
Quantum Biology: An Update and Perspective

 ,
,  , ,
, ,  , ,
, ,  , , ,
, , , 
 , ,
, ,  and add
Show full author list
and add
Show full author list
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Quantum Tunnelling and the Importance of Protein Dynamics in Enzymatic H-transfer Reactions
2.1. Introduction
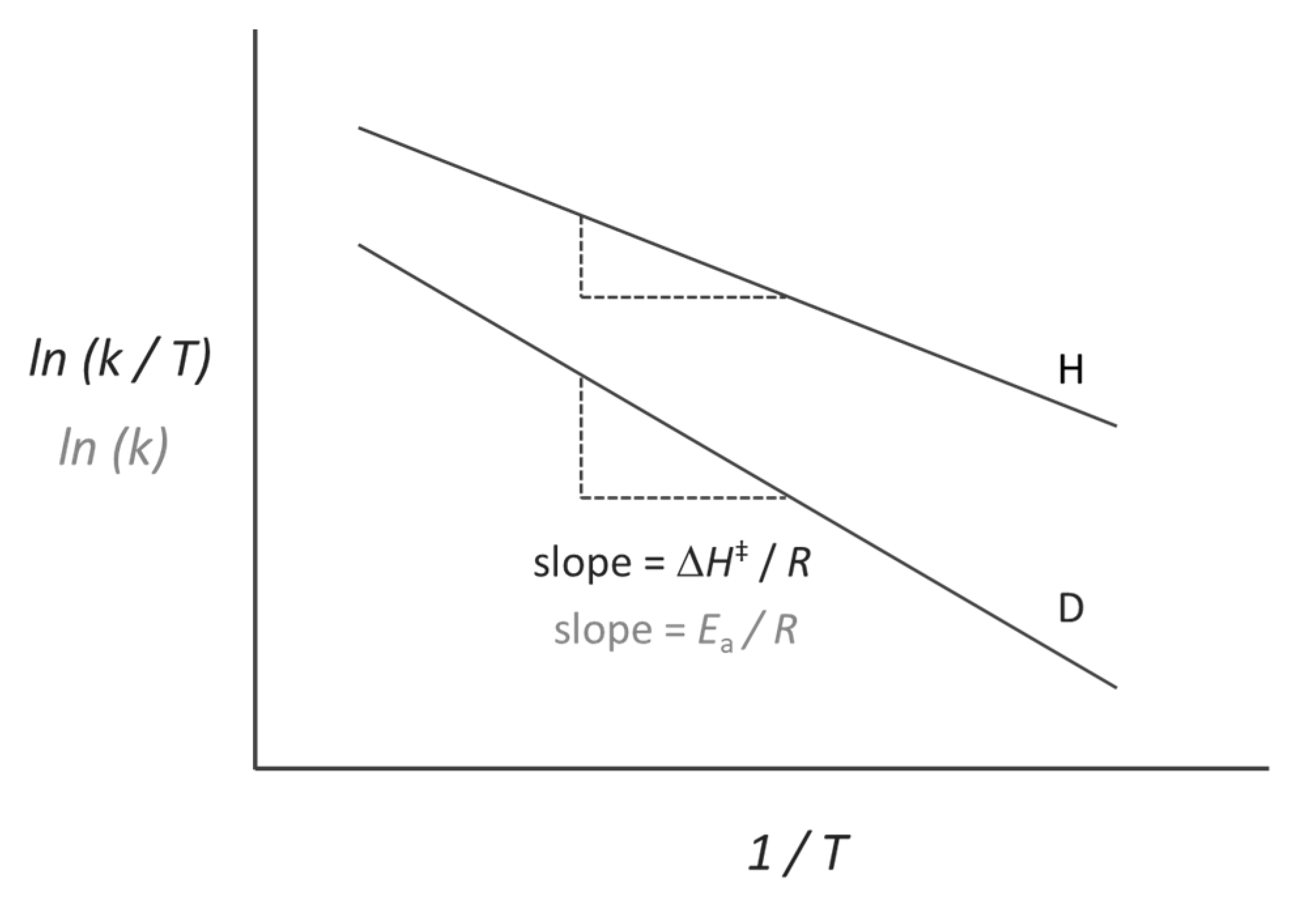
2.2. Temperature-Dependent KIEs and the Role of Fast Dynamics
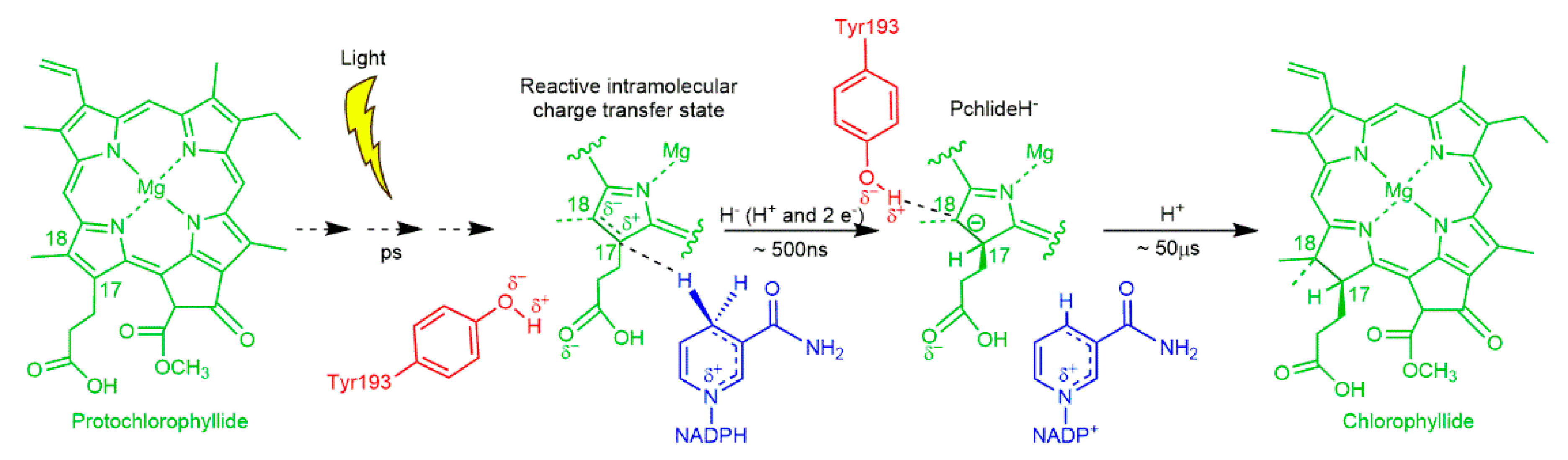
2.3. A Paradigm Model System—Light-Activated Enzymatic H-Transfer Chemistry
2.4. Conclusions
3. Quantum Effects in Photosynthesis
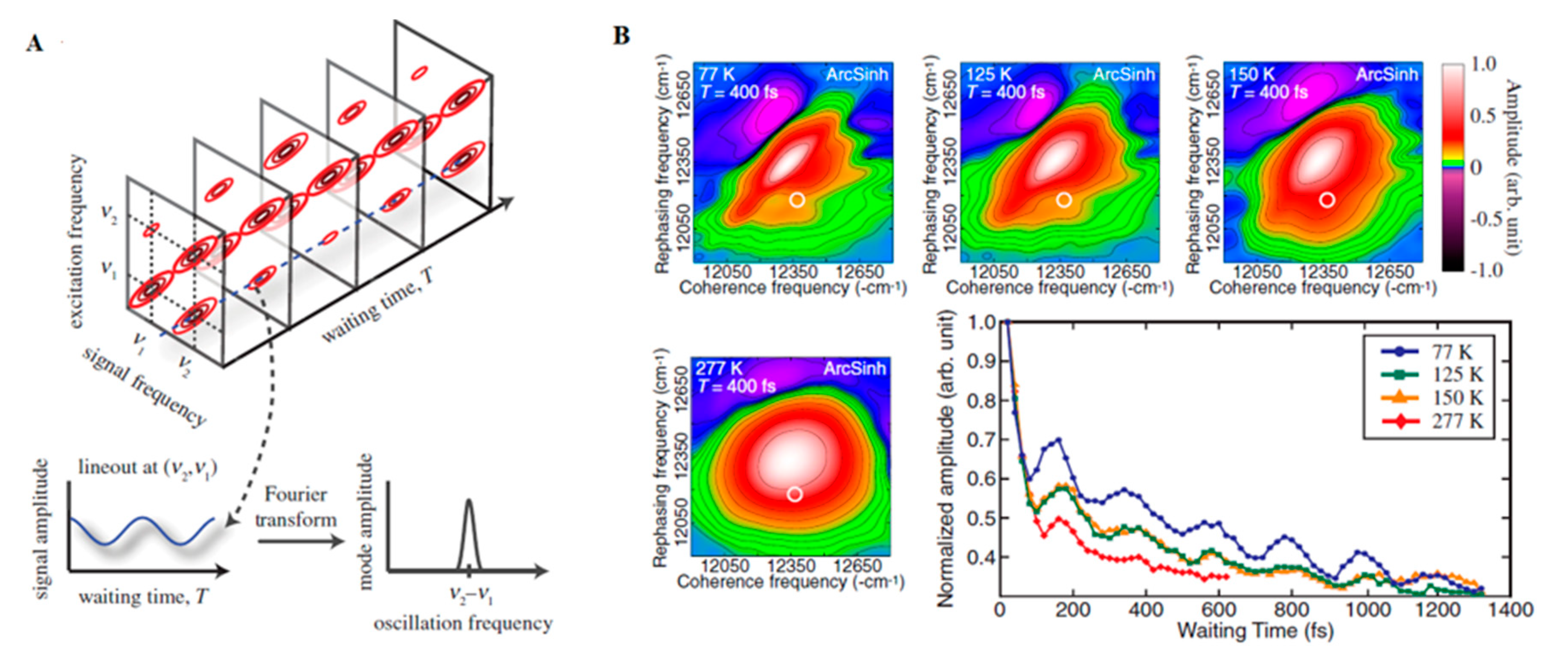
3.1. Theory and Experimental Studies
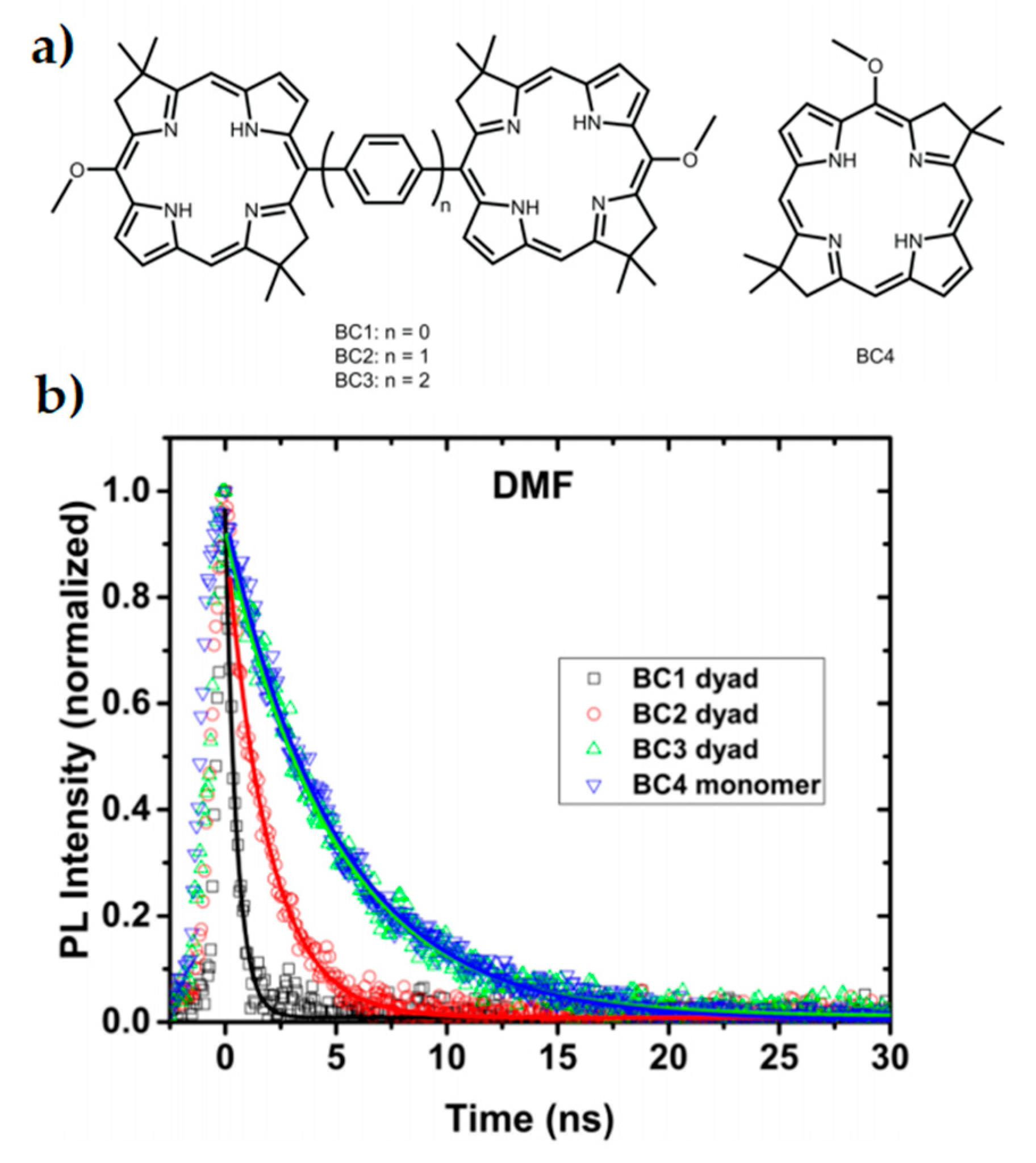
3.2. Bio-Inspired Synthetic Light Harvesting Systems
3.3. Conclusions
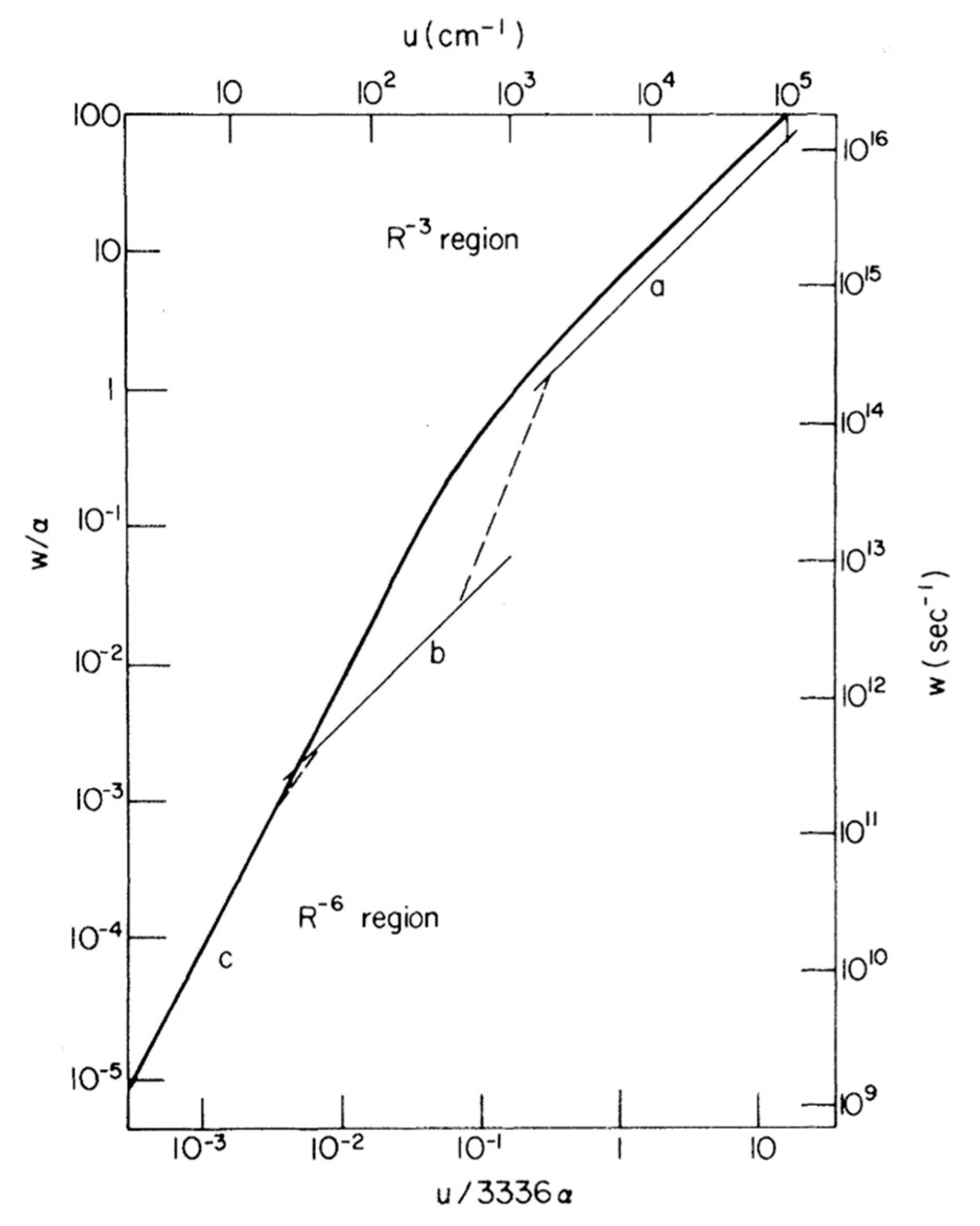
4. Magnetic Field Effects on Spin-Dependent Reactions in Biology: Candidates and Constraints
4.1. The Question of Thermodynamics
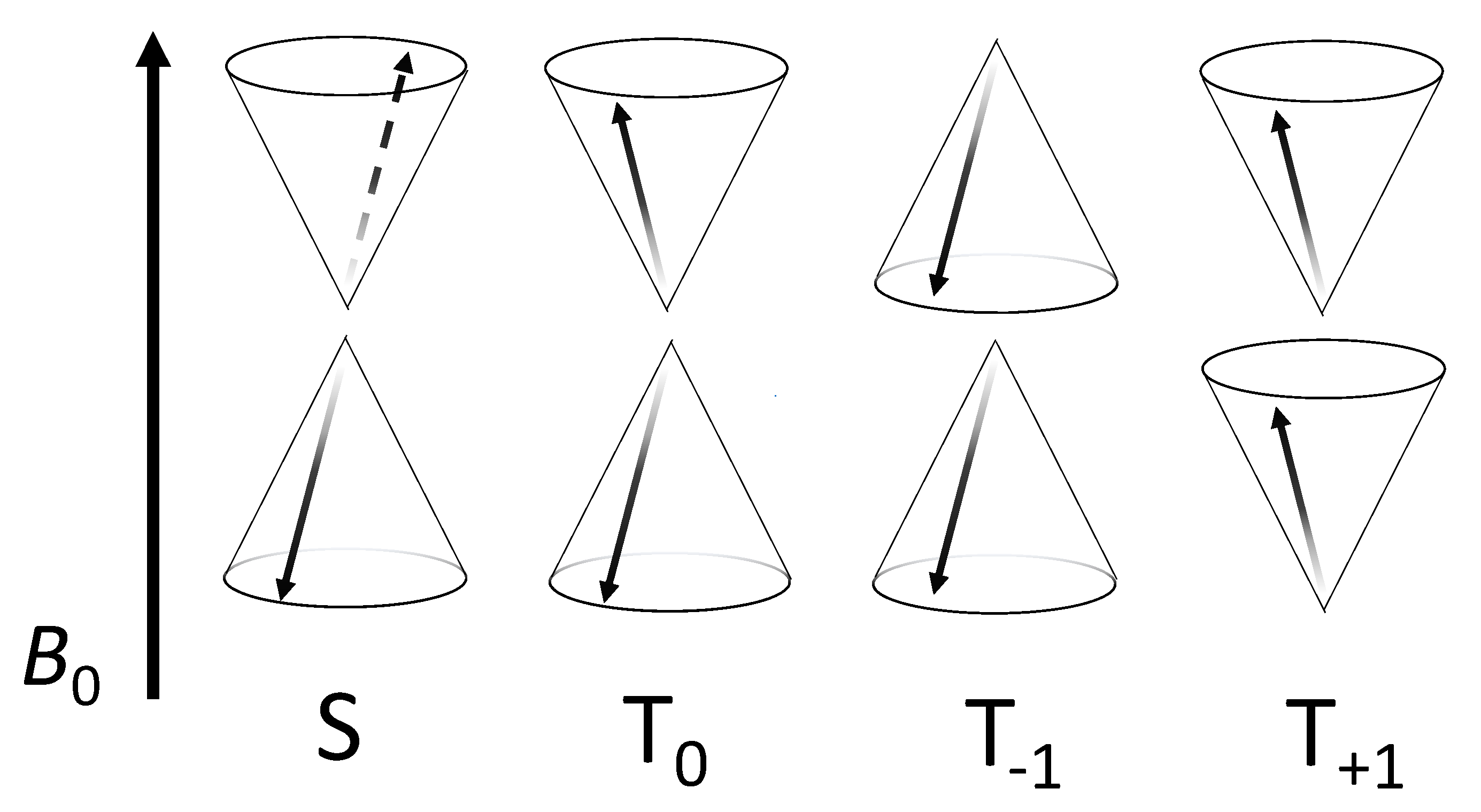
4.2. Is Spin-Correlation Common in Biology?
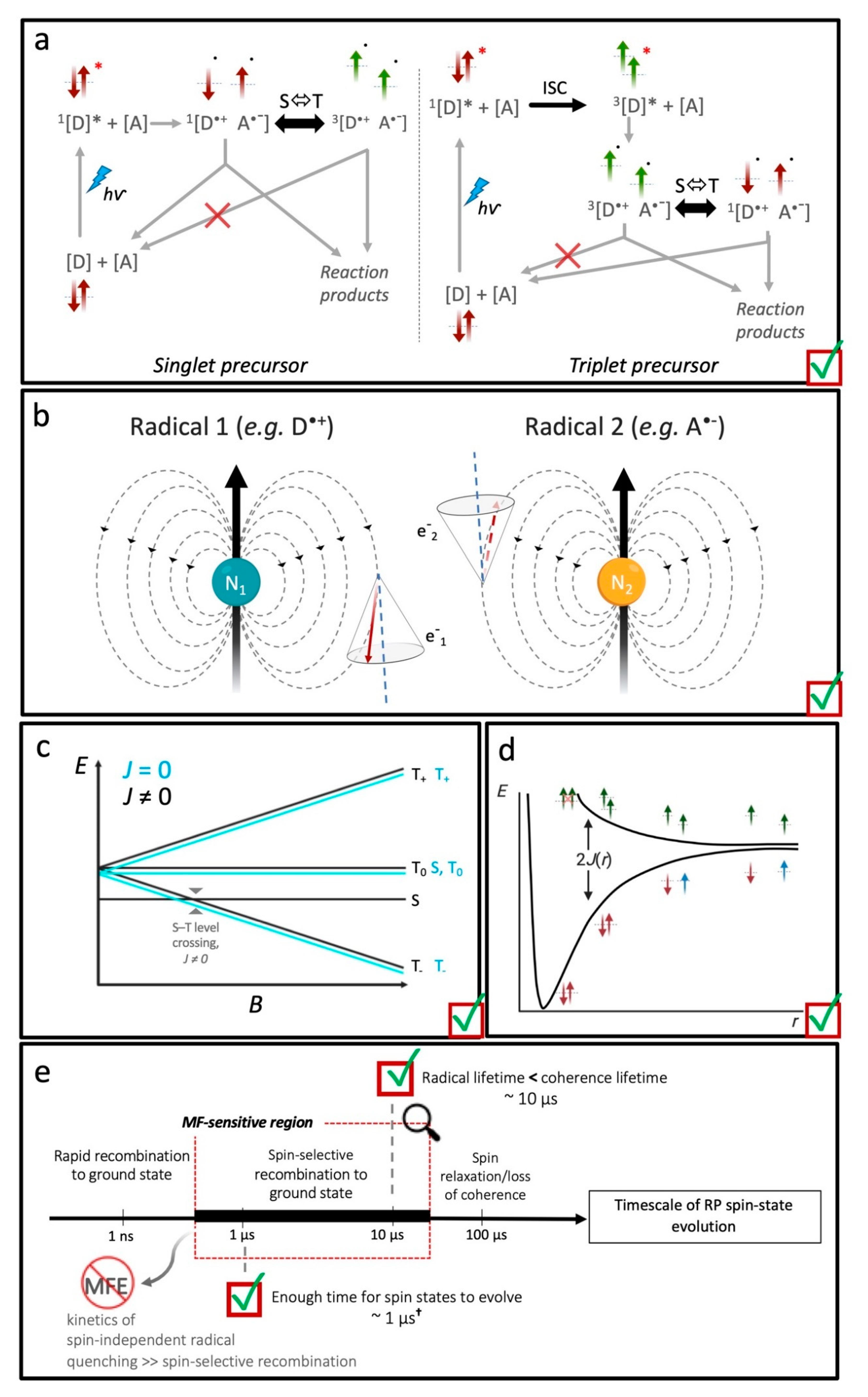
4.3. What Drives Coherent Spin-State Evolution and What Coherence Lifetimes Are Necessary?
4.4. What Is the Likely Influence of Inter-Radical Interactions?
4.5. Are Spin-Selective Reactions Widespread in Biology?
4.6. Conclusions and Outlook
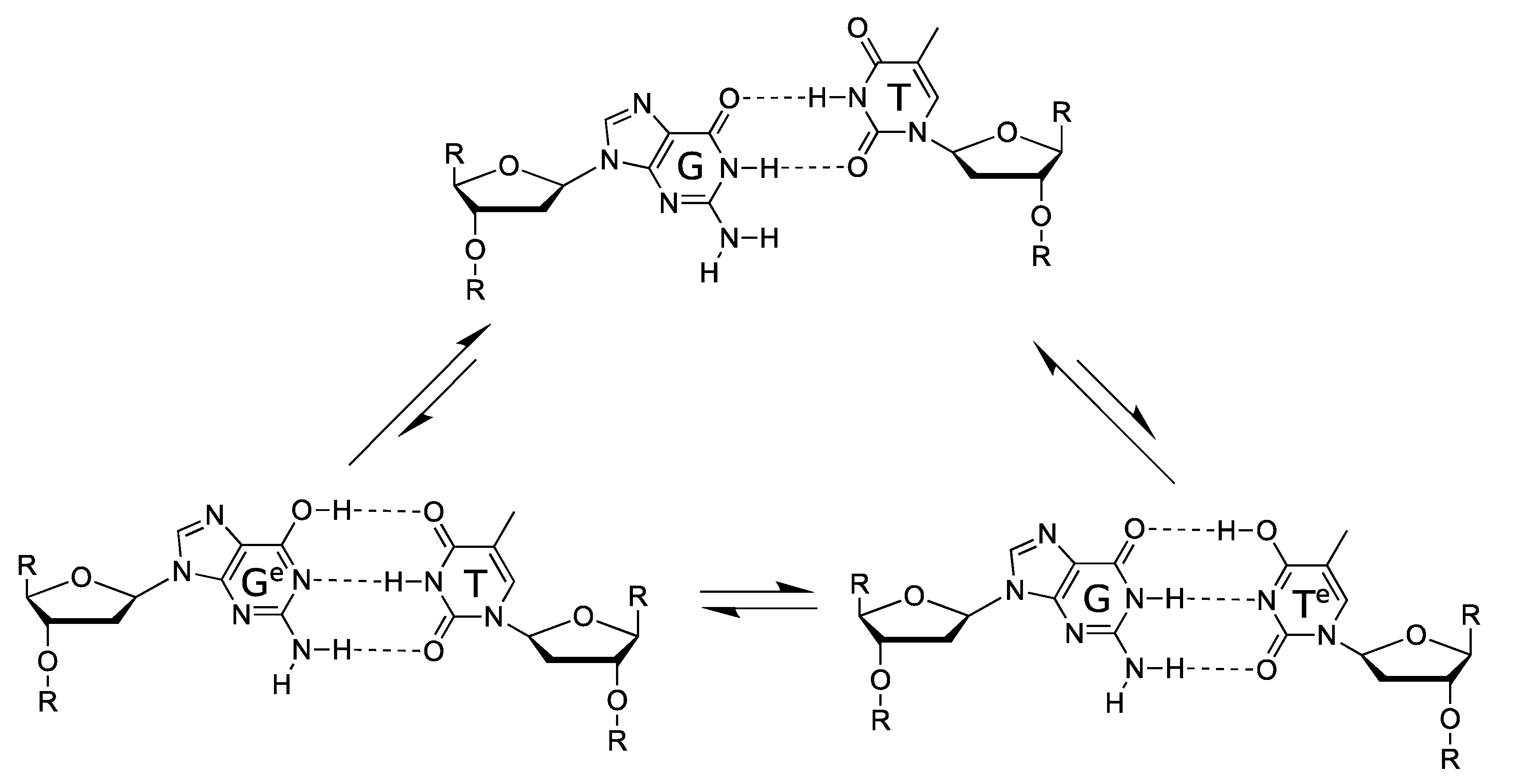
5. Proton Tunnelling in DNA
5.1. DNA Replication
5.2. Spontaneous Mutagenesis
5.3. In Silico Studies of Proton Tunnelling
5.4. Conclusions
6. Fluorescent Protein as a Novel Model System for Quantum Biology
6.1. Excitation Energy Transfer
6.2. Fluorescent Protein
6.3. Förster Resonance Energy Transfer
6.4. Anomalous Photophysical Properties of Fluorescent Proteins
6.5. Generation of Polarisation-Entangled Photon Pairs in an Ensemble of eGFP Molecules
6.6. Exciton Coupling in Homodimers of Yellow Fluorescent Protein VenusA206
6.7. Conclusions and Outlook
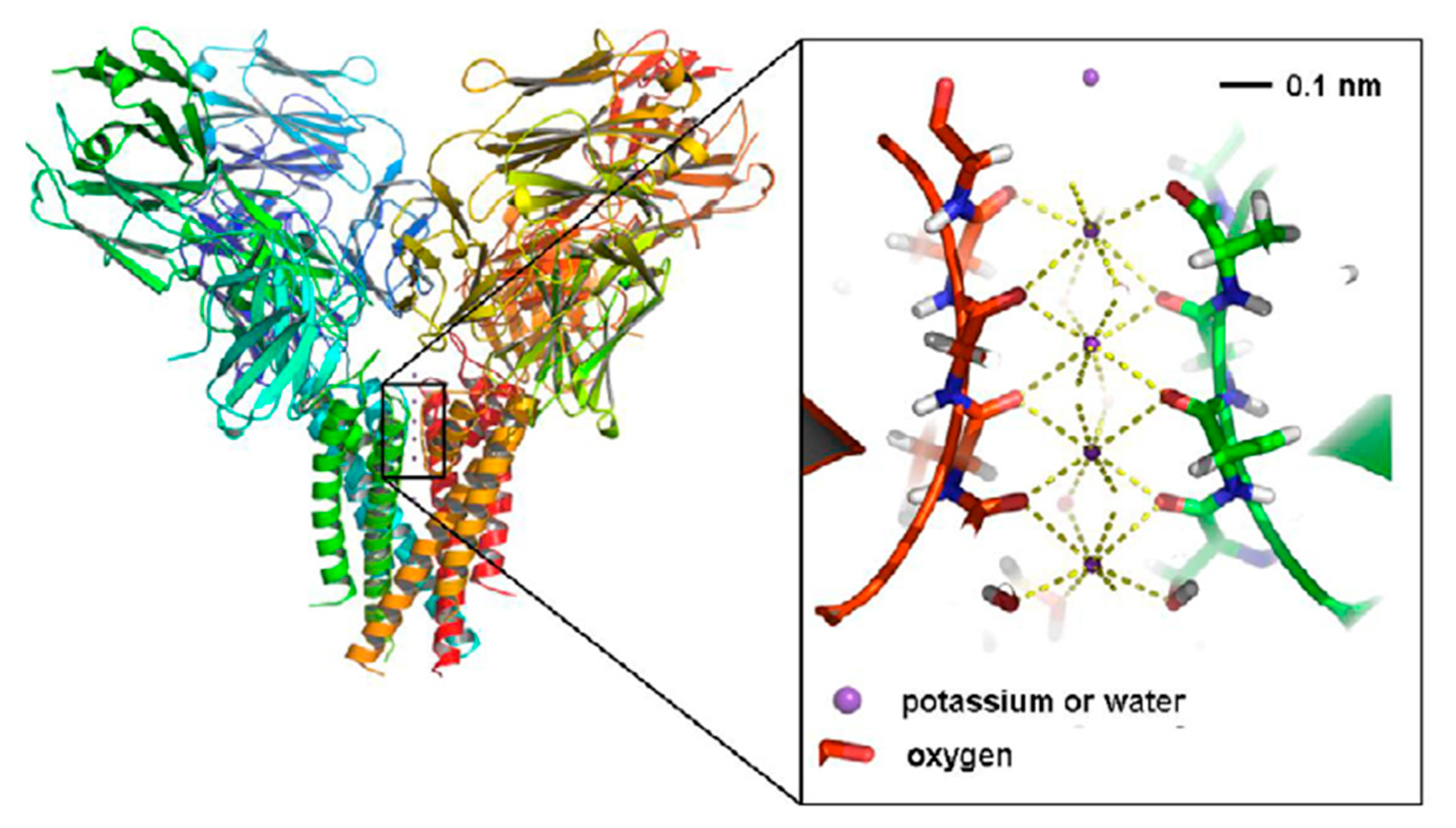
7. Quantum Coherence in Neuronal Ion Channels
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- McFadden, J.; Al-Khalili, J. The origins of quantum biology. Proc. R. Soc. A 2018, 474, 20180674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrodinger, E. What Is Life; Cambridge University Press: Cambridge, UK, 1944. [Google Scholar]
- Melkikh, A.V.; Khrennikov, A. Nontrivial quantum and quantum-like effects in biosystems: Unsolved questions and paradoxes. Prog. Biophys. Mol. Biol. 2015, 119, 137–161. [Google Scholar] [CrossRef] [PubMed]
- Tegmark, M. Importance of quantum decoherence in brain processes. Phys. Rev. E 2000, 61, 4194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKemmish, L.K.; Reimers, J.R.; McKenzie, R.H.; Mark, A.E.; Hush, N.S. Penrose-Hameroff orchestrated objective-reduction proposal for human consciousness is not biologically feasible. Phys. Rev. E 2009, 80, 021912. [Google Scholar] [CrossRef] [Green Version]
- Duan, H.-G.; Prokhorenko, V.I.; Cogdell, R.J.; Ashraf, K.; Stevens, A.L.; Thorwart, M.; Miller, R.D. Nature does not rely on long-lived electronic quantum coherence for photosynthetic energy transfer. Proc. Natl. Acad. Sci. USA 2017, 114, 8493–8498. [Google Scholar] [CrossRef] [Green Version]
- Wilkins, D.M.; Dattani, N.S. Why quantum coherence is not important in the Fenna–Matthews–Olsen complex. J. Chem. Theory Comput. 2015, 11, 3411–3419. [Google Scholar] [CrossRef] [Green Version]
- Lambert, N.; Chen, Y.-N.; Cheng, Y.-C.; Li, C.-M.; Chen, G.-Y.; Nori, F. Quantum biology. Nat. Phys. 2013, 9, 10–18. [Google Scholar] [CrossRef]
- Mohseni, M.; Omar, Y.; Engel, G.S.; Plenio, M.B. Quantum Effects in Biology; Cambridge University Press: Cambridge, UK, 2014. [Google Scholar]
- McFadden, J.; Al-Khalili, J. Life on the Edge: The Coming of Age of Quantum Biology; Broadway Books: Portland, OR, USA, 2016. [Google Scholar]
- Brookes, J.C. Quantum effects in biology: Golden rule in enzymes, olfaction, photosynthesis and magnetodetection. Proc. R. Soc. A Math. Phys. Eng. Sci. 2017, 473, 20160822. [Google Scholar] [CrossRef] [Green Version]
- Scholes, G.D.; Fleming, G.R.; Chen, L.X.; Aspuru-Guzik, A.; Buchleitner, A.; Coker, D.F.; Engel, G.S.; Van Grondelle, R.; Ishizaki, A.; Jonas, D.M. Using coherence to enhance function in chemical and biophysical systems. Nature 2017, 543, 647–656. [Google Scholar] [CrossRef]
- Marais, A.; Adams, B.; Ringsmuth, A.K.; Ferretti, M.; Gruber, J.M.; Hendrikx, R.; Schuld, M.; Smith, S.L.; Sinayskiy, I.; Krüger, T.P. The future of quantum biology. J. R. Soc. Interface 2018, 15, 20180640. [Google Scholar] [CrossRef] [Green Version]
- Bahnson, B.J.; Colby, T.D.; Chin, J.K.; Goldstein, B.M.; Klinman, J.P. A link between protein structure and enzyme catalyzed hydrogen tunneling. Proc. Natl. Acad. Sci. USA 1997, 94, 12797–12802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basran, J.; Sutcliffe, M.J.; Scrutton, N.S. Enzymatic H-transfer requires vibration-driven extreme tunneling. Biochemistry 1999, 38, 3218–3222. [Google Scholar] [CrossRef] [PubMed]
- Alhambra, C.; Corchado, J.; Snchez, M.L.; Garcia-Viloca, M.; Gao, J.; Truhlar, D.G. Canonical Variational Theory for Enzyme Kinetics with the Protein Mean Force and Multidimensional Quantum Mechanical Tunneling Dynamics. Theory and Application to Liver Alcohol Dehydrogenase. J. Phys. Chem. B 2001, 105, 11326–11340. [Google Scholar] [CrossRef]
- Billeter, S.R.; Webb, S.P.; Agarwal, P.K.; Iordanov, T.; Hammes-Schiffer, S. Hydride transfer in liver alcohol dehydrogenase: Quantum dynamics, kinetic isotope effects, and role of enzyme motion. J. Am. Chem. Soc. 2001, 123, 11262–11272. [Google Scholar] [CrossRef] [PubMed]
- Knapp, M.J.; Klinman, J.P. Environmentally coupled hydrogen tunneling. Linking catalysis to dynamics. Eur. J. Biochem. 2002, 269, 3113–3121. [Google Scholar] [CrossRef]
- Maglia, G.; Allemann, R.K. Evidence for environmentally coupled hydrogen tunneling during dihydrofolate reductase catalysis. J. Am. Chem. Soc. 2003, 125, 13372–13373. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Viloca, M.; Gao, J.; Karplus, M.; Truhlar, D.G. How enzymes work: Analysis by modern rate theory and computer simulations. Science 2004, 303, 186–195. [Google Scholar] [CrossRef]
- Liang, Z.X.; Klinman, J.P. Structural bases of hydrogen tunneling in enzymes: Progress and puzzles. Curr. Opin. Struct. Biol. 2004, 14, 648–655. [Google Scholar] [CrossRef]
- Liang, Z.X.; Lee, T.; Resing, K.A.; Ahn, N.G.; Klinman, J.P. Thermal-activated protein mobility and its correlation with catalysis in thermophilic alcohol dehydrogenase. Proc. Natl. Acad. Sci. USA 2004, 101, 9556–9561. [Google Scholar] [CrossRef] [Green Version]
- Masgrau, L.; Roujeinikova, A.; Johannissen, L.O.; Hothi, P.; Basran, J.; Ranaghan, K.E.; Mulholland, A.J.; Sutcliffe, M.J.; Scrutton, N.S.; Leys, D. Atomic description of an enzyme reaction dominated by proton tunneling. Science 2006, 312, 237–241. [Google Scholar] [CrossRef] [Green Version]
- Nagel, Z.D.; Klinman, J.P. Tunneling and dynamics in enzymatic hydride transfer. Chem. Rev. 2006, 106, 3095–3118. [Google Scholar] [CrossRef] [PubMed]
- Olsson, M.H.; Parson, W.W.; Warshel, A. Dynamical contributions to enzyme catalysis: Critical tests of a popular hypothesis. Chem. Rev. 2006, 106, 1737–1756. [Google Scholar] [CrossRef] [PubMed]
- Pu, J.Z.; Gao, J.L.; Truhlar, D.G. Multidimensional tunneling, recrossing, and the transmission coefficient for enzymatic reactions. Chem. Rev. 2006, 106, 3140–3169. [Google Scholar] [CrossRef] [Green Version]
- Hay, S.; Pudney, C.R.; Scrutton, N.S. Structural and mechanistic aspects of flavoproteins: Probes of hydrogen tunnelling. FEBS J. 2009, 276, 3930–3941. [Google Scholar] [CrossRef]
- Major, D.T.; Heroux, A.; Orville, A.M.; Valley, M.P.; Fitzpatrick, P.F.; Gao, J.L. Differential quantum tunneling contributions in nitroalkane oxidase catalyzed and the uncatalyzed proton transfer reaction. Proc. Natl. Acad. Sci. USA 2009, 106, 20734–20739. [Google Scholar] [CrossRef] [Green Version]
- Kamerlin, S.C.L.; Mavri, J.; Warshel, A. Examining the case for the effect of barrier compression on tunneling, vibrationally enhanced catalysis, catalytic entropy and related issues. FEBS Lett. 2010, 584, 2759–2766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamerlin, S.C.L.; Warshel, A. An analysis of all the relevant facts and arguments indicates that enzyme catalysis does not involve large contributions from nuclear tunneling. J. Phys. Org. Chem. 2010, 23, 677–684. [Google Scholar] [CrossRef] [Green Version]
- Smirnov, A.Y.; Mourokh, L.G.; Nori, F. Kinetics of proton pumping in cytochrome c oxidase. J. Chem. Phys. 2009, 130, 06B620. [Google Scholar] [CrossRef] [Green Version]
- Smirnov, A.Y.; Savel’ev, S.E.; Nori, F. Diffusion-controlled generation of a proton-motive force across a biomembrane. Phys. Rev. E 2009, 80, 011916. [Google Scholar] [CrossRef] [Green Version]
- Smirnov, A.Y.; Nori, F. Modeling the Q-cycle mechanism of transmembrane energy conversion. Phys. Biol. 2012, 9, 016011. [Google Scholar] [CrossRef] [Green Version]
- Hay, S.; Johannissen, L.O.; Sutcliffe, M.J.; Scrutton, N.S. Barrier compression and its contribution to both classical and quantum mechanical aspects of enzyme catalysis. Biophys. J. 2010, 98, 121–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johannissen, L.O.; Hay, S.; Scrutton, N.S. Nuclear quantum tunnelling in enzymatic reactions--an enzymologist’s perspective. Phys. Chem. Chem. Phys. 2015, 17, 30775–30782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johannissen, L.O.; Iorgu, A.I.; Scrutton, N.S.; Hay, S. What are the signatures of tunnelling in enzyme-catalysed reactions? Faraday Discuss. 2019, 221, 367–378. [Google Scholar] [CrossRef] [PubMed]
- Eyring, H.; Stearn, A.E. The Application of the Theory of Absolute Reacton Rates to Proteins. Chem. Rev. 1939, 24, 253–270. [Google Scholar] [CrossRef]
- Truhlar, D.G.; Garrett, B.C.; Klippenstein, S.J. Current status of transition-state theory. J. Phys. Chem. 1996, 100, 12771–12800. [Google Scholar] [CrossRef]
- Warshel, A.; Sharma, P.K.; Kato, M.; Xiang, Y.; Liu, H.; Olsson, M.H. Electrostatic basis for enzyme catalysis. Chem. Rev. 2006, 106, 3210–3235. [Google Scholar] [CrossRef]
- Gandour, R. Transition States of Biochemical Processes; Springer Science & Business Media: Berlin, Germany, 2013. [Google Scholar]
- Bell, R.P. The Tunnel Effect in Chemistry; Chapman and Hall: London, UK, 1980; pp. 51–140. [Google Scholar]
- Rickert, K.W.; Klinman, J.P. Nature of hydrogen transfer in soybean lipoxygenase 1: Separation of primary and secondary isotope effects. Biochemistry 1999, 38, 12218–12228. [Google Scholar] [CrossRef]
- Faulder, P.F.; Tresadern, G.; Chohan, K.K.; Scrutton, N.S.; Sutcliffe, M.J.; Hillier, I.H.; Burton, N.A. QM/MM studies show substantial tunneling for the hydrogen-transfer reaction in methylamine dehydrogenase. J. Am. Chem. Soc. 2001, 123, 8604–8605. [Google Scholar] [CrossRef]
- Truhlar, D.G.; Gao, J.L.; Garcia-Viloca, M.; Alhambra, C.; Corchado, J.; Sanchez, M.L.; Poulsen, T.D. Ensemble-averaged variational transition state theory with optimized multidimensional tunneling for enzyme kinetics and other condensed-phase reactions. Int. J. Quant. Chem. 2004, 100, 1136–1152. [Google Scholar] [CrossRef]
- Hwang, J.K.; Warshel, A. A Quantized Classical Path Approach for Calculations of Quantum-Mechanical Rate Constants. J. Phys. Chem. 1993, 97, 10053–10058. [Google Scholar] [CrossRef]
- Mavri, J.; Liu, H.; Olsson, M.H.; Warshel, A. Simulation of tunneling in enzyme catalysis by combining a biased propagation approach and the quantum classical path method: Application to lipoxygenase. J. Phys. Chem. B 2008, 112, 5950–5954. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Viloca, M.; Truhlar, D.G.; Gao, J. Reaction-Path Energetics and Kinetics of the Hydride Transfer Reaction Catalyzed by Dihydrofolate Reductase. Biochemistry 2003, 42, 13558–13575. [Google Scholar] [CrossRef] [PubMed]
- Pang, J.; Pu, J.; Gao, J.; Truhlar, D.G.; Allemann, R.K. Hydride Transfer Reaction Catalyzed by Hyperthermophilic Dihydrofolate Reductase Is Dominated by Quantum Mechanical Tunneling and Is Promoted by Both Inter- and Intramonomeric Correlated Motions. J. Am. Chem. Soc. 2006, 128, 8015–8023. [Google Scholar] [CrossRef] [PubMed]
- Alhambra, C.; Gao, J.; Corchado, J.C.; Villà, J.; Truhlar, D.G. Quantum Mechanical Dynamical Effects in an Enzyme-Catalyzed Proton Transfer Reaction. J. Am. Chem. Soc. 1999, 121, 2253–2258. [Google Scholar] [CrossRef]
- Tresadern, G.; McNamara, J.P.; Mohr, M.; Wang, H.; Burton, N.A.; Hillier, I.H. Calculations of hydrogen tunnelling and enzyme catalysis: A comparison of liver alcohol dehydrogenase, methylamine dehydrogenase and soybean lipoxygenase. Chem. Phys. Lett. 2002, 358, 489–494. [Google Scholar] [CrossRef]
- Garcia-Viloca, M.; Alhambra, C.; Truhlar, D.G.; Gao, J. Hydride transfer catalyzed by xylose isomerase: Mechanism and quantum effects. J. Comput. Chem. 2003, 24, 177–190. [Google Scholar] [CrossRef]
- Doll, K.M.; Bender, B.R.; Finke, R.G. The first experimental test of the hypothesis that enzymes have evolved to enhance hydrogen tunneling. J. Am. Chem. Soc. 2003, 125, 10877–10884. [Google Scholar] [CrossRef]
- Doll, K.M.; Finke, R.G. A compelling experimental test of the hypothesis that enzymes have evolved to enhance quantum mechanical tunneling in hydrogen transfer reactions: The beta-neopentylcobalamin system combined with prior adocobalamin data. Inorg. Chem. 2003, 42, 4849–4856. [Google Scholar] [CrossRef]
- Feierberg, I.; Luzhkov, V.; Aqvist, J. Computer simulation of primary kinetic isotope effects in the proposed rate-limiting step of the glyoxalase I catalyzed reaction. J. Biol. Chem. 2000, 275, 22657–22662. [Google Scholar] [CrossRef] [Green Version]
- Williams, I.H. Quantum catalysis? A comment on tunnelling contributions for catalysed and uncatalysed reactions. J. Phys. Org. Chem. 2010, 23, 685–689. [Google Scholar] [CrossRef] [Green Version]
- Olsson, M.H.M.; Mavri, J.; Warshel, A. Transition state theory can be used in studies of enzyme catalysis: Lessons from simulations of tunnelling and dynamical effects in lipoxygenase and other systems. Phil. Trans. Roy. Soc. B Biol. Sci. 2006, 361, 1417–1432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sikorski, R.S.; Wang, L.; Markham, K.A.; Rajagopalan, P.T.R.; Benkovic, S.J.; Kohen, A. Tunneling and coupled motion in the Escherichia coli dihydrofolate reductase catalysis. J. Am. Chem. Soc. 2004, 126, 4778–4779. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Damo, S.M.; Lee, S.Y.; Wemmer, D.; Klinman, J.P. Structure and hydride transfer mechanism of a moderate thermophilic dihydrofolate reductase from Bacillus stearothermophilus and comparison to its mesophilic and hyperthermophilic homologues. Biochemistry 2005, 44, 11428–11439. [Google Scholar] [CrossRef] [PubMed]
- Benner, S.A.; Sassi, S.O.; Gaucher, E.A. Molecular paleoscience: Systems biology from the past. Adv. Enzymol. Relat. Areas. Mol. Biol. 2007, 75, 1. [Google Scholar] [PubMed]
- Hall, B.G. Simple and accurate estimation of ancestral protein sequences. Proc. Natl. Acad. Sci. USA 2006, 103, 5431–5436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez-Jimenez, R.; Ingles-Prieto, A.; Zhao, Z.M.; Sanchez-Romero, I.; Alegre-Cebollada, J.; Kosuri, P.; Garcia-Manyes, S.; Kappock, T.J.; Tanokura, M.; Holmgren, A.; et al. Single-molecule paleoenzymology probes the chemistry of resurrected enzymes. Nat. Struct. Mol. Biol. 2011, 18, 592–596. [Google Scholar] [CrossRef] [Green Version]
- Thornton, J.W. Resurrecting ancient genes: Experimental analysis of extinct molecules. Nat Rev Genet 2004, 5, 366–375. [Google Scholar] [CrossRef]
- Devault, D.O.N.; Parkes, J.H.; Chance, B. Electron Tunnelling in Cytochromes. Nature 1967, 215, 642–644. [Google Scholar] [CrossRef]
- Liu, H.; Warshel, A. Origin of the temperature dependence of isotope effects in enzymatic reactions: The case of dihydrofolate reductase. J. Phys. Chem. B 2007, 111, 7852–7861. [Google Scholar] [CrossRef]
- Kohen, A. Dihydrofolate reductase as a model for studies of enzyme dynamics and catalysis. F1000Research 2015, 4, F1000 Faculty Rev-1464. [Google Scholar] [CrossRef] [Green Version]
- Hammes-Schiffer, S. Hydrogen tunneling and protein motion in enzyme reactions. Acc. Chem. Res. 2006, 39, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Klinman, J.P.; Kohen, A. Hydrogen tunneling links protein dynamics to enzyme catalysis. Annu. Rev. Biochem. 2013, 82, 471–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klinman, J.P.; Offenbacher, A.R. Understanding Biological Hydrogen Transfer Through the Lens of Temperature Dependent Kinetic Isotope Effects. Acc. Chem. Res. 2018, 51, 1966–1974. [Google Scholar] [CrossRef] [PubMed]
- Hay, S.; Pudney, C.; Hothi, P.; Johannissen, L.O.; Masgrau, L.; Pang, J.; Leys, D.; Sutcliffe, M.J.; Scrutton, N.S. Atomistic insight into the origin of the temperature-dependence of kinetic isotope effects and H-tunnelling in enzyme systems is revealed through combined experimental studies and biomolecular simulation. Biochem. Soc. Trans. 2008, 36, 16–21. [Google Scholar] [CrossRef] [Green Version]
- Offenbacher, A.R.; Sharma, A.; Doan, P.E.; Klinman, J.P.; Hoffman, B.M. The Soybean Lipoxygenase-Substrate Complex: Correlation between the Properties of Tunneling-Ready States and ENDOR-Detected Structures of Ground States. Biochemistry 2020, 59, 901–910. [Google Scholar] [CrossRef]
- Hu, S.; Soudackov, A.V.; Hammes-Schiffer, S.; Klinman, J.P. Enhanced Rigidification within a Double Mutant of Soybean Lipoxygenase Provides Experimental Support for Vibronically Nonadiabatic Proton-Coupled Electron Transfer Models. ACS Catal. 2017, 7, 3569–3574. [Google Scholar] [CrossRef]
- Kuznetsov, A.M.; Ulstrup, J. Proton and hydrogen atom tunnelling in hydrolytic and redox enzyme catalysis. Can. J. Chem. 1999, 77, 1085–1096. [Google Scholar] [CrossRef]
- Knapp, M.J.; Rickert, K.; Klinman, J.P. Temperature-dependent isotope effects in soybean lipoxygenase- 1: Correlating hydrogen tunneling with protein dynamics. J. Am. Chem. Soc. 2002, 124, 3865–3874. [Google Scholar] [CrossRef]
- Johannissen, L.O.; Irebo, T.; Sjöin, M.; Johansson, O.; Hammarström, L. The kinetic effect of internal hydrogen bonds on proton-coupled electron transfer from phenols: A theoretical analysis with modeling of experimental data. J. Phys. Chem. B 2009, 113, 16214–16225. [Google Scholar] [CrossRef]
- Hatcher, E.; Soudackov, A.; Hammes-Schiffer, S. Nonadiabatic proton-coupled electron transfer reactions: Impact of donor-acceptor vibrations, reorganization energies, and couplings on dynamics and rates. J. Phys. Chem. B 2005, 109, 18565–18574. [Google Scholar] [CrossRef]
- Meyer, M.P.; Tomchick, D.R.; Klinman, J.P. Enzyme structure and dynamics affect hydrogen tunneling: The impact of a remote side chain (I553) in soybean lipoxygenase-1. Proc. Natl. Acad. Sci. USA 2008, 105, 1146–1151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pudney, C.R.; Johannissen, L.O.; Sutcliffe, M.J.; Hay, S.; Scrutton, N.S. Direct analysis of donor-acceptor distance and relationship to isotope effects and the force constant for barrier compression in enzymatic H-tunneling reactions. J. Am. Chem. Soc. 2010, 132, 11329–11335. [Google Scholar] [CrossRef] [PubMed]
- Hatcher, E.; Soudackov, A.V.; Hammes-Schiffer, S. Proton-coupled electron transfer in soybean lipoxygenase: Dynamical behavior and temperature dependence of kinetic isotope effects. J. Am. Chem. Soc. 2007, 129, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Johannissen, L.O.; Hay, S.; Scrutton, N.S.; Sutcliffe, M.J. Proton tunneling in aromatic amine dehydrogenase is driven by a short-range sub-picosecond promoting vibration: Consistency of simulation and theory with experiment. J. Phys. Chem. B 2007, 111, 2631–2638. [Google Scholar] [CrossRef]
- Johannissen, L.O.; Scrutton, N.S.; Sutcliffe, M.J. How does pressure affect barrier compression and isotope effects in an enzymatic hydrogen tunneling reaction? Angew. Chem. Int. Ed. Engl. 2011, 50, 2129–2132. [Google Scholar] [CrossRef]
- Hay, S.; Johannissen, L.O.; Hothi, P.; Sutcliffe, M.J.; Scrutton, N.S. Pressure effects on enzyme-catalyzed quantum tunneling events arise from protein-specific structural and dynamic changes. J. Am. Chem. Soc. 2012, 134, 9749–9754. [Google Scholar] [CrossRef]
- Hay, S.; Pudney, C.R.; Sutcliffe, M.J.; Scrutton, N.S. Probing active site geometry using high pressure and secondary isotope effects in an enzyme-catalysed ‘deep’ H-tunnelling reaction. J. Phys. Org. Chem. 2010, 23, 696–701. [Google Scholar] [CrossRef] [Green Version]
- Hay, S.; Scrutton, N.S. Incorporation of hydrostatic pressure into models of hydrogen tunneling highlights a role for pressure-modulated promoting vibrations. Biochemistry 2008, 47, 9880–9887. [Google Scholar] [CrossRef]
- Hay, S.; Sutcliffe, M.J.; Scrutton, N.S. Promoting motions in enzyme catalysis probed by pressure studies of kinetic isotope effects. Proc. Natl. Acad. Sci. USA 2007, 104, 507–512. [Google Scholar] [CrossRef] [Green Version]
- Pudney, C.R.; McGrory, T.; Lafite, P.; Pang, J.Y.; Hay, S.; Leys, D.; Sutcliffe, M.J.; Scrutton, N.S. Parallel pathways and free-energy landscapes for enzymatic hydride transfer probed by hydrostatic pressure. ChemBioChem 2009, 10, 1379–1384. [Google Scholar] [CrossRef]
- Isaacs, N.S. Isotope Effects in Organic Chemistry; Buncel, E., Lee, C.C., Eds.; Elsevier: London, UK, 1984; Volume 6, pp. 67–105. [Google Scholar]
- Northrop, D.B. Unusual origins of isotope effects in enzyme-catalysed reactions. Phil. Trans. R. Soc. B 2006, 361, 1341–1349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isaacs, N.S.; Javaid, K.; Rannala, E. Pressure Effects on Proton Tunnelling. Nature 1977, 268, 372. [Google Scholar] [CrossRef] [PubMed]
- Northrop, D.B. Effects of high pressure on isotope effects and hydrogen tunneling. J. Am. Chem. Soc. 1999, 121, 3521–3524. [Google Scholar] [CrossRef]
- Hoeven, R.; Heyes, D.J.; Hay, S.; Scrutton, N.S. Does the pressure dependence of kinetic isotope effects report usefully on dynamics in enzyme H-transfer reactions? FEBS J. 2015, 282, 3243–3255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ledward, D.A. Effects of pressure on protein structure. High Press. Res. 2000, 19, 391–400. [Google Scholar] [CrossRef]
- Ruiz-Pernia, J.J.; Behiry, E.; Luk, L.Y.P.; Loveridge, E.J.; Tunon, I.; Moliner, V.; Allemann, R.K. Minimization of dynamic effects in the evolution of dihydrofolate reductase. Chem. Sci. 2016, 7, 3248–3255. [Google Scholar] [CrossRef] [Green Version]
- Warshel, A.; Bora, R.P. Perspective: Defining and quantifying the role of dynamics in enzyme catalysis. J. Chem. Phys. 2016, 144, 180901. [Google Scholar] [CrossRef]
- Warshel, A. Electrostatic origin of the catalytic power of enzymes and the role of preorganized active sites. J. Biol. Chem. 1998, 273, 27035–27038. [Google Scholar] [CrossRef] [Green Version]
- Schramm, V.L.; Schwartz, S.D. Promoting Vibrations and the Function of Enzymes. Emerging Theoretical and Experimental Convergence. Biochemistry 2018, 57, 3299–3308. [Google Scholar] [CrossRef]
- Johannissen, L.O.; Scrutton, N.S.; Sutcliffe, M.J. The enzyme aromatic amine dehydrogenase induces a substrate conformation crucial for promoting vibration that significantly reduces the effective potential energy barrier to proton transfer. J. R. Soc. Interface 2008, 5 (Suppl. 3), S225–S232. [Google Scholar] [CrossRef]
- Scrutton, N.S.; Groot, M.L.; Heyes, D.J. Excited state dynamics and catalytic mechanism of the light-driven enzyme protochlorophyllide oxidoreductase. Phys. Chem. Chem. Phys. 2012, 14, 8818–8824. [Google Scholar] [CrossRef] [PubMed]
- Heyes, D.J.; Hunter, C.N. Making light work of enzyme catalysis: Protochlorophyllide oxidoreductase. Trends Biochem. Sci. 2005, 30, 642–649. [Google Scholar] [CrossRef] [PubMed]
- Menon, B.R.; Waltho, J.P.; Scrutton, N.S.; Heyes, D.J. Cryogenic and laser photoexcitation studies identify multiple roles for active site residues in the light-driven enzyme protochlorophyllide oxidoreductase. J. Biol. Chem. 2009, 284, 18160–18166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heyes, D.J.; Sakuma, M.; de Visser, S.P.; Scrutton, N.S. Nuclear quantum tunneling in the light-activated enzyme protochlorophyllide oxidoreductase. J. Biol. Chem. 2009, 284, 3762–3767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heyes, D.J.; Sakuma, M.; Scrutton, N.S. Solvent-slaved protein motions accompany proton but not hydride tunneling in light-activated protochlorophyllide oxidoreductase. Angew. Chem. Int. Ed. Engl. 2009, 48, 3850–3853. [Google Scholar] [CrossRef]
- Hoeven, R.; Hardman, S.J.; Heyes, D.J.; Scrutton, N.S. Cross-Species Analysis of Protein Dynamics Associated with Hydride and Proton Transfer in the Catalytic Cycle of the Light-Driven Enzyme Protochlorophyllide Oxidoreductase. Biochemistry 2016, 55, 903–913. [Google Scholar] [CrossRef] [PubMed]
- Heyes, D.J.; Hardman, S.J.; Hedison, T.M.; Hoeven, R.; Greetham, G.M.; Towrie, M.; Scrutton, N.S. Excited-state charge separation in the photochemical mechanism of the light-driven enzyme protochlorophyllide oxidoreductase. Angew. Chem. Int. Ed. Engl. 2015, 54, 1512–1515. [Google Scholar] [CrossRef] [Green Version]
- Menon, B.R.; Hardman, S.J.; Scrutton, N.S.; Heyes, D.J. Multiple active site residues are important for photochemical efficiency in the light-activated enzyme protochlorophyllide oxidoreductase (POR). J. Photochem. Photobiol. B 2016, 161, 236–243. [Google Scholar] [CrossRef]
- Menon, B.R.; Davison, P.A.; Hunter, C.N.; Scrutton, N.S.; Heyes, D.J. Mutagenesis alters the catalytic mechanism of the light-driven enzyme protochlorophyllide oxidoreductase. J. Biol. Chem. 2010, 285, 2113–2119. [Google Scholar] [CrossRef] [Green Version]
- Heyes, D.J.; Levy, C.; Sakuma, M.; Robertson, D.L.; Scrutton, N.S. A twin-track approach has optimized proton and hydride transfer by dynamically coupled tunneling during the evolution of protochlorophyllide oxidoreductase. J. Biol. Chem. 2011, 286, 11849–11854. [Google Scholar] [CrossRef] [Green Version]
- Heyes, D.J.; Heathcote, P.; Rigby, S.E.; Palacios, M.A.; van Grondelle, R.; Hunter, C.N. The first catalytic step of the light-driven enzyme protochlorophyllide oxidoreductase proceeds via a charge transfer complex. J. Biol. Chem. 2006, 281, 26847–26853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garrone, A.; Archipowa, N.; Zipfel, P.F.; Hermann, G.; Dietzek, B. Plant Protochlorophyllide Oxidoreductases A and B: Catalytic Efficiency and Initial Reaction Steps. J. Biol. Chem. 2015, 290, 28530–28539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Archipowa, N.; Kutta, R.J.; Heyes, D.J.; Scrutton, N.S. Stepwise Hydride Transfer in a Biological System: Insights into the Reaction Mechanism of the Light-Dependent Protochlorophyllide Oxidoreductase. Angew. Chem. 2018, 130, 2712–2716. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Heyes, D.J.; Feng, L.; Sun, W.; Johannissen, L.O.; Liu, H.; Levy, C.W.; Li, X.; Yang, J.; Yu, X.; et al. Structural basis for enzymatic photocatalysis in chlorophyll biosynthesis. Nature 2019, 574, 722–725. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.S.; Zhang, W.L.; Wang, Q.; Li, Y.S.; Wang, X.; Zhang, M.; Liu, L. Crystal structures of cyanobacterial light-dependent protochlorophyllide oxidoreductase. Proc. Natl. Acad. Sci. USA 2020, 117, 8455–8461. [Google Scholar] [CrossRef] [PubMed]
- Bassham, J.A.; Calvin, M. The path of carbon in photosynthesis. In Die CO2-Assimilation/The Assimilation of Carbon Dioxide; Springer: Berlin/Heidelberg, Germany, 1960; pp. 884–922. [Google Scholar]
- Bolton, J.R.; Hall, D.O. The maximum efficiency of photosynthesis. Photochem. Photobiol. 1991, 53, 545–548. [Google Scholar] [CrossRef]
- Zhu, X.-G.; Long, S.P.; Ort, D.R. What is the maximum efficiency with which photosynthesis can convert solar energy into biomass? Curr. Opin. Biotechnol. 2008, 19, 153–159. [Google Scholar] [CrossRef]
- Chen, G.-Y.; Lambert, N.; Li, C.-M.; Chen, Y.-N.; Nori, F. Rerouting excitation transfers in the Fenna-Matthews-Olson complex. Phys. Rev. E 2013, 88, 032120. [Google Scholar] [CrossRef] [Green Version]
- Panitchayangkoon, G.; Voronine, D.V.; Abramavicius, D.; Caram, J.R.; Lewis, N.H.; Mukamel, S.; Engel, G.S. Direct evidence of quantum transport in photosynthetic light-harvesting complexes. Proc. Natl. Acad. Sci. USA 2011, 108, 20908–20912. [Google Scholar] [CrossRef] [Green Version]
- Förster, T. Transfer mechanisms of electronic excitation energy. Radiat. Res. Suppl. 1960, 2, 326–339. [Google Scholar] [CrossRef]
- Perrin, F. Théorie quantique des transferts d’activation entre molécules de même espèce. Cas des solutions fluorescentes. Ann. Phys. 1932, 10, 283–314. [Google Scholar] [CrossRef]
- Forster, T. Energiewanderung und fluoreszenz. Naturwissenschaften 1946, 33, 166–175. [Google Scholar] [CrossRef]
- Alden, R.; Johnson, E.; Nagarajan, V.; Parson, W.; Law, C.; Cogdell, R. Calculations of Spectroscopic Properties of the LH2 Bacteriochlorophyll− Protein Antenna Complex from Rhodopseudomonas Acidophila. J. Phys. Chem. B 1997, 101, 4667–4680. [Google Scholar] [CrossRef]
- Chachisvilis, M.; Kühn, O.; Pullerits, T.; Sundström, V. Excitons in photosynthetic purple bacteria: Wavelike motion or incoherent hopping? J. Phys. Chem. B 1997, 101, 7275–7283. [Google Scholar] [CrossRef]
- Jang, S.; Newton, M.D.; Silbey, R.J. Multichromophoric Förster resonance energy transfer from B800 to B850 in the light harvesting complex 2: Evidence for subtle energetic optimization by purple bacteria. J. Phys. Chem. B 2007, 111, 6807–6814. [Google Scholar] [CrossRef] [PubMed]
- Olbrich, C.; Jansen, T.L.; Liebers, J.; Aghtar, M.; Strümpfer, J.; Schulten, K.; Knoester, J.; Kleinekathöfer, U. From atomistic modeling to excitation transfer and two-dimensional spectra of the FMO light-harvesting complex. J. Phys. Chem. B 2011, 115, 8609–8621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sauer, K.; Cogdell, R.J.; Prince, S.M.; Freer, A.; Isaacs, N.W.; Scheer, H. Structure-based calculations of the optical spectra of the LH2 bacteriochlorophyll-protein complex from Rhodopseudomonas acidophila. Photochem. Photobiol. 1996, 64, 564–576. [Google Scholar] [CrossRef]
- Scholes, G.D.; Gould, I.R.; Cogdell, R.J.; Fleming, G.R. Ab initio molecular orbital calculations of electronic couplings in the LH2 bacterial light-harvesting complex of Rps. acidophila. J. Phys. Chem. B 1999, 103, 2543–2553. [Google Scholar] [CrossRef]
- Blankenship, R.E. Molecular Mechanisms of Photosynthesis; John Wiley & Sons: Hoboken, NJ, USA, 2014. [Google Scholar]
- Fleming, G.R.; van Grondelle, R. Femtosecond spectroscopy of photosynthetic light-harvesting systems. Curr. Opin. Struct. Biol. 1997, 7, 738–748. [Google Scholar] [CrossRef]
- Van Amerongen, H.; Van Grondelle, R. Photosynthetic Excitons; World Scientific: Singapore, 2000. [Google Scholar]
- Nakamura, Y.; Aratani, N.; Osuka, A. Cyclic porphyrin arrays as artificial photosynthetic antenna: Synthesis and excitation energy transfer. Chem. Soc. Rev. 2007, 36, 831–845. [Google Scholar] [CrossRef]
- Fassioli, F.; Dinshaw, R.; Arpin, P.C.; Scholes, G.D. Photosynthetic light harvesting: Excitons and coherence. J. R. Soc. Interface 2014, 11, 20130901. [Google Scholar] [CrossRef] [PubMed]
- Jumper, C.C.; Rafiq, S.; Wang, S.; Scholes, G.D. From coherent to vibronic light harvesting in photosynthesis. Curr. Opin. Chem. Biol. 2018, 47, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.-P. The electronic couplings in electron transfer and excitation energy transfer. Acc. Chem. Res. 2009, 42, 509–518. [Google Scholar] [CrossRef] [PubMed]
- Seibt, J.; Mančal, T. Ultrafast energy transfer with competing channels: Non-equilibrium Förster and Modified Redfield theories. J. Chem. Phys. 2017, 146, 174109. [Google Scholar] [CrossRef] [PubMed]
- Tao, M.-J.; Zhang, N.-N.; Wen, P.-Y.; Deng, F.-G.; Ai, Q.; Long, G.-L. Coherent and incoherent theories for photosynthetic energy transfer. Sci. Bull. 2020, 65, 318–328. [Google Scholar] [CrossRef] [Green Version]
- Ishizaki, A.; Tanimura, Y. Quantum dynamics of system strongly coupled to low-temperature colored noise bath: Reduced hierarchy equations approach. J. Phys. Soc. Jpn. 2005, 74, 3131–3134. [Google Scholar] [CrossRef] [Green Version]
- Ishizaki, A.; Fleming, G.R. Theoretical examination of quantum coherence in a photosynthetic system at physiological temperature. Proc. Natl. Acad. Sci. USA 2009, 106, 17255–17260. [Google Scholar] [CrossRef] [Green Version]
- Lambert, N.; Ahmed, S.; Cirio, M.; Nori, F. Modelling the ultra-strongly coupled spin-boson model with unphysical modes. Nat. Commun. 2019, 10, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Prior, J.; Chin, A.W.; Huelga, S.F.; Plenio, M.B. Efficient simulation of strong system-environment interactions. Phys. Rev. Lett. 2010, 105, 050404. [Google Scholar] [CrossRef]
- Chin, A.W.; Rivas, Á.; Huelga, S.F.; Plenio, M.B. Exact mapping between system-reservoir quantum models and semi-infinite discrete chains using orthogonal polynomials. J. Math. Phys. 2010, 51, 092109. [Google Scholar] [CrossRef] [Green Version]
- Chin, A.W.; Huelga, S.F.; Plenio, M.B. Chain representations of open quantum systems and their numerical simulation with time-adaptive density matrix renormalisation group methods. In Semiconductors and Semimetals; Elsevier: Amsterdam, The Netherlands, 2011; Volume 85, pp. 115–143. [Google Scholar]
- Chin, A.; Prior, J.; Rosenbach, R.; Caycedo-Soler, F.; Huelga, S.F.; Plenio, M.B. The role of non-equilibrium vibrational structures in electronic coherence and recoherence in pigment–protein complexes. Nat. Phys. 2013, 9, 113–118. [Google Scholar] [CrossRef]
- Gelzinis, A.; Augulis, R.; Butkus, V.; Robert, B.; Valkunas, L. Two-dimensional spectroscopy for non-specialists. Biochim. Biophys. Acta Bioenerg. 2019, 1860, 271–285. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Allodi, M.A.; Engel, G.S. Quantum coherences reveal excited-state dynamics in biophysical systems. Nat. Rev. Chem. 2019, 3, 477–490. [Google Scholar] [CrossRef]
- Lewis, K.L.; Ogilvie, J.P. Probing photosynthetic energy and charge transfer with two-dimensional electronic spectroscopy. J. Phys. Chem. Lett. 2012, 3, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Ostroumov, E.E.; Mulvaney, R.M.; Cogdell, R.J.; Scholes, G.D. Broadband 2D electronic spectroscopy reveals a carotenoid dark state in purple bacteria. Science 2013, 340, 52–56. [Google Scholar] [CrossRef] [Green Version]
- Chin, A.W.; Huelga, S.F.; Plenio, M.B. Quantum metrology in non-Markovian environments. Phys. Rev. Lett. 2012, 109, 233601. [Google Scholar] [CrossRef] [Green Version]
- Baghbanzadeh, S.; Kassal, I. Distinguishing the roles of energy funnelling and delocalization in photosynthetic light harvesting. Phys. Chem. Chem. Phys. 2016, 18, 7459–7467. [Google Scholar] [CrossRef] [Green Version]
- Engel, G.S.; Calhoun, T.R.; Read, E.L.; Ahn, T.-K.; Mančal, T.; Cheng, Y.-C.; Blankenship, R.E.; Fleming, G.R. Evidence for wavelike energy transfer through quantum coherence in photosynthetic systems. Nature 2007, 446, 782–786. [Google Scholar] [CrossRef]
- Sarovar, M.; Ishizaki, A.; Fleming, G.R.; Whaley, K.B. Quantum entanglement in photosynthetic light-harvesting complexes. Nat. Phys. 2010, 6, 462–467. [Google Scholar] [CrossRef]
- Cao, J.; Cogdell, R.J.; Coker, D.F.; Duan, H.-G.; Hauer, J.; Kleinekathöfer, U.; Jansen, T.L.; Mančal, T.; Miller, R.D.; Ogilvie, J.P. Quantum biology revisited. Sci. Adv. 2020, 6, eaaz4888. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.; Cheng, Y.-C.; Fleming, G.R. Coherence dynamics in photosynthesis: Protein protection of excitonic coherence. Science 2007, 316, 1462–1465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryu, I.S.; Dong, H.; Fleming, G.R. Role of electronic-vibrational mixing in enhancing vibrational coherences in the ground electronic states of photosynthetic bacterial reaction center. J. Phys. Chem. B 2014, 118, 1381–1388. [Google Scholar] [CrossRef] [PubMed]
- Ma, F.; Romero, E.; Jones, M.R.; Novoderezhkin, V.I.; van Grondelle, R. Both electronic and vibrational coherences are involved in primary electron transfer in bacterial reaction center. Nat. Commun. 2019, 10, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Christensson, N.; Kauffmann, H.F.; Pullerits, T.; Mancal, T. Origin of long-lived coherences in light-harvesting complexes. J. Phys. Chem. B 2012, 116, 7449–7454. [Google Scholar] [CrossRef]
- Thyrhaug, E.; Tempelaar, R.; Alcocer, M.J.; Žídek, K.; Bína, D.; Knoester, J.; Jansen, T.L.; Zigmantas, D. Identification and characterization of diverse coherences in the Fenna–Matthews–Olson complex. Nat. Chem. 2018, 10, 780–786. [Google Scholar] [CrossRef]
- Dean, J.C.; Mirkovic, T.; Toa, Z.S.; Oblinsky, D.G.; Scholes, G.D. Vibronic enhancement of algae light harvesting. Chem 2016, 1, 858–872. [Google Scholar] [CrossRef] [Green Version]
- Tiwari, V.; Peters, W.K.; Jonas, D.M. Electronic resonance with anticorrelated pigment vibrations drives photosynthetic energy transfer outside the adiabatic framework. Proc. Natl. Acad. Sci. USA 2013, 110, 1203–1208. [Google Scholar] [CrossRef] [Green Version]
- Paleček, D.; Edlund, P.; Westenhoff, S.; Zigmantas, D. Quantum coherence as a witness of vibronically hot energy transfer in bacterial reaction center. Sci. Adv. 2017, 3, e1603141. [Google Scholar] [CrossRef] [Green Version]
- Calhoun, T.R.; Ginsberg, N.S.; Schlau-Cohen, G.S.; Cheng, Y.-C.; Ballottari, M.; Bassi, R.; Fleming, G.R. Quantum coherence enabled determination of the energy landscape in light-harvesting complex II. J. Phys. Chem. B 2009, 113, 16291–16295. [Google Scholar] [CrossRef]
- Collini, E.; Wong, C.Y.; Wilk, K.E.; Curmi, P.M.; Brumer, P.; Scholes, G.D. Coherently wired light-harvesting in photosynthetic marine algae at ambient temperature. Nature 2010, 463, 644–647. [Google Scholar] [CrossRef]
- Romero, E.; Augulis, R.; Novoderezhkin, V.I.; Ferretti, M.; Thieme, J.; Zigmantas, D.; Van Grondelle, R. Quantum coherence in photosynthesis for efficient solar-energy conversion. Nat. Phys. 2014, 10, 676–682. [Google Scholar] [CrossRef]
- Panitchayangkoon, G.; Hayes, D.; Fransted, K.A.; Caram, J.R.; Harel, E.; Wen, J.; Blankenship, R.E.; Engel, G.S. Long-lived quantum coherence in photosynthetic complexes at physiological temperature. Proc. Natl. Acad. Sci. USA 2010, 107, 12766–12770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karki, K.J.; Chen, J.; Sakurai, A.; Shi, Q.; Gardiner, A.T.; Kühn, O.; Cogdell, R.J.; Pullerits, T. Before Förster. Initial excitation in photosynthetic light harvesting. Chem. Sci. 2019, 10, 7923–7928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irgen-Gioro, S.; Gururangan, K.; Saer, R.G.; Blankenship, R.E.; Harel, E. Electronic coherence lifetimes of the Fenna–Matthews–Olson complex and light harvesting complex II. Chem. Sci. 2019, 10, 10503–10509. [Google Scholar] [CrossRef] [PubMed]
- Hildner, R.; Brinks, D.; Nieder, J.B.; Cogdell, R.J.; van Hulst, N.F. Quantum coherent energy transfer over varying pathways in single light-harvesting complexes. Science 2013, 340, 1448–1451. [Google Scholar] [CrossRef] [PubMed]
- Chin, A.; Huelga, S.F.; Plenio, M.B. Coherence and decoherence in biological systems: Principles of noise-assisted transport and the origin of long-lived coherences. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2012, 370, 3638–3657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harel, E.; Engel, G.S. Quantum coherence spectroscopy reveals complex dynamics in bacterial light-harvesting complex 2 (LH2). Proc. Natl. Acad. Sci. USA 2012, 109, 706–711. [Google Scholar] [CrossRef] [Green Version]
- Fischer, M.; Gutiérrez-Medina, B.; Raizen, M. Observation of the quantum Zeno and anti-Zeno effects in an unstable system. Phys. Rev. Lett. 2001, 87, 040402. [Google Scholar] [CrossRef] [Green Version]
- Rebentrost, P.; Mohseni, M.; Kassal, I.; Lloyd, S.; Aspuru-Guzik, A. Environment-assisted quantum transport. New J. Phys. 2009, 11, 033003. [Google Scholar] [CrossRef]
- Mohseni, M.; Shabani, A.; Lloyd, S.; Rabitz, H. Energy-scales convergence for optimal and robust quantum transport in photosynthetic complexes. J. Chem. Phys. 2014, 140, 01B609_601. [Google Scholar] [CrossRef] [Green Version]
- Jang, S.J. Robust and Fragile Quantum Effects in the Transfer Kinetics of Delocalized Excitons between B850 Units of LH2 Complexes. J. Phys. Chem. Lett. 2018, 9, 6576–6583. [Google Scholar] [CrossRef] [PubMed]
- Prokhorenko, V.I.; Nagy, A.M.; Waschuk, S.A.; Brown, L.S.; Birge, R.R.; Miller, R.D. Coherent control of retinal isomerization in bacteriorhodopsin. Science 2006, 313, 1257–1261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bickel-Sandkötter, S.; Gärtner, W.; Dane, M. Conversion of energy in halobacteria: ATP synthesis and phototaxis. Arch. Microbiol. 1996, 166, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Oren, A. The order halobacteriales. Prokaryotes 2006, 3, 113–164. [Google Scholar]
- Frydrych, M.; Silfsten, P.; Parkkinen, S.; Parkkinen, J.; Jaaskelainen, T. Color sensitive retina based on bacteriorhodopsin. Biosystems 2000, 54, 131–140. [Google Scholar] [CrossRef]
- Hampp, N. Bacteriorhodopsin as a photochromic retinal protein for optical memories. Chem. Rev. 2000, 100, 1755–1776. [Google Scholar] [CrossRef] [PubMed]
- Margesin, R.; Schinner, F. Potential of halotolerant and halophilic microorganisms for biotechnology. Extremophiles 2001, 5, 73–83. [Google Scholar] [CrossRef]
- Rakovich, A.; Sukhanova, A.; Bouchonville, N.; Lukashev, E.; Oleinikov, V.; Artemyev, M.; Lesnyak, V.; Gaponik, N.; Molinari, M.; Troyon, M. Resonance energy transfer improves the biological function of bacteriorhodopsin within a hybrid material built from purple membranes and semiconductor quantum dots. Nano Lett. 2010, 10, 2640–2648. [Google Scholar] [CrossRef]
- Ghosh, P.K.; Smirnov, A.Y.; Nori, F. Modeling light-driven proton pumps in artificial photosynthetic reaction centers. J. Chem. Phys. 2009, 131, 07B610. [Google Scholar] [CrossRef] [Green Version]
- Proppe, A.H.; Li, Y.C.; Aspuru-Guzik, A.; Berlinguette, C.P.; Chang, C.J.; Cogdell, R.; Doyle, A.G.; Flick, J.; Gabor, N.M.; van Grondelle, R. Bioinspiration in light harvesting and catalysis. Nat. Rev. Mater. 2020, 5, 828–846. [Google Scholar] [CrossRef]
- Lee, S.H.; Matula, A.J.; Hu, G.; Troiano, J.L.; Karpovich, C.J.; Crabtree, R.H.; Batista, V.S.; Brudvig, G.W. Strongly coupled phenazine–porphyrin dyads: Light-harvesting molecular assemblies with broad absorption coverage. ACS Appl. Mater. Interfaces 2019, 11, 8000–8008. [Google Scholar] [CrossRef] [PubMed]
- Prinz, J.-H.; Wu, H.; Sarich, M.; Keller, B.; Senne, M.; Held, M.; Chodera, J.D.; Schütte, C.; Noé, F. Markov models of molecular kinetics: Generation and validation. J. Chem. Phys. 2011, 134, 174105. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, P.K.; Smirnov, A.Y.; Nori, F. Quantum effects in energy and charge transfer in an artificial photosynthetic complex. J. Chem. Phys. 2011, 134, 06B611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roscioli, J.D.; Ghosh, S.; LaFountain, A.M.; Frank, H.A.; Beck, W.F. Structural Tuning of Quantum Decoherence and Coherent Energy Transfer in Photosynthetic Light Harvesting. J. Phys. Chem. Lett. 2018, 9, 5071–5077. [Google Scholar] [CrossRef]
- Delor, M.; Dai, J.; Roberts, T.D.; Rogers, J.R.; Hamed, S.M.; Neaton, J.B.; Geissler, P.L.; Francis, M.B.; Ginsberg, N.S. Exploiting chromophore–protein interactions through linker engineering to tune photoinduced dynamics in a biomimetic light-harvesting platform. J. Am. Chem. Soc. 2018, 140, 6278–6287. [Google Scholar] [CrossRef]
- Wang, L.; Griffin, G.B.; Zhang, A.; Zhai, F.; Williams, N.E.; Jordan, R.F.; Engel, G.S. Controlling quantum-beating signals in 2D electronic spectra by packing synthetic heterodimers on single-walled carbon nanotubes. Nat. Chem. 2017, 9, 219. [Google Scholar] [CrossRef]
- Freixas, V.; Tretiak, S.; Makhov, D.V.; Shalashilin, D.V.; Fernandez-Alberti, S. Vibronic Quantum Beating between Electronic Excited States in a Heterodimer. J. Phys. Chem. B 2020, 124, 3992–4001. [Google Scholar] [CrossRef]
- McCleese, C.; Yu, Z.; Esemoto, N.N.; Kolodziej, C.; Maiti, B.; Bhandari, S.; Dunietz, B.D.; Burda, C.; Ptaszek, M. Excitonic interactions in bacteriochlorin homo-dyads enable charge transfer: A new approach to the artificial photosynthetic special pair. J. Phys. Chem. B 2018, 122, 4131–4140. [Google Scholar] [CrossRef]
- Tiwari, V.; Matutes, Y.A.; Konar, A.; Yu, Z.; Ptaszek, M.; Bocian, D.F.; Holten, D.; Kirmaier, C.; Ogilvie, J.P. Strongly coupled bacteriochlorin dyad studied using phase-modulated fluorescence-detected two-dimensional electronic spectroscopy. Opt. Express 2018, 26, 22327–22341. [Google Scholar] [CrossRef]
- Shoji, S.; Tamiaki, H. Supramolecular light-harvesting antenna system by co-aggregates of zinc (bacterio) chlorophyll-a derivatives with biomimetic chlorosomal self-assemblies. Dye. Pigment. 2019, 160, 514–518. [Google Scholar] [CrossRef]
- Shoji, S.; Nomura, Y.; Tamiaki, H. Heterodimers of zinc and free-base chlorophyll derivatives co-assembled in biomimetic chlorosomal J-aggregates. Photochem. Photobiol. Sci. 2019, 18, 555–562. [Google Scholar] [CrossRef] [PubMed]
- Pandya, R.; Chen, R.Y.; Cheminal, A.; Thomas, T.; Thampi, A.; Tanoh, A.; Richter, J.; Shivanna, R.; Deschler, F.; Schnedermann, C. Observation of Vibronic-Coupling-Mediated Energy Transfer in Light-Harvesting Nanotubes Stabilized in a Solid-State Matrix. J. Phys. Chem. Lett. 2018, 9, 5604–5611. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.; Ham, S.; Lee, S.H.; Hong, Y.; Kim, D. Enhancement of exciton transport in porphyrin aggregate nanostructures by controlling the hierarchical self-assembly. Nanoscale 2018, 10, 16438–16446. [Google Scholar] [CrossRef]
- Lloyd, S.; Mohseni, M. Symmetry-enhanced supertransfer of delocalized quantum states. New J. Phys. 2010, 12, 075020. [Google Scholar] [CrossRef]
- Chuang, C.; Lee, C.K.; Moix, J.M.; Knoester, J.; Cao, J. Quantum diffusion on molecular tubes: Universal scaling of the 1D to 2D transition. Phys. Rev. Lett. 2016, 116, 196803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, J.; Paleček, D.; Caycedo-Soler, F.; Lincoln, C.N.; Prior, J.; Von Berlepsch, H.; Huelga, S.F.; Plenio, M.B.; Zigmantas, D.; Hauer, J. Vibronic origin of long-lived coherence in an artificial molecular light harvester. Nat. Commun. 2015, 6, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butkus, V.; Alster, J.; Bašinskaitė, E.; Augulis, R.N.; Neuhaus, P.; Valkunas, L.; Anderson, H.L.; Abramavicius, D.; Zigmantas, D. Discrimination of diverse coherences allows identification of electronic transitions of a molecular nanoring. J. Phys. Chem. Lett. 2017, 8, 2344–2349. [Google Scholar] [CrossRef]
- Cheng, Y.-C.; Fleming, G.R. Dynamics of light harvesting in photosynthesis. Annu. Rev. Phys. Chem. 2009, 60, 241–262. [Google Scholar] [CrossRef] [Green Version]
- Calhoun, T.R.; Fleming, G.R. Quantum coherence in photosynthetic complexes. Phys. Status Solidi (b) 2011, 248, 833–838. [Google Scholar] [CrossRef]
- van Grondelle, R.; Novoderezhkin, V.I. Quantum effects in photosynthesis. Procedia Chem. 2011, 3, 198–210. [Google Scholar] [CrossRef] [Green Version]
- Lambrev, P.H.; Akhtar, P.; Tan, H.-S. Insights into the mechanisms and dynamics of energy transfer in plant light-harvesting complexes from two-dimensional electronic spectroscopy. Biochim. Biophys. Acta Bioenerg. 2020, 1861, 148050. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.-Y.; Lambert, N.; Shih, Y.-A.; Liu, M.-H.; Chen, Y.-N.; Nori, F. Plasmonic bio-sensing for the Fenna-Matthews-Olson complex. Sci. Rep. 2017, 7, 39720. [Google Scholar] [CrossRef] [PubMed]
- Li, C.-M.; Lambert, N.; Chen, Y.-N.; Chen, G.-Y.; Nori, F. Witnessing quantum coherence: From solid-state to biological systems. Sci. Rep. 2012, 2, 885. [Google Scholar] [CrossRef] [Green Version]
- Yuen-Zhou, J.; Krich, J.J.; Aspuru-Guzik, A. A witness for coherent electronic vs vibronic-only oscillations in ultrafast spectroscopy. J. Chem. Phys. 2012, 136, 234501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, A.S.; Yuen-Zhou, J.; Aspuru-Guzik, A.; Krich, J.J. Practical witness for electronic coherences. J. Chem. Phys. 2014, 141, 244109. [Google Scholar] [CrossRef] [Green Version]
- Halliwell, B.; Gutteridge, J.M. Free Radicals in Biology and Medicine; Oxford University Press: New York, NY, USA, 2015. [Google Scholar]
- Kannan, K.; Jain, S.K. Oxidative stress and apoptosis. Pathophysiology 2000, 7, 153–163. [Google Scholar] [CrossRef]
- Ryter, S.W.; Kim, H.P.; Hoetzel, A.; Park, J.W.; Nakahira, K.; Wang, X.; Choi, A.M. Mechanisms of cell death in oxidative stress. Antioxid. Redox Signal. 2007, 9, 49–89. [Google Scholar] [CrossRef]
- Chen, Y.; Zhou, Z.; Min, W. Mitochondria, oxidative stress and innate immunity. Front. Physiol. 2018, 9, 1487. [Google Scholar] [CrossRef] [Green Version]
- Shadel, G.S.; Horvath, T.L. Mitochondrial ROS signaling in organismal homeostasis. Cell 2015, 163, 560–569. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, Y.J.; Forman, H.J.; Sevanian, A. Oxidants as stimulators of signal transduction. Free Radic. Biol. Med. 1997, 22, 269–285. [Google Scholar] [CrossRef]
- Buckel, W.; Golding, B.T. Radical enzymes. Encycl. Radic. Chem. Biol. Mater. 2012. [Google Scholar] [CrossRef]
- Conrad, K.S.; Manahan, C.C.; Crane, B.R. Photochemistry of flavoprotein light sensors. Nat. Chem. Biol. 2014, 10, 801–809. [Google Scholar] [CrossRef] [PubMed]
- Steiner, U.E.; Ulrich, T. Magnetic field effects in chemical kinetics and related phenomena. Chem. Rev. 1989, 89, 51–147. [Google Scholar] [CrossRef] [Green Version]
- Hore, P.J.; Mouritsen, H. The radical-pair mechanism of magnetoreception. Annu. Rev. Biophys. 2016, 45, 299–344. [Google Scholar] [CrossRef] [PubMed]
- Lukzen, N.N.; Ivanov, K.L.; Sadovsky, V.M.; Sagdeev, R.Z. Magnetic field effect on recombination of radicals diffusing on a two-dimensional plane. J. Chem. Phys. 2020, 152, 034103. [Google Scholar] [CrossRef]
- Sampson, C.; Keens, R.H.; Kattnig, D.R. On the magnetosensitivity of lipid peroxidation: Two-versus three-radical dynamics. Phys. Chem. Chem. Phys. 2019, 21, 13526–13538. [Google Scholar] [CrossRef] [Green Version]
- Brocklehurst, B.; McLauchlan, K.A. Free radical mechanism for the effects of environmental electromagnetic fields on biological systems. Int. J. Radiat. Biol. 1996, 69, 3–24. [Google Scholar] [CrossRef]
- Jones, A.R. Magnetic field effects in proteins. Mol. Phys. 2016, 114, 1691–1702. [Google Scholar] [CrossRef]
- Ghodbane, S.; Lahbib, A.; Sakly, M.; Abdelmelek, H. Bioeffects of static magnetic fields: Oxidative stress, genotoxic effects, and cancer studies. Biomed Res. Int. 2013, 2013, 602987. [Google Scholar] [CrossRef] [Green Version]
- Mattsson, M.-O.; Simkó, M. Grouping of experimental conditions as an approach to evaluate effects of extremely low-frequency magnetic fields on oxidative response in in vitro studies. Front. Public Health 2014, 2, 132. [Google Scholar] [CrossRef] [Green Version]
- Grissom, C.B. Magnetic field effects in biology: A survey of possible mechanisms with emphasis on radical-pair recombination. Chem. Rev. 1995, 95, 3–24. [Google Scholar] [CrossRef]
- Muheim, R.; Boström, J.; Åkesson, S.; Liedvogel, M. Sensory mechanisms of animal orientation and navigation. In Animal Movement across Scales; Oxford University Press: Oxford, UK, 2014; pp. 179–194. [Google Scholar]
- Fay, T.P.; Lindoy, L.P.; Manolopoulos, D.E.; Hore, P. How quantum is radical pair magnetoreception? Faraday Discuss. 2019, 221, 77–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woodward, J. Radical pairs in solution. Prog. React. Kinet. Mech. 2002, 27, 165–207. [Google Scholar] [CrossRef]
- Rodgers, C.T. Magnetic field effects in chemical systems. Pure Appl. Chem. 2009, 81, 19–43. [Google Scholar] [CrossRef] [Green Version]
- Maeda, K.; Henbest, K.B.; Cintolesi, F.; Kuprov, I.; Rodgers, C.T.; Liddell, P.A.; Gust, D.; Timmel, C.R.; Hore, P.J. Chemical compass model of avian magnetoreception. Nature 2008, 453, 387–390. [Google Scholar] [CrossRef]
- Rodgers, C.T.; Hore, P.J. Chemical magnetoreception in birds: The radical pair mechanism. Proc. Natl. Acad. Sci. USA 2009, 106, 353–360. [Google Scholar] [CrossRef] [Green Version]
- Anikeeva, P.; Jasanoff, A. Magnetogenetics: Problems on the back of an envelope. Elife 2016, 5, e19569. [Google Scholar] [CrossRef]
- Meister, M. Physical limits to magnetogenetics. Elife 2016, 5, e17210. [Google Scholar] [CrossRef]
- Timmel, C.R.; Henbest, K.B. A study of spin chemistry in weak magnetic fields. Philos. Trans. R. Soc. Lond. Ser. A Math. Phys. Eng. Sci. 2004, 362, 2573–2589. [Google Scholar] [CrossRef] [Green Version]
- Biskup, T.; Schleicher, E.; Okafuji, A.; Link, G.; Hitomi, K.; Getzoff, E.D.; Weber, S. Direct observation of a photoinduced radical pair in a cryptochrome blue-light photoreceptor. Angew. Chem. Int. Ed. 2009, 48, 404–407. [Google Scholar] [CrossRef] [Green Version]
- Wiltschko, W.; Munro, U.; Ford, H.; Wiltschko, R. Red light disrupts magnetic orientation of migratory birds. Nature 1993, 364, 525–527. [Google Scholar] [CrossRef]
- Phillips, J.; Sayeed, O. Wavelength-dependent effects of light on magnetic compass orientation in Drosophila melanogaster. J. Comp. Physiol. A 1993, 172, 303–308. [Google Scholar] [CrossRef] [PubMed]
- Kutta, R.J.; Magerl, K.; Kensy, U.; Dick, B. A search for radical intermediates in the photocycle of LOV domains. Photochem. Photobiol. Sci. 2015, 14, 288–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, S. Light-driven enzymatic catalysis of DNA repair: A review of recent biophysical studies on photolyase. Biochim. Biophys. Acta Bioenerg. 2005, 1707, 1–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hay, S.; Brenner, S.; Khara, B.; Quinn, A.M.; Rigby, S.E.; Scrutton, N.S. Nature of the energy landscape for gated electron transfer in a dynamic redox protein. J. Am. Chem. Soc. 2010, 132, 9738–9745. [Google Scholar] [CrossRef] [PubMed]
- Silverman, R.B. Radical ideas about monoamine oxidase. Acc. Chem. Res. 1995, 28, 335–342. [Google Scholar] [CrossRef]
- Brown, K.L. Chemistry and enzymology of vitamin B12. Chem. Rev. 2005, 105, 2075–2150. [Google Scholar] [CrossRef] [PubMed]
- Marsh, E.N.G.; Patterson, M.D.P.; Li, L. Adenosyl radical: Reagent and catalyst in enzyme reactions. Chembiochem A Eur. J. Chem. Biol. 2010, 11, 604. [Google Scholar] [CrossRef] [Green Version]
- Jones, A.R. The photochemistry and photobiology of vitamin B 12. Photochem. Photobiol. Sci. 2017, 16, 820–834. [Google Scholar] [CrossRef]
- Jones, A.R.; Woodward, J.R.; Scrutton, N.S. Continuous wave photolysis magnetic field effect investigations with free and protein-bound alkylcobalamins. J. Am. Chem. Soc. 2009, 131, 17246–17253. [Google Scholar] [CrossRef]
- Lukinović, V.; Woodward, J.R.; Marrafa, T.C.; Shanmugam, M.; Heyes, D.J.; Hardman, S.J.; Scrutton, N.S.; Hay, S.; Fielding, A.J.; Jones, A.R. Photochemical Spin Dynamics of the Vitamin B12 Derivative, Methylcobalamin. J. Phys. Chem. B 2019, 123, 4663–4672. [Google Scholar] [CrossRef] [PubMed]
- Kipriyanov Jr, A.; Doktorov, A.; Purtov, P. Magnetic field effects on bistability and bifurcation phenomena in lipid peroxidation. Bioelectromagnetics 2015, 36, 485–493. [Google Scholar] [CrossRef] [PubMed]
- Buchachenko, A.L.; Kouznetsov, D.A.; Shishkov, A.V. Spin biochemistry: Magnetic isotope effect in the reaction of creatine kinase with CH3HgCl. J. Phys. Chem. A 2004, 108, 707–710. [Google Scholar] [CrossRef]
- Buchachenko, A.L.; Kouznetsov, D.A.; Orlova, M.A.; Markarian, A.A. Magnetic isotope effect of magnesium in phosphoglycerate kinase phosphorylation. Proc. Natl. Acad. Sci. USA 2005, 102, 10793–10796. [Google Scholar] [CrossRef] [Green Version]
- Buchachenko, A.L.; Kouznetsov, D.A.; Breslavskaya, N.N.; Orlova, M.A. Magnesium isotope effects in enzymatic phosphorylation. J. Phys. Chem. B 2008, 112, 2548–2556. [Google Scholar] [CrossRef]
- Buchachenko, A.L.; Kuznetsov, D.A. Magnetic field affects enzymatic ATP synthesis. J. Am. Chem. Soc. 2008, 130, 12868–12869. [Google Scholar] [CrossRef]
- Crotty, D.; Silkstone, G.; Poddar, S.; Ranson, R.; Prina-Mello, A.; Wilson, M.T.; Coey, J. Reexamination of magnetic isotope and field effects on adenosine triphosphate production by creatine kinase. Proc. Natl. Acad. Sci. USA 2012, 109, 1437–1442. [Google Scholar] [CrossRef] [Green Version]
- Usselman, R.J.; Chavarriaga, C.; Castello, P.R.; Procopio, M.; Ritz, T.; Dratz, E.A.; Singel, D.J.; Martino, C.F. The quantum biology of reactive oxygen species partitioning impacts cellular bioenergetics. Sci. Rep. 2016, 6, 38543. [Google Scholar] [CrossRef] [Green Version]
- Usselman, R.J.; Hill, I.; Singel, D.J.; Martino, C.F. Spin biochemistry modulates reactive oxygen species (ROS) production by radio frequency magnetic fields. PLoS ONE 2014, 9, e93065. [Google Scholar] [CrossRef] [Green Version]
- Evans, E.W.; Dodson, C.A.; Maeda, K.; Biskup, T.; Wedge, C.; Timmel, C.R. Magnetic field effects in flavoproteins and related systems. Interface Focus 2013, 3, 20130037. [Google Scholar] [CrossRef] [Green Version]
- Evans, E.W.; Kattnig, D.R.; Henbest, K.B.; Hore, P.; Mackenzie, S.R.; Timmel, C.R. Sub-millitesla magnetic field effects on the recombination reaction of flavin and ascorbic acid radicals. J. Chem. Phys. 2016, 145, 085101. [Google Scholar] [CrossRef] [PubMed]
- Ikeya, N.; Woodward, J.R. Cellular autofluorescence is magnetic field sensitive. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar]
- Jones, A.R.; Hardman, S.J.; Hay, S.; Scrutton, N.S. Is there a dynamic protein contribution to the substrate trigger in coenzyme B12-dependent ethanolamine ammonia lyase? Angew. Chem. Int. Ed. 2011, 50, 10843–10846. [Google Scholar] [CrossRef] [PubMed]
- Hughes, J.A.; Hardman, S.J.; Scrutton, N.S.; Graham, D.M.; Woodward, J.R.; Jones, A.R. Observation of the Δ g mechanism resulting from the ultrafast spin dynamics that follow the photolysis of coenzyme B12. J. Chem. Phys. 2019, 151, 201102. [Google Scholar] [CrossRef] [PubMed]
- Musewald, C.; Gilch, P.; Hartwich, G.; Pöllinger-Dammer, F.; Scheer, H.; Michel-Beyerle, M.E. Magnetic field dependence of ultrafast intersystem-crossing: A triplet mechanism on the picosecond time scale? J. Am. Chem. Soc. 1999, 121, 8876–8881. [Google Scholar] [CrossRef]
- Maeda, K.; Robinson, A.J.; Henbest, K.B.; Hogben, H.J.; Biskup, T.; Ahmad, M.; Schleicher, E.; Weber, S.; Timmel, C.R.; Hore, P.J. Magnetically sensitive light-induced reactions in cryptochrome are consistent with its proposed role as a magnetoreceptor. Proc. Natl. Acad. Sci. USA 2012, 109, 4774–4779. [Google Scholar] [CrossRef] [Green Version]
- Sheppard, D.M.; Li, J.; Henbest, K.B.; Neil, S.R.; Maeda, K.; Storey, J.; Schleicher, E.; Biskup, T.; Rodriguez, R.; Weber, S. Millitesla magnetic field effects on the photocycle of an animal cryptochrome. Sci. Rep. 2017, 7, 42228. [Google Scholar] [CrossRef] [Green Version]
- Sherrard, R.M.; Morellini, N.; Jourdan, N.; El-Esawi, M.; Arthaut, L.-D.; Niessner, C.; Rouyer, F.; Klarsfeld, A.; Doulazmi, M.; Witczak, J. Low-intensity electromagnetic fields induce human cryptochrome to modulate intracellular reactive oxygen species. Plos Biol. 2018, 16, e2006229. [Google Scholar] [CrossRef] [Green Version]
- Wiltschko, R.; Ahmad, M.; Nießner, C.; Gehring, D.; Wiltschko, W. Light-dependent magnetoreception in birds: The crucial step occurs in the dark. J. R. Soc. Interface 2016, 13, 20151010. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.A.; Lau, J.C.; Hogben, H.J.; Biskup, T.; Kattnig, D.R.; Hore, P. Alternative radical pairs for cryptochrome-based magnetoreception. J. R. Soc. Interface 2014, 11, 20131063. [Google Scholar] [CrossRef] [Green Version]
- Procopio, M.; Ritz, T. Radical-pair based avian magnetoreception. APS 2014, 2014, J10. 009. [Google Scholar]
- Hogben, H.J.; Efimova, O.; Wagner-Rundell, N.; Timmel, C.R.; Hore, P. Possible involvement of superoxide and dioxygen with cryptochrome in avian magnetoreception: Origin of Zeeman resonances observed by in vivo EPR spectroscopy. Chem. Phys. Lett. 2009, 480, 118–122. [Google Scholar] [CrossRef]
- Worster, S.; Kattnig, D.R.; Hore, P. Spin relaxation of radicals in cryptochrome and its role in avian magnetoreception. J. Chem. Phys. 2016, 145, 035104. [Google Scholar] [CrossRef] [PubMed]
- Kattnig, D.R.; Sowa, J.K.; Solov’yov, I.A.; Hore, P. Electron spin relaxation can enhance the performance of a cryptochrome-based magnetic compass sensor. New J. Phys. 2016, 18, 063007. [Google Scholar] [CrossRef]
- Kattnig, D.R.; Solov’yov, I.A.; Hore, P. Electron spin relaxation in cryptochrome-based magnetoreception. Phys. Chem. Chem. Phys. 2016, 18, 12443–12456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karogodina, T.Y.; Dranov, I.G.; Sergeeva, S.V.; Stass, D.V.; Steiner, U.E. Kinetic Magnetic-Field Effect Involving the Small Biologically Relevant Inorganic Radicals NO and O2−. ChemPhysChem 2011, 12, 1714–1728. [Google Scholar] [CrossRef] [PubMed]
- Uhrig, G.S. Exact results on dynamical decoupling by π pulses in quantum information processes. New J. Phys. 2008, 10, 083024. [Google Scholar] [CrossRef]
- Keens, R.H.; Bedkihal, S.; Kattnig, D.R. Magnetosensitivity in dipolarly coupled three-spin systems. Phys. Rev. Lett. 2018, 121, 096001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babcock, N.S.; Kattnig, D.R. Electron–Electron Dipolar Interaction Poses a Challenge to the Radical Pair Mechanism of Magnetoreception. J. Phys. Chem. Lett. 2020, 11, 2414–2421. [Google Scholar] [CrossRef]
- Kattnig, D.R. Radical-pair-based magnetoreception amplified by radical scavenging: Resilience to spin relaxation. J. Phys. Chem. B 2017, 121, 10215–10227. [Google Scholar] [CrossRef] [Green Version]
- Kattnig, D.R.; Hore, P. The sensitivity of a radical pair compass magnetoreceptor can be significantly amplified by radical scavengers. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Endres, S.; Granzin, J.; Circolone, F.; Stadler, A.; Krauss, U.; Drepper, T.; Svensson, V.; Knieps-Grünhagen, E.; Wirtz, A.; Cousin, A. Structure and function of a short LOV protein from the marine phototrophic bacterium Dinoroseobacter shibae. BMC Microbiol. 2015, 15, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mancia, F.; Smith, G.; Evans, P. Crystal structure of substrate complexes of methylmalonyl-CoA mutase. Biochemistry 1999, 38, 7999–8005. [Google Scholar] [CrossRef] [PubMed]
- Tollinger, M.; Konrat, R.; Hilbert, B.H.; Marsh, E.N.G.; Kräutler, B. How a protein prepares for B12 binding: Structure and dynamics of the B12-binding subunit of glutamate mutase from Clostridium tetanomorphum. Structure 1998, 6, 1021–1033. [Google Scholar] [CrossRef] [Green Version]
- Bender, G.n.; Poyner, R.R.; Reed, G.H. Identification of the substrate radical intermediate derived from ethanolamine during catalysis by ethanolamine ammonia-lyase. Biochemistry 2008, 47, 11360–11366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Efimova, O.; Hore, P. Role of exchange and dipolar interactions in the radical pair model of the avian magnetic compass. Biophys. J. 2008, 94, 1565–1574. [Google Scholar] [CrossRef] [Green Version]
- Nohr, D.; Paulus, B.; Rodriguez, R.; Okafuji, A.; Bittl, R.; Schleicher, E.; Weber, S. Determination of Radical–Radical Distances in Light-Active Proteins and Their Implication for Biological Magnetoreception. Angew. Chem. Int. Ed. 2017, 56, 8550–8554. [Google Scholar] [CrossRef] [Green Version]
- Nohr, D.; Franz, S.; Rodriguez, R.; Paulus, B.; Essen, L.-O.; Weber, S.; Schleicher, E. Extended electron-transfer in animal cryptochromes mediated by a tetrad of aromatic amino acids. Biophys. J. 2016, 111, 301–311. [Google Scholar] [CrossRef] [Green Version]
- Zoltowski, B.D.; Vaidya, A.T.; Top, D.; Widom, J.; Young, M.W.; Crane, B.R. Structure of full-length Drosophila cryptochrome. Nature 2011, 480, 396–399. [Google Scholar] [CrossRef]
- Levy, C.; Zoltowski, B.D.; Jones, A.R.; Vaidya, A.T.; Top, D.; Widom, J.; Young, M.W.; Scrutton, N.S.; Crane, B.R.; Leys, D. Updated structure of Drosophila cryptochrome. Nature 2013, 495, E3–E4. [Google Scholar] [CrossRef] [Green Version]
- Müller, P.; Yamamoto, J.; Martin, R.; Iwai, S.; Brettel, K. Discovery and functional analysis of a 4th electron-transferring tryptophan conserved exclusively in animal cryptochromes and (6-4) photolyases. Chem. Commun. 2015, 51, 15502–15505. [Google Scholar] [CrossRef] [PubMed]
- Chagovetz, A.M.; Grissom, C.B. Magnetic field effects in adenosylcob (III) alamin photolysis: Relevance to B12 enzymes. J. Am. Chem. Soc. 1993, 115, 12152–12157. [Google Scholar] [CrossRef]
- Jones, A.R.; Hay, S.; Woodward, J.R.; Scrutton, N.S. Magnetic field effect studies indicate reduced geminate recombination of the radical pair in substrate-bound adenosylcobalamin-dependent ethanolamine ammonia lyase. J. Am. Chem. Soc. 2007, 129, 15718–15727. [Google Scholar] [CrossRef] [PubMed]
- Henbest, K.B.; Maeda, K.; Hore, P.; Joshi, M.; Bacher, A.; Bittl, R.; Weber, S.; Timmel, C.R.; Schleicher, E. Magnetic-field effect on the photoactivation reaction of Escherichia coli DNA photolyase. Proc. Natl. Acad. Sci. USA 2008, 105, 14395–14399. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Wang, L.; Zhong, D. Photolyase: Dynamics and electron-transfer mechanisms of DNA repair. Arch. Biochem. Biophys. 2017, 632, 158–174. [Google Scholar] [CrossRef]
- Hore, P. A DNA-Based Magnetic Sensor; ACS Publications: Washington, DC, USA, 2018. [Google Scholar]
- Zwang, T.J.; Tse, E.C.; Zhong, D.; Barton, J.K. A compass at weak magnetic fields using thymine dimer repair. ACS Cent. Sci. 2018, 4, 405–412. [Google Scholar] [CrossRef]
- Messiha, H.L.; Wongnate, T.; Chaiyen, P.; Jones, A.R.; Scrutton, N.S. Magnetic field effects as a result of the radical pair mechanism are unlikely in redox enzymes. J. R. Soc. Interface 2015, 12, 20141155. [Google Scholar] [CrossRef]
- Ritz, T.; Thalau, P.; Phillips, J.B.; Wiltschko, R.; Wiltschko, W. Resonance effects indicate a radical-pair mechanism for avian magnetic compass. Nature 2004, 429, 177–180. [Google Scholar] [CrossRef]
- Engels, S.; Schneider, N.-L.; Lefeldt, N.; Hein, C.M.; Zapka, M.; Michalik, A.; Elbers, D.; Kittel, A.; Hore, P.; Mouritsen, H. Anthropogenic electromagnetic noise disrupts magnetic compass orientation in a migratory bird. Nature 2014, 509, 353–356. [Google Scholar] [CrossRef]
- Hiscock, H.G.; Mouritsen, H.; Manolopoulos, D.E.; Hore, P. Disruption of magnetic compass orientation in migratory birds by radiofrequency electromagnetic fields. Biophys. J. 2017, 113, 1475–1484. [Google Scholar] [CrossRef] [Green Version]
- Kavokin, K. The puzzle of magnetic resonance effect on the magnetic compass of migratory birds. Bioelectromagn. J. Bioelectromagn. Soc. Soc. Phys. Regul. Biol. Med. Eur. Bioelectromagn. Assoc. 2009, 30, 402–410. [Google Scholar] [CrossRef] [PubMed]
- Hiscock, H.G.; Worster, S.; Kattnig, D.R.; Steers, C.; Jin, Y.; Manolopoulos, D.E.; Mouritsen, H.; Hore, P. The quantum needle of the avian magnetic compass. Proc. Natl. Acad. Sci. USA 2016, 113, 4634–4639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watson, J.; Crick, F. The structure of DNA. Cold Spring Harbor Symp. Quant. Biol. 1953, 18, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.D. Grover’s Algorithm in Natural Settings. arXiv 2020, arXiv:2001.00214. [Google Scholar]
- Marletto, C. Constructor theory of life. J. R. Soc. Interface 2015, 12, 20141226. [Google Scholar] [CrossRef]
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA damage, repair, and mutagenesis. Environ. Mol. Mutagenes. 2017, 58, 235–263. [Google Scholar] [CrossRef] [Green Version]
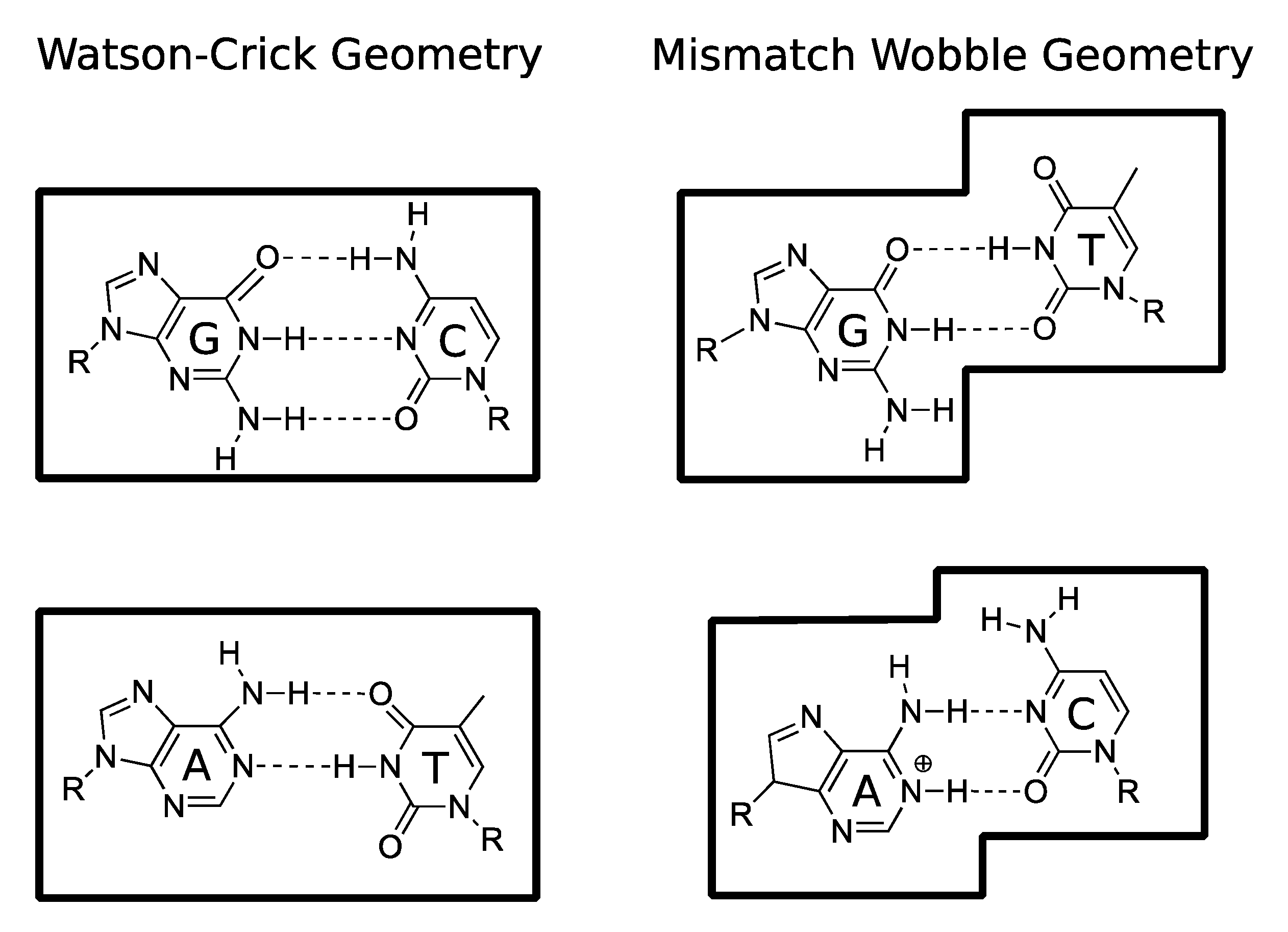
- Wang, W.; Hellinga, H.W.; Beese, L.S. Structural evidence for the rare tautomer hypothesis of spontaneous mutagenesis. Proc. Natl. Acad. Sci. USA 2011, 108, 17644–17648. [Google Scholar] [CrossRef] [Green Version]
- Bebenek, K.; Pedersen, L.C.; Kunkel, T.A. Replication infidelity via a mismatch with Watson–Crick geometry. Proc. Natl. Acad. Sci. USA 2011, 108, 1862–1867. [Google Scholar] [CrossRef] [Green Version]
- Koag, M.-C.; Lee, S. Insights into the effect of minor groove interactions and metal cofactors on mutagenic replication by human DNA polymerase β. Biochem. J. 2018, 475, 571–585. [Google Scholar] [CrossRef]
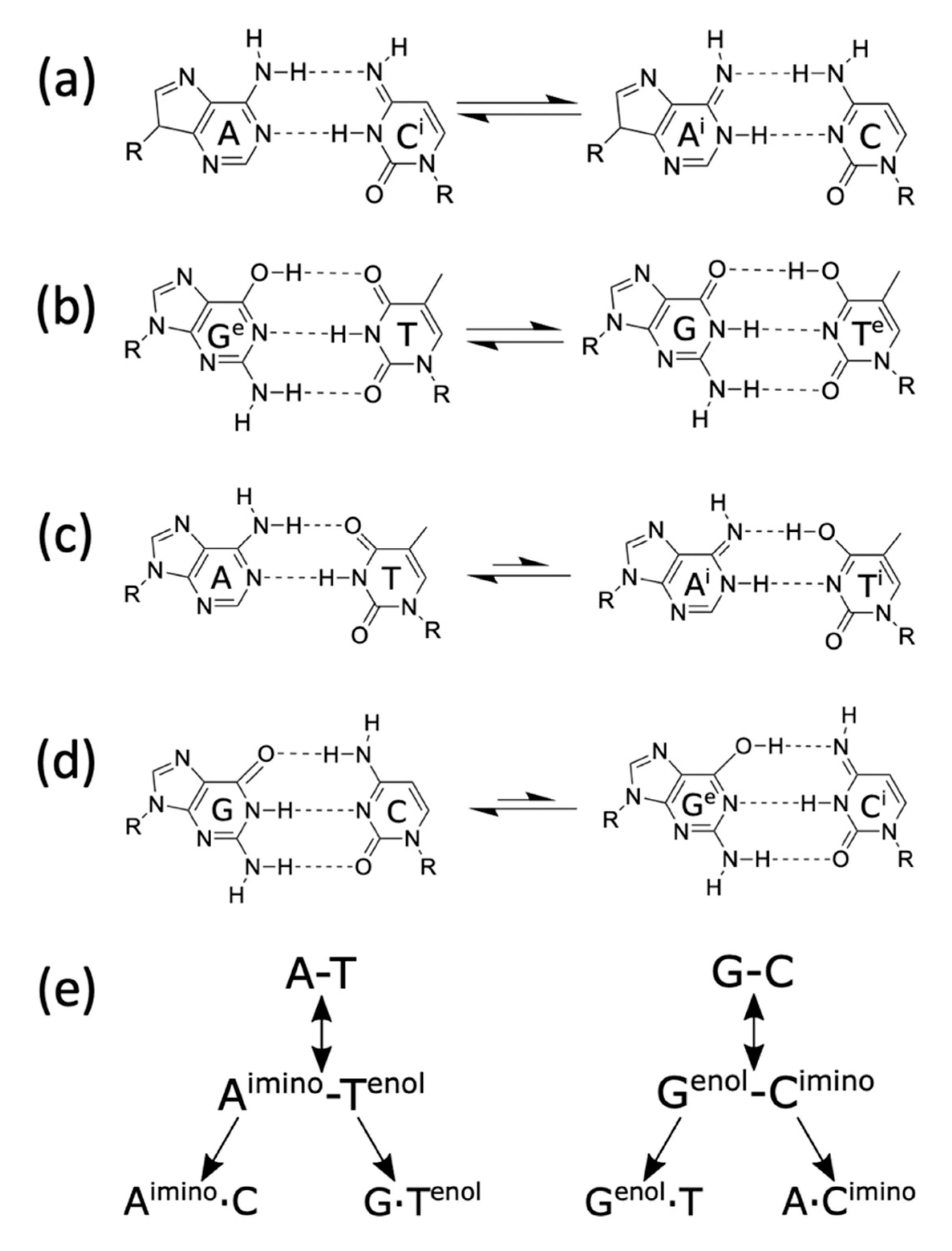
- Kimsey, I.J.; Petzold, K.; Sathyamoorthy, B.; Stein, Z.W.; Al-Hashimi, H.M. Visualizing transient Watson–Crick-like mispairs in DNA and RNA duplexes. Nature 2015, 519, 315–320. [Google Scholar] [CrossRef] [Green Version]
- Kimsey, I.J.; Szymanski, E.S.; Zahurancik, W.J.; Shakya, A.; Xue, Y.; Chu, C.-C.; Sathyamoorthy, B.; Suo, Z.; Al-Hashimi, H.M. Dynamic basis for dG• dT misincorporation via tautomerization and ionization. Nature 2018, 554, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Löwdin, P.-O. Proton tunneling in DNA and its biological implications. Rev. Mod. Phys. 1963, 35, 724. [Google Scholar] [CrossRef]
- Mueller, S.H.; Spenkelink, L.M.; van Oijen, A.M. When proteins play tag: The dynamic nature of the replisome. Biophys. Rev. 2019, 11, 641–651. [Google Scholar] [CrossRef] [PubMed]
- Tolosa, S.; Sansón, J.; Hidalgo, A. Mechanisms for guanine–cytosine tautomeric equilibrium in solution via steered molecular dynamic simulations. J. Mol. Liq. 2018, 251, 308–316. [Google Scholar] [CrossRef]
- Tolosa, S.; Sansón, J.; Hidalgo, A. Mechanisms of the TA to CG transition studied by SMD simulations: Deamination vs. tautomerisation. J. Mol. Liq. 2020, 308, 113036. [Google Scholar] [CrossRef]
- Li, P.; Rangadurai, A.; Al-Hashimi, H.M.; Hammes-Schiffer, S. Environmental Effects on Guanine-Thymine Mispair Tautomerization Explored with Quantum Mechanical/Molecular Mechanical Free Energy Simulations. J. Am. Chem. Soc. 2020, 142, 11183–11191. [Google Scholar] [CrossRef] [PubMed]
- Maximoff, S.N.; Kamerlin, S.C.L.; Florián, J. DNA polymerase λ active site favors a mutagenic mispair between the enol form of deoxyguanosine triphosphate substrate and the keto form of thymidine template: A free energy perturbation study. J. Phys. Chem. B 2017, 121, 7813–7822. [Google Scholar] [CrossRef] [Green Version]
- Rangadurai, A.; Szymanski, E.S.; Kimsey, I.; Shi, H.; Al-Hashimi, H.M. Probing conformational transitions towards mutagenic Watson–Crick-like G· T mismatches using off-resonance sugar carbon R 1ρ relaxation dispersion. J. Biomol. NMR 2020, 74, 457–471. [Google Scholar] [CrossRef]
- Burke, K. Perspective on density functional theory. J. Chem. Phys. 2012, 136, 150901. [Google Scholar] [CrossRef]
- Cramer, C.J. Essentials of Computational Chemistry: Theories and Models; John Wiley & Sons: Hoboken, NJ, USA, 2013. [Google Scholar]
- Jensen, F. Introduction to Computational Chemistry; John Wiley & Sons: Hoboken, NJ, USA, 2017. [Google Scholar]
- Brovarets’, O.h.O.; Hovorun, D.M. Proton tunneling in the A∙ T Watson-Crick DNA base pair: Myth or reality? J. Biomol. Struct. Dyn. 2015, 33, 2716–2720. [Google Scholar] [CrossRef]
- Brovarets’, O.h.O.; Tsiupa, K.S.; Hovorun, D.M. Novel pathway for mutagenic tautomerization of classical A∙ T DNA base pairs via sequential proton transfer through quasi-orthogonal transition states: A QM/QTAIM investigation. PLoS ONE 2018, 13, e0199044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brovarets’, O.h.O.; Hovorun, D.M. How many tautomerization pathways connect Watson–Crick-like G*· T DNA base mispair and wobble mismatches? J. Biomol. Struct. Dyn. 2015, 33, 2297–2315. [Google Scholar] [CrossRef] [PubMed]
- Brovarets, O.h.O.; Tsiupa, K.S.; Dinets, A.; Hovorun, D.M. Unexpected routes of the mutagenic tautomerization of the T nucleobase in the classical A· T DNA base pairs: A QM/QTAIM comprehensive view. Front. Chem. 2018, 6, 532. [Google Scholar] [CrossRef] [PubMed]
- Brovarets, O.h.O.; Hovorun, D.M. Can tautomerization of the A· T Watson–Crick base pair via double proton transfer provoke point mutations during DNA replication? A comprehensive QM and QTAIM analysis. J. Biomol. Struct. Dyn. 2014, 32, 127–154. [Google Scholar] [CrossRef] [PubMed]
- Brovarets’, O.h.O.; Hovorun, D.M. Why the tautomerization of the G· C Watson–Crick base pair via the DPT does not cause point mutations during DNA replication? QM and QTAIM comprehensive analysis. J. Biomol. Struct. Dyn. 2014, 32, 1474–1499. [Google Scholar] [CrossRef] [PubMed]
- Villani, G. Theoretical Investigation of the Coupling between Hydrogen-Atom Transfer and Stacking Interaction in Adenine–Thymine Dimers. ChemPhysChem 2013, 14, 1256–1263. [Google Scholar] [CrossRef]
- Soler-Polo, D.; Mendieta-Moreno, J.I.; Trabada, D.G.; Mendieta, J.; Ortega, J. Proton Transfer in Guanine-Cytosine Base Pairs in B-DNA. J. Chem. Theory Comput. 2019, 15, 6984–6991. [Google Scholar] [CrossRef] [PubMed]
- Roßbach, S.; Ochsenfeld, C. Influence of coupling and embedding schemes on QM size convergence in QM/MM approaches for the example of a proton transfer in DNA. J. Chem. Theory Comput. 2017, 13, 1102–1107. [Google Scholar] [CrossRef]
- Brovarets’, O.h.O.; Hovorun, D.M. Atomistic mechanisms of the double proton transfer in the H-bonded nucleobase pairs: QM/QTAIM computational lessons. J. Biomol. Struct. Dyn. 2019, 37, 1880–1907. [Google Scholar] [CrossRef]
- Jurečka, P.; Šponer, J.; Černý, J.; Hobza, P. Benchmark database of accurate (MP2 and CCSD (T) complete basis set limit) interaction energies of small model complexes, DNA base pairs, and amino acid pairs. Phys. Chem. Chem. Phys. 2006, 8, 1985–1993. [Google Scholar] [CrossRef]
- van der Wijst, T.; Guerra, C.F.; Swart, M.; Bickelhaupt, F.M. Performance of various density functionals for the hydrogen bonds in DNA base pairs. Chem. Phys. Lett. 2006, 426, 415–421. [Google Scholar] [CrossRef]
- Bende, A. Hydrogen bonding in the urea dimers and adenine–thymine DNA base pair: Anharmonic effects in the intermolecular H-bond and intramolecular H-stretching vibrations. Theor. Chem. Acc. 2010, 125, 253–268. [Google Scholar] [CrossRef]
- Godbeer, A. Quantum Tunnelling Effect in DNA Base Pair Mutation; University of Surrey: Guildford, UK, 2014. [Google Scholar]
- Godbeer, A.; Al-Khalili, J.; Stevenson, P. Environment-induced dephasing versus von Neumann measurements in proton tunneling. Phys. Rev. A 2014, 90, 012102. [Google Scholar] [CrossRef] [Green Version]
- Godbeer, A.; Al-Khalili, J.; Stevenson, P. Modelling proton tunnelling in the adenine–thymine base pair. Phys. Chem. Chem. Phys. 2015, 17, 13034–13044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pohl, R.; Socha, O.; Slavíček, P.; Šála, M.; Hodgkinson, P.; Dračínský, M. Proton transfer in guanine–cytosine base pair analogues studied by NMR spectroscopy and PIMD simulations. Faraday Discuss. 2018, 212, 331–344. [Google Scholar] [CrossRef] [Green Version]
- Fang, W.; Chen, J.; Rossi, M.; Feng, Y.; Li, X.-Z.; Michaelides, A. Inverse temperature dependence of nuclear quantum effects in DNA base pairs. J. Phys. Chem. Lett. 2016, 7, 2125–2131. [Google Scholar] [CrossRef] [Green Version]
- Nelson, P.C. The role of quantum decoherence in FRET. Biophys. J. 2018, 115, 167–172. [Google Scholar] [CrossRef] [Green Version]
- Clegg, R.M. The history of FRET. In Reviews in Fluorescence 2006; Springer: Berlin/Heidelberg, Germany, 2006; pp. 1–45. [Google Scholar]
- Kenkre, V.; Knox, R. Theory of fast and slow excitation transfer rates. Phys. Rev. Lett. 1974, 33, 803. [Google Scholar] [CrossRef]
- Förster, T. Delocalized Excitation and Excitation Transfer; Sinanoglu, O., Ed.; Modern Quantum Chemistry. Istanbul Lectures 3; Academic Press: New York, NY, USA; London, UK, 1965. [Google Scholar]
- Cinelli, R.A.; Tozzini, V.; Pellegrini, V.; Beltram, F.; Cerullo, G.; Zavelani-Rossi, M.; De Silvestri, S.; Tyagi, M.; Giacca, M. Coherent dynamics of photoexcited green fluorescent proteins. Phys. Rev. Lett. 2001, 86, 3439. [Google Scholar] [CrossRef] [Green Version]
- Jung, G.; Ma, Y.; Prall, B.S.; Fleming, G.R. Ultrafast fluorescence depolarisation in the yellow fluorescent protein due to its dimerisation. ChemPhysChem 2005, 6, 1628–1632. [Google Scholar] [CrossRef]
- Shi, S.; Kumar, P.; Lee, K.F. Generation of photonic entanglement in green fluorescent proteins. Nat. Commun. 2017, 8, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Puhl III, H.L.; Chen, E.; Taumoefolau, G.H.; Nguyen, T.A.; Kliger, D.S.; Blank, P.S.; Vogel, S.S. VenusA206 Dimers Behave Coherently at Room Temperature. Biophys. J. 2019, 116, 1918–1930. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, C.P.; Steude, A.; Tropf, L.; Schubert, M.; Kronenberg, N.M.; Ostermann, K.; Höfling, S.; Gather, M.C. An exciton-polariton laser based on biologically produced fluorescent protein. Sci. Adv. 2016, 2, e1600666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sánchez-Mosteiro, G.; Koopman, M.; van Dijk, E.M.; Hernando, J.; van Hulst, N.F.; García-Parajó, M.F. Photon antibunching proves emission from a single subunit in the autofluorescent protein DsRed. ChemPhysChem 2004, 5, 1782–1785. [Google Scholar] [CrossRef] [PubMed]
- Visser, N.V.; Hink, M.A.; Borst, J.W.; van der Krogt, G.N.; Visser, A.J. Circular dichroism spectroscopy of fluorescent proteins. FEBS Lett. 2002, 521, 31–35. [Google Scholar] [CrossRef] [Green Version]
- Koushik, S.V.; Blank, P.S.; Vogel, S.S. Anomalous surplus energy transfer observed with multiple FRET acceptors. PLoS ONE 2009, 4, e8031. [Google Scholar] [CrossRef]
- Lounis, B.; Deich, J.; Rosell, F.; Boxer, S.G.; Moerner, W. Photophysics of Ds Red, a red fluorescent protein, from the ensemble to the single-molecule level. J. Phys. Chem. B 2001, 105, 5048–5054. [Google Scholar] [CrossRef]
- Shi, S.; Thomas, A.; Corzo, N.V.; Kumar, P.; Huang, Y.; Lee, K.F. Broadband photon pair generation in green fluorescent proteins through spontaneous four-wave mixing. Sci. Rep. 2016, 6, 24344. [Google Scholar] [CrossRef] [Green Version]
- Tsien, R.Y. The green fluorescent protein. Annu. Rev. Biochem. 1998, 67, 509–544. [Google Scholar] [CrossRef]
- Rodriguez, E.A.; Campbell, R.E.; Lin, J.Y.; Lin, M.Z.; Miyawaki, A.; Palmer, A.E.; Shu, X.; Zhang, J.; Tsien, R.Y. The growing and glowing toolbox of fluorescent and photoactive proteins. Trends Biochem. Sci. 2017, 42, 111–129. [Google Scholar] [CrossRef] [Green Version]
- Gross, L.A.; Baird, G.S.; Hoffman, R.C.; Baldridge, K.K.; Tsien, R.Y. The structure of the chromophore within DsRed, a red fluorescent protein from coral. Proc. Natl. Acad. Sci. USA 2000, 97, 11990–11995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaner, N.C.; Lambert, G.G.; Chammas, A.; Ni, Y.; Cranfill, P.J.; Baird, M.A.; Sell, B.R.; Allen, J.R.; Day, R.N.; Israelsson, M. A bright monomeric green fluorescent protein derived from Branchiostoma lanceolatum. Nat. Methods 2013, 10, 407–409. [Google Scholar] [CrossRef] [PubMed]
- Ward, W.; Cormier, M. Energy transfer protein in coelenterate bioluminescence. J. Biol. Chem. 1979, 254, 781–788. [Google Scholar] [CrossRef]
- Arpino, J.A.; Rizkallah, P.J.; Jones, D.D. Crystal structure of enhanced green fluorescent protein to 1.35 Å resolution reveals alternative conformations for Glu222. PLoS ONE 2012, 7, e47132. [Google Scholar] [CrossRef] [Green Version]
- Taghizadeh, R.R.; Sherley, J.L. CFP and YFP, but not GFP, provide stable fluorescent marking of rat hepatic adult stem cells. J. Biomed. Biotechnol. 2008, 2008, 453590. [Google Scholar] [CrossRef]
- Chudakov, D.M.; Lukyanov, S.; Lukyanov, K.A. Fluorescent proteins as a toolkit for in vivo imaging. Trends Biotechnol. 2005, 23, 605–613. [Google Scholar] [CrossRef]
- Hebisch, E.; Knebel, J.; Landsberg, J.; Frey, E.; Leisner, M. High variation of fluorescence protein maturation times in closely related Escherichia coli strains. PLoS ONE 2013, 8, e75991. [Google Scholar] [CrossRef]
- Gather, M.C.; Yun, S.H. Bio-optimized energy transfer in densely packed fluorescent protein enables near-maximal luminescence and solid-state lasers. Nat. Commun. 2014, 5, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Specht, E.A.; Braselmann, E.; Palmer, A.E. A critical and comparative review of fluorescent tools for live-cell imaging. Annu. Rev. Physiol. 2017, 79, 93–117. [Google Scholar] [CrossRef]
- Balleza, E.; Kim, J.M.; Cluzel, P. Systematic characterization of maturation time of fluorescent proteins in living cells. Nat. Methods 2018, 15, 47–51. [Google Scholar] [CrossRef]
- Sarkar, P.; Koushik, S.V.; Vogel, S.S.; Gryczynski, I.; Gryczynski, Z.K. Photophysical properties of Cerulean and Venus fluorescent proteins. J. Biomed. Opt. 2009, 14, 034047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilmore, J.; McKenzie, R.H. Spin boson models for quantum decoherence of electronic excitations of biomolecules and quantum dots in a solvent. J. Phys. Condens. Matter 2005, 17, 1735. [Google Scholar] [CrossRef]
- Goedhart, J.; Von Stetten, D.; Noirclerc-Savoye, M.; Lelimousin, M.; Joosen, L.; Hink, M.A.; Van Weeren, L.; Gadella, T.W.; Royant, A. Structure-guided evolution of cyan fluorescent proteins towards a quantum yield of 93%. Nat. Commun. 2012, 3, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Algar, W.R.; Hildebrandt, N.; Vogel, S.S.; Medintz, I.L. FRET as a biomolecular research tool—understanding its potential while avoiding pitfalls. Nat. Methods 2019, 16, 815–829. [Google Scholar] [CrossRef]
- Siegel, R.M.; Chan, F.K.-M.; Zacharias, D.A.; Swofford, R.; Holmes, K.L.; Tsien, R.Y.; Lenardo, M.J. Measurement of molecular interactions in living cells by fluorescence resonance energy transfer between variants of the green fluorescent protein. Sci. Signal. 2000, 2000, pl1. [Google Scholar] [CrossRef]
- Stryer, L.; Haugland, R.P. Energy transfer: A spectroscopic ruler. Proc. Natl. Acad. Sci. USA 1967, 58, 719. [Google Scholar] [CrossRef] [Green Version]
- Gilmore, J.; McKenzie, R.H. Quantum dynamics of electronic excitations in biomolecular chromophores: Role of the protein environment and solvent. J. Phys. Chem. A 2008, 112, 2162–2176. [Google Scholar] [CrossRef] [Green Version]
- Borst, J.W.; Hink, M.A.; van Hoek, A.; Visser, A.J. Effects of refractive index and viscosity on fluorescence and anisotropy decays of enhanced cyan and yellow fluorescent proteins. J. Fluoresc. 2005, 15, 153–160. [Google Scholar] [CrossRef] [Green Version]
- Geddes, C.D.; Lakowicz, J.R. Reviews in Fluorescence 2006; Springer: Berlin/Heidelberg, Germany, 2005. [Google Scholar]
- Berova, N.; Di Bari, L.; Pescitelli, G. Application of electronic circular dichroism in configurational and conformational analysis of organic compounds. Chem. Soc. Rev. 2007, 36, 914–931. [Google Scholar] [CrossRef]
- Grishina, I.B.; Woody, R.W. Contributions of tryptophan side chains to the circular dichroism of globular proteins: Exciton couplets and coupled oscillators. Faraday Discuss. 1994, 99, 245–262. [Google Scholar] [CrossRef]
- Ilagan, R.P.; Rhoades, E.; Gruber, D.F.; Kao, H.T.; Pieribone, V.A.; Regan, L. A new bright green-emitting fluorescent protein–engineered monomeric and dimeric forms. FEBS J. 2010, 277, 1967–1978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stuart, H. Quantum computation in brain microtubules? The Penrose–Hameroff ‘Orch OR ‘model of consciousness. Philos. Trans. R. Soc. Lond. Ser. A Math. Phys. Eng. Sci. 1998, 356, 1869–1896. [Google Scholar] [CrossRef] [Green Version]
- Penrose, R.; Hameroff, S. Consciousness in the universe: Neuroscience, quantum space-time geometry and Orch OR theory. J. Cosmol. 2011, 14, 1–17. [Google Scholar]
- Hameroff, S.R.; Penrose, R. Consciousness in the universe an updated review of the” Orch OR” theory. In Biophysics of Consciousness: A Foundational Approach; World Scientific Publishing Co. Pte. Ltd.: Singapore, 2016; pp. 517–599. [Google Scholar]
- Bezanilla, F. Voltage-gated ion channels. In Biological Membrane Ion Channels; Springer: Berlin/Heidelberg, Germany, 2007; pp. 81–118. [Google Scholar]
- Vaziri, A.; Plenio, M.B. Quantum coherence in ion channels: Resonances, transport and verification. New J. Phys. 2010, 12, 085001. [Google Scholar] [CrossRef]
- Doyle, D.A.; Cabral, J.M.; Pfuetzner, R.A.; Kuo, A.; Gulbis, J.M.; Cohen, S.L.; Chait, B.T.; MacKinnon, R. The structure of the potassium channel: Molecular basis of K+ conduction and selectivity. Science 1998, 280, 69–77. [Google Scholar] [CrossRef] [Green Version]
- MacKinnon, R. Potassium channels and the atomic basis of selective ion conduction (Nobel Lecture). Angew. Chem. Int. Ed. 2004, 43, 4265–4277. [Google Scholar] [CrossRef]
- Seoh, S.-A.; Sigg, D.; Papazian, D.M.; Bezanilla, F. Voltage-sensing residues in the S2 and S4 segments of the Shaker K+ channel. Neuron 1996, 16, 1159–1167. [Google Scholar] [CrossRef] [Green Version]
- Allen, T.W.; Kuyucak, S.; Chung, S.-H. Molecular dynamics study of the KcsA potassium channel. Biophys. J. 1999, 77, 2502–2516. [Google Scholar] [CrossRef] [Green Version]
- Baker, K.A.; Tzitzilonis, C.; Kwiatkowski, W.; Choe, S.; Riek, R. Conformational dynamics of the KcsA potassium channel governs gating properties. Nat. Struct. Mol. Biol. 2007, 14, 1089–1095. [Google Scholar] [CrossRef]
- Chung, S.-H.; Allen, T.W.; Kuyucak, S. Conducting-state properties of the KcsA potassium channel from molecular and Brownian dynamics simulations. Biophys. J. 2002, 82, 628–645. [Google Scholar] [CrossRef] [Green Version]
- Roux, B.; Schulten, K. Computational studies of membrane channels. Structure 2004, 12, 1343–1351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Summhammer, J.; Sulyok, G.; Bernroider, G. Quantum dynamics and non-local effects behind ion transition states during permeation in membrane channel proteins. Entropy 2018, 20, 558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ficek, Z.; Swain, S. Quantum Interference and Coherence: Theory and Experiments; Springer Science & Business Media: Berlin, Germany, 2005; Volume 100. [Google Scholar]
- Salari, V.; Moradi, N.; Sajadi, M.; Fazileh, F.; Shahbazi, F. Quantum decoherence time scales for ionic superposition states in ion channels. Phys. Rev. E 2015, 91, 032704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganim, Z.; Tokmakoff, A.; Vaziri, A. Vibrational excitons in ionophores: Experimental probes for quantum coherence-assisted ion transport and selectivity in ion channels. New J. Phys. 2011, 13, 113030. [Google Scholar] [CrossRef]
- Bernroider, G.; Summhammer, J. Can quantum entanglement between ion transition states effect action potential initiation? Cogn. Comput. 2012, 4, 29–37. [Google Scholar] [CrossRef]
- Qaswal, A.B. Quantum Electrochemical Equilibrium: Quantum Version of the Goldman–Hodgkin–Katz Equation. Quantum Rep. 2020, 2, 17. [Google Scholar] [CrossRef]
- Barjas Qaswal, A. Quantum tunneling of ions through the closed voltage-gated channels of the biological membrane: A mathematical model and implications. Quantum Rep. 2019, 1, 19. [Google Scholar] [CrossRef] [Green Version]
- Qaswal, A.B. Lithium stabilizes the mood of bipolar patients by depolarizing the neuronal membrane via quantum tunneling through the sodium channels. Clin. Psychopharmacol. Neurosci. 2020, 18, 214. [Google Scholar] [CrossRef]
- Alrabayah, M.; Qaswal, A.B.; Suleiman, A.; Khreesha, L. Role of Potassium Ions Quantum Tunneling in the Pathophysiology of Phantom Limb Pain. Brain Sci. 2020, 10, 241. [Google Scholar] [CrossRef] [Green Version]
- Wen, L.; Zhang, X.; Tian, Y.; Jiang, L. Quantum-confined superfluid: From nature to artificial. Sci. China Mater. 2018, 61, 1027–1032. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Jiang, L. Quantum-confined ion superfluid in nerve signal transmission. Nano Res. 2019, 12, 1219–1221. [Google Scholar] [CrossRef]
- Fröhlich, F.; McCormick, D.A. Endogenous electric fields may guide neocortical network activity. Neuron 2010, 67, 129–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, C.; Shivacharan, R.S.; Zhang, M.; Durand, D.M. Can neural activity propagate by endogenous electrical field? J. Neurosci. 2015, 35, 15800–15811. [Google Scholar] [CrossRef] [PubMed]
- Francis, J.T.; Gluckman, B.J.; Schiff, S.J. Sensitivity of neurons to weak electric fields. J. Neurosci. 2003, 23, 7255–7261. [Google Scholar] [CrossRef]
- Jedlicka, P. Revisiting the Quantum Brain Hypothesis: Toward Quantum (Neuro)biology? Front. Mol. Neurosci. 2017, 10, 366. [Google Scholar] [CrossRef]
- Jedlicka, P. Quantum stochasticity and (the end of) neurodeterminism. Quantum Phys. Meets Philos. Mind 2014, 183–197. [Google Scholar]
- Anastassiou, C.A.; Koch, C. Ephaptic coupling to endogenous electric field activity: Why bother? Curr. Opin. Neurobiol. 2015, 31, 95–103. [Google Scholar] [CrossRef]












Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, Y.; Bertagna, F.; D’Souza, E.M.; Heyes, D.J.; Johannissen, L.O.; Nery, E.T.; Pantelias, A.; Sanchez-Pedreño Jimenez, A.; Slocombe, L.; Spencer, M.G.; et al. Quantum Biology: An Update and Perspective. Quantum Rep. 2021, 3, 80-126. https://doi.org/10.3390/quantum3010006
Kim Y, Bertagna F, D’Souza EM, Heyes DJ, Johannissen LO, Nery ET, Pantelias A, Sanchez-Pedreño Jimenez A, Slocombe L, Spencer MG, et al. Quantum Biology: An Update and Perspective. Quantum Reports. 2021; 3(1):80-126. https://doi.org/10.3390/quantum3010006
Chicago/Turabian StyleKim, Youngchan, Federico Bertagna, Edeline M. D’Souza, Derren J. Heyes, Linus O. Johannissen, Eveliny T. Nery, Antonio Pantelias, Alejandro Sanchez-Pedreño Jimenez, Louie Slocombe, Michael G. Spencer, and et al. 2021. "Quantum Biology: An Update and Perspective" Quantum Reports 3, no. 1: 80-126. https://doi.org/10.3390/quantum3010006
APA StyleKim, Y., Bertagna, F., D’Souza, E. M., Heyes, D. J., Johannissen, L. O., Nery, E. T., Pantelias, A., Sanchez-Pedreño Jimenez, A., Slocombe, L., Spencer, M. G., Al-Khalili, J., Engel, G. S., Hay, S., Hingley-Wilson, S. M., Jeevaratnam, K., Jones, A. R., Kattnig, D. R., Lewis, R., Sacchi, M., ... McFadden, J. (2021). Quantum Biology: An Update and Perspective. Quantum Reports, 3(1), 80-126. https://doi.org/10.3390/quantum3010006