] |
| River | # Private Alleles per River | Ssa197 | SSOSL85 | SSOSL311 | SSOSL417 | Sssp2210 | SSspG7 | SsaF43 | SSOSL438 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Navia | 3 | - | - | *214 | 1.0% | *173 | 1.0% | *209 | 1.0% | - | - | - | - | - | - | - | - |
| Narcea | 1 | - | - | - | - | - | - | *185 | 1.0% | - | - | - | - | - | - | - | - |
| Sella | 5 | *151 | 2.0% | - | - | - | - | *159 | 1.0% | - | - | *106 | 2.0% | - | - | - | - |
| *110 | 7.1% | ||||||||||||||||
| *138 | 1.0% | ||||||||||||||||
| Deva-Cares | 6 | - | - | - | - | *123 | 1.1% | *189 | 2.2% | *158 | 3.3% | - | - | - | - | *128 | 1.1% |
| *171 | 1.1% | *199 | 1.1% | - | - | - | - | ||||||||||
| Nansa | 1 | - | - | *216 | 3.2% | - | - | - | - | - | - | - | - | - | - | - | - |
| Pas | 1 | - | - | - | - | - | - | - | - | - | - | - | - | *128 | 6.4% | - | - |
| Asón | 3 | - | - | *204 | 1.0% | *127 | 1.0% | - | - | *138 | 3.1% | - | - | - | - | - | - |
| Bidasoa | 8 | - | - | - | - | *137 | 2.5% | *175 | 2.9% | *170 | 1.3% | - | - | *112 | 1.3% | *142 | 1.4% |
| *149 | 6.3% | *183 | 5.7% | ||||||||||||||
| *163 | 3.8% | ||||||||||||||||
| # Private alleles per locus | 1 | 3 | 7 | 7 | 3 | 3 | 2 | 2 | |||||||||
| Total alleles | 14 | 15 | 19 | 18 | 13 | 15 | 9 | 7 | |||||||||
| Navia | Narcea | Sella | Deva-Cares | Nansa | Pas | Asón | |
|---|---|---|---|---|---|---|---|
| Narcea | 0.019 | ||||||
| Sella | 0.054 | 0.039 | |||||
| Deva-Cares | 0.059 | 0.040 | 0.008 | ||||
| Nansa | 0.064 | 0.049 | 0.049 | 0.035 | |||
| Pas | 0.085 | 0.079 | 0.098 | 0.082 | 0.083 | ||
| Asón | 0.052 | 0.048 | 0.036 | 0.044 | 0.052 | 0.085 | |
| Bidasoa | 0.033 | 0.037 | 0.043 | 0.052 | 0.071 | 0.067 | 0.052 |
| River | SMM | M-ratio | Mc | |||
|---|---|---|---|---|---|---|
| Ne = 50 | Ne = 100 | Ne = 500 | Ne = 1000 | |||
| Navia | p = 0.981 | 0.771 | 0.754 | 0.744 | 0.701 | 0.685 |
| Narcea | p = 0.680 | 0.781 | 0.754 | 0.744 | 0.701 | 0.685 |
| Sella | p = 0.422 | 0.777 | 0.758 | 0.746 | 0.701 | 0.683 |
| Deva-Cares | p = 0.875 | 0.782 | 0.756 | 0.748 | 0.703 | 0.680 |
| Nansa | p = 0.273 | 0.761 | 0.754 | 0.744 | 0.701 | 0.685 |
| Pas | p = 0.273 | 0.802 | 0.758 | 0.749 | 0.703 | 0.799 |
| Asón | p = 0.809 | 0.776 | 0.754 | 0.744 | 0.701 | 0.685 |
| Bidasoa | p = 0.727 | 0.765 | 0.756 | 0.749 | 0.700 | 0.677 |
| River | Nc | Migrants Removed | Nb | Nb(adj) | Ne(adj) | Nb(adj)/Nc | Ne(adj)/Nc |
|---|---|---|---|---|---|---|---|
| [CI 95%] | [CI 95%] | [CI 95%] | |||||
| Navia | 701 | 1 | 320 [162–∞] | 285 [145–∞] | 667 [338–∞] | 0.407 | 0.951 |
| Narcea | 1298 | 2 | 156 [80–∞] | 139 [71–∞] | 325 [166–∞] | 0.107 | 0.250 |
| Deva-Cares + Sella | 2238 | 2 | 1167 [161–∞] | 1040 [144–∞] | 2431 [336–∞] | 0.465 | 1.086 |
| Nansa | 124 | 0 | 95 [54–275] | 85 [48–245] | 197 [111–572] | 0.681 | 1.592 |
| Pas | 136 | 1 | 86 [48–259] | 77 [43–230] | 180 [100–538] | 0.566 | 1.323 |
| Asón | 1051 | 2 | 645 [197–∞] | 575 [175–∞] | 1344 [410–∞] | 0.547 | 1.278 |
| Bidasoa | 102 | 1 | 331 [83–∞] | 295 [74–∞] | 689 [173–∞] | 2.890 | 6.753 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Almodóvar, A.; Nicola, G.G.; Ayllón, D.; Leal, S.; Marchán, D.F.; Elvira, B. A Benchmark for Atlantic Salmon Conservation: Genetic Diversity and Structure in a Southern European Glacial Refuge before the Climate Changed. Fishes 2023, 8, 321. https://doi.org/10.3390/fishes8060321
Almodóvar A, Nicola GG, Ayllón D, Leal S, Marchán DF, Elvira B. A Benchmark for Atlantic Salmon Conservation: Genetic Diversity and Structure in a Southern European Glacial Refuge before the Climate Changed. Fishes. 2023; 8(6):321. https://doi.org/10.3390/fishes8060321
Chicago/Turabian StyleAlmodóvar, Ana, Graciela G. Nicola, Daniel Ayllón, Sheila Leal, Daniel F. Marchán, and Benigno Elvira. 2023. "A Benchmark for Atlantic Salmon Conservation: Genetic Diversity and Structure in a Southern European Glacial Refuge before the Climate Changed" Fishes 8, no. 6: 321. https://doi.org/10.3390/fishes8060321
APA StyleAlmodóvar, A., Nicola, G. G., Ayllón, D., Leal, S., Marchán, D. F., & Elvira, B. (2023). A Benchmark for Atlantic Salmon Conservation: Genetic Diversity and Structure in a Southern European Glacial Refuge before the Climate Changed. Fishes, 8(6), 321. https://doi.org/10.3390/fishes8060321