
Hybrid Quantum-Classical Eigensolver without Variation or Parametric Gates


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Background
Mapping to Qubits & Computational Basis
3. Constructing an Effective Matrix Representation for a Hamiltonian by Qubit Measurement
3.1. Effective Hamiltonian and Circuit Representation
3.2. Implementing Measurements
3.3. Preparing the Computational Basis
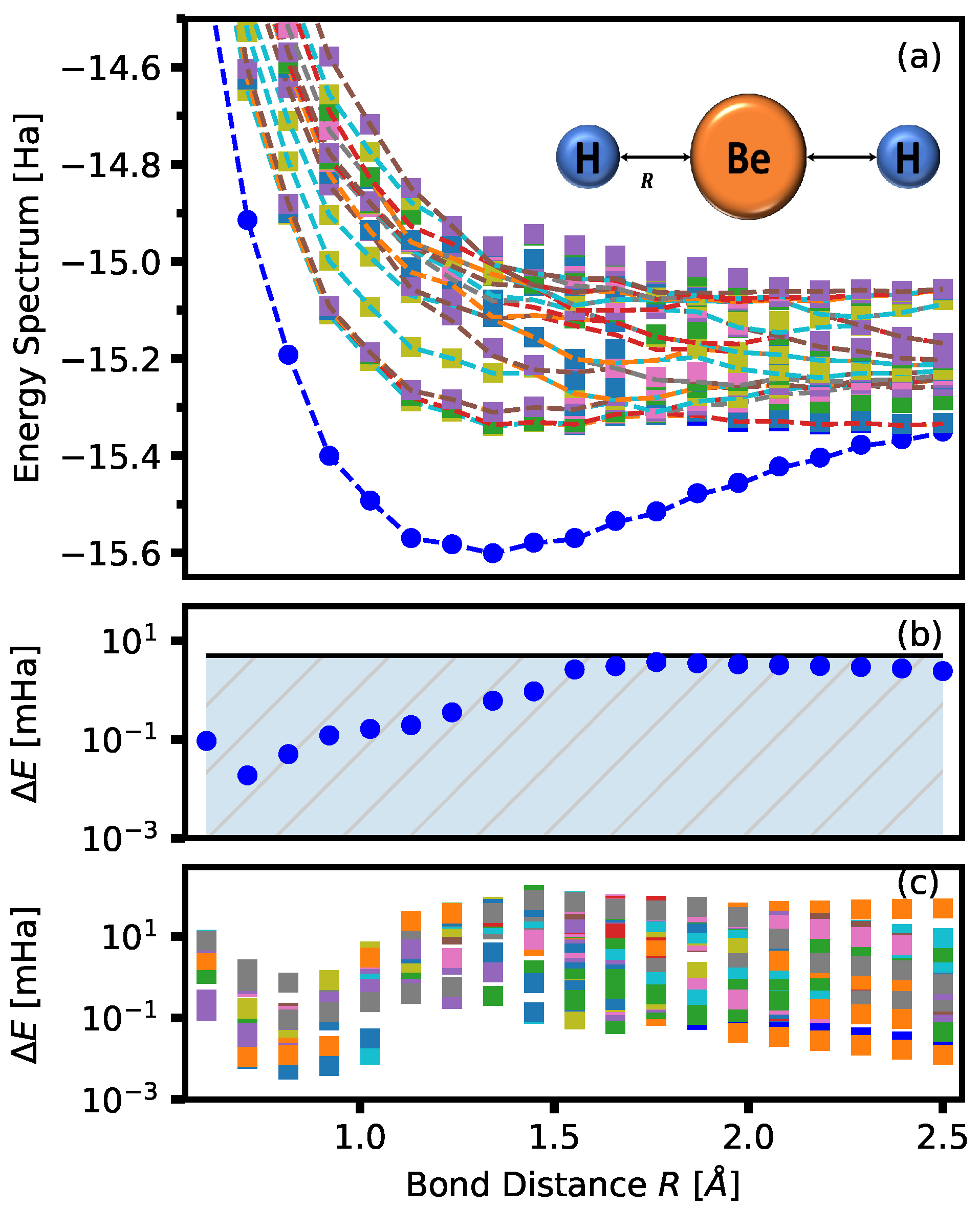
4. Numerical Demonstration: LiH and BeH
Density of States
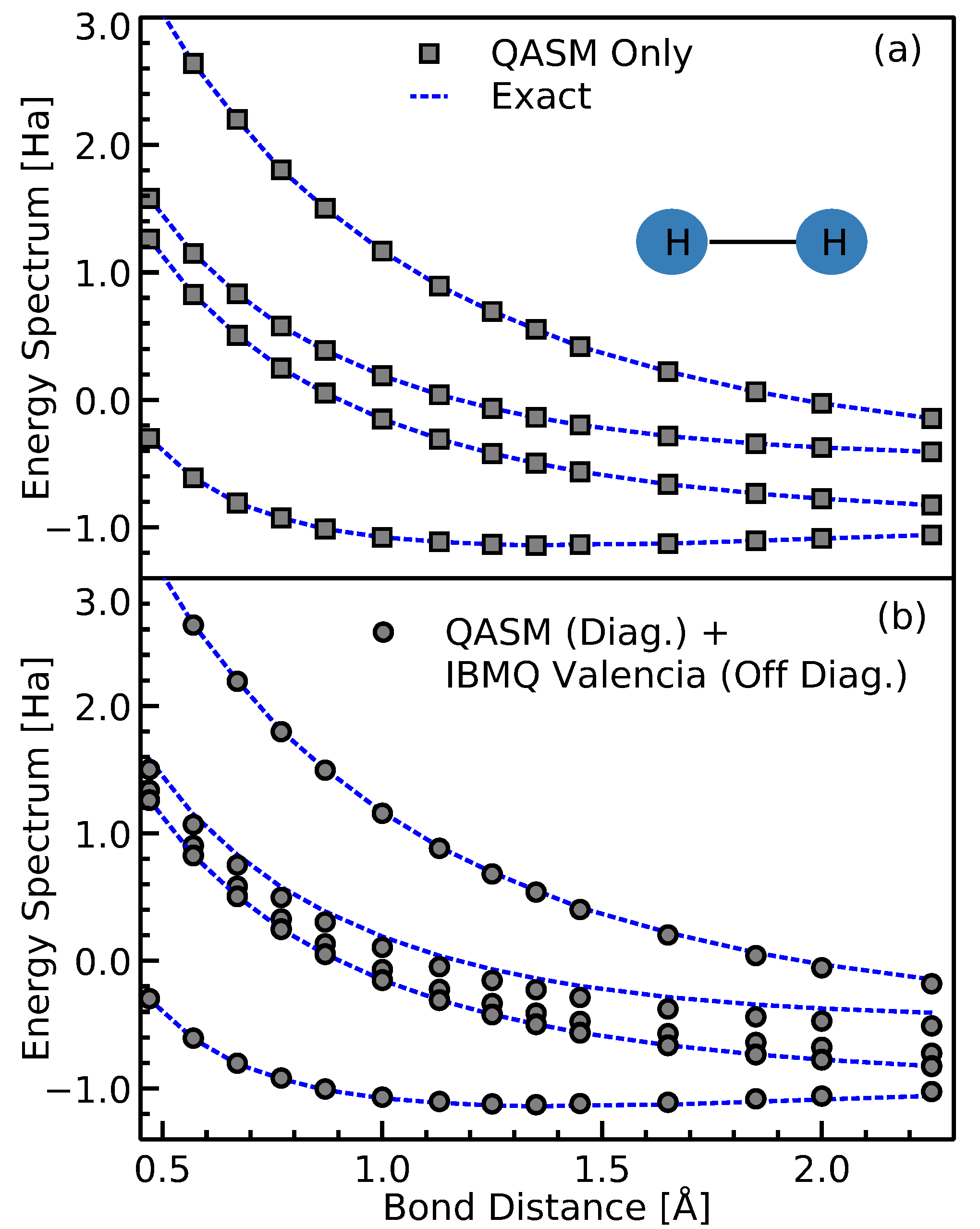
5. Hardware Demonstration: H
6. Discussion and Summary
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Preskill, J. Quantum Computing in the NISQ era and beyond. Quantum 2018, 2, 79. [Google Scholar] [CrossRef]
- McArdle, S.; Endo, S.; Aspuru-Guzik, A.; Benjamin, S.C.; Yuan, X. Quantum computational chemistry. Rev. Mod. Phys. 2020, 92, 015003. [Google Scholar] [CrossRef] [Green Version]
- Yuan, X.; Endo, S.; Zhao, Q.; Li, Y.; Benjamin, S.C. Theory of variational quantum simulation. Quantum 2019, 3, 191. [Google Scholar] [CrossRef]
- Peruzzo, A.; McClean, J.; Shadbolt, P.; Yung, M.H.; Zhou, X.Q.; Love, P.J.; Aspuru-Guzik, A.; O’brien, J.L. A variational eigenvalue solver on a photonic quantum processor. Nat. Commun. 2014, 5, 4213. [Google Scholar] [CrossRef] [Green Version]
- McClean, J.R.; Romero, J.; Babbush, R.; Aspuru-Guzik, A. The theory of variational hybrid quantum-classical algorithms. New J. Phys. 2016, 18, 023023. [Google Scholar] [CrossRef]
- Kandala, A.; Mezzacapo, A.; Temme, K.; Takita, M.; Brink, M.; Chow, J.M.; Gambetta, J.M. Hardware-efficient variational quantum eigensolver for small molecules and quantum magnets. Nature 2017, 549, 242–246. [Google Scholar] [CrossRef]
- Barkoutsos, P.K.; Gonthier, J.F.; Sokolov, I.; Moll, N.; Salis, G.; Fuhrer, A.; Ganzhorn, M.; Egger, D.J.; Troyer, M.; Mezzacapo, A.; et al. Quantum algorithms for electronic structure calculations: Particle-hole Hamiltonian and optimized wave-function expansions. Phys. Rev. A 2018, 98, 022322. [Google Scholar] [CrossRef] [Green Version]
- Jones, T.; Endo, S.; McArdle, S.; Yuan, X.; Benjamin, S.C. Variational quantum algorithms for discovering Hamiltonian spectra. Phys. Rev. A 2019, 99, 062304. [Google Scholar] [CrossRef] [Green Version]
- Nakanishi, K.M.; Mitarai, K.; Fujii, K. Subspace-search variational quantum eigensolver for excited states. Phys. Rev. Res. 2019, 1, 033062. [Google Scholar] [CrossRef] [Green Version]
- Parrish, R.M.; Hohenstein, E.G.; McMahon, P.L.; Martínez, T.J. Quantum Computation of Electronic Transitions Using a Variational Quantum Eigensolver. Phys. Rev. Lett. 2019, 122, 230401. [Google Scholar] [CrossRef] [Green Version]
- Higgott, O.; Wang, D.; Brierley, S. Variational Quantum Computation of Excited States. Quantum 2019, 3, 156. [Google Scholar] [CrossRef]
- Jouzdani, P.; Bringuier, S.; Kostuk, M. A Method of Determining Excited-States for Quantum Computation. arXiv 2019, arXiv:1908.05238. [Google Scholar]
- McClean, J.R.; Boixo, S.; Smelyanskiy, V.N.; Babbush, R.; Neven, H. Barren plateaus in quantum neural network training landscapes. Nat. Commun. 2018, 9, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parrish, R.M.; Iosue, J.T.; Ozaeta, A.; McMahon, P.L. A Jacobi Diagonalization and Anderson Acceleration Algorithm For Variational Quantum Algorithm Parameter Optimization. arXiv 2019, arXiv:1904.03206. [Google Scholar]
- Romero, J.; Babbush, R.; McClean, J.R.; Hempel, C.; Love, P.; Aspuru-Guzik, A. Strategies for quantum computing molecular energies using the unitary coupled cluster ansatz. arXiv 2017, arXiv:1701.02691. [Google Scholar] [CrossRef] [Green Version]
- Herasymenko, Y.; O’Brien, T.E. A diagrammatic approach to variational quantum ansatz construction. arXiv 2019, arXiv:1907.081576. [Google Scholar]
- IBM-Q team. IBM-Q 5 Qubit Valencia Backend, Specification v1.3.1. 2020. Available online: https://quantum-computing.ibm.com (accessed on 15 October 2020).
- Reine, S.; Helgaker, T.; Lindh, R. Multi-electron integrals. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 290–303. [Google Scholar] [CrossRef]
- Fradkin, E. Jordan-Wigner transformation for quantum-spin systems in two dimensions and fractional statistics. Phys. Rev. Lett. 1989, 63, 322. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, M.A.; Chuang, I. Quantum Computation and Quantum Information; American Cambridge University Press: Cambridge, UK, 2016. [Google Scholar]
- Steudtner, M.; Wehner, S. Quantum codes for quantum simulation of fermions on a square lattice of qubits. Phys. Rev. A 2019, 99, 022308. [Google Scholar] [CrossRef] [Green Version]
- Wright, K.; Beck, K.M.; Debnath, S.; Amini, J.M.; Nam, Y.; Grzesiak, N.; Chen, J.S.; Pisenti, N.C.; Chmielewski, M.; Collins, C.; et al. Benchmarking an 11-qubit quantum computer. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Knill, E.; Ortiz, G.; Somma, R.D. Optimal quantum measurements of expectation values of observables. Phys. Rev. A 2007, 75, 012328. [Google Scholar] [CrossRef] [Green Version]
- Dobšíček, M.; Johansson, G.; Shumeiko, V.; Wendin, G. Arbitrary accuracy iterative quantum phase estimation algorithm using a single ancillary qubit: A two-qubit benchmark. Phys. Rev. A 2007, 76, 030306. [Google Scholar] [CrossRef] [Green Version]
- Mitarai, K.; Fujii, K. Methodology for replacing indirect measurements with direct measurements. Phys. Rev. Res. 2019, 1, 013006. [Google Scholar] [CrossRef] [Green Version]
- Jensen, F. Introduction to Computational Chemistry; John Wiley & Sons: Hoboken, NJ, USA, 2017. [Google Scholar]
- Bartlett, R.J.; Musiał, M. Coupled-cluster theory in quantum chemistry. Rev. Mod. Phys. 2007, 79, 291. [Google Scholar] [CrossRef] [Green Version]
- Wecker, D.; Hastings, M.B.; Troyer, M. Progress towards practical quantum variational algorithms. Phys. Rev. A 2015, 92, 042303. [Google Scholar] [CrossRef] [Green Version]
- Janke, W. Monte Carlo Simulations of Spin Systems. In Computational Physics: Selected Methods Simple Exercises Serious Applications; Springer: Berlin/Heidelberg, Germany, 1996; pp. 10–43. [Google Scholar] [CrossRef] [Green Version]
- Parrish, R.M.; Burns, L.A.; Smith, D.G.A.; Simmonett, A.C.; DePrince, A.E.; Hohenstein, E.G.; Bozkaya, U.; Sokolov, A.Y.; Di Remigio, R.; Richard, R.M.; et al. Psi4 1.1: An Open-Source Electronic Structure Program Emphasizing Automation, Advanced Libraries, and Interoperability. J. Chem. Theory Comput. 2017, 13, 3185–3197. [Google Scholar] [CrossRef]
- McClean, J.R.; Sung, K.J.; Kivlichan, I.D.; Cao, Y.; Dai, C.; Fried, E.S.; Gidney, C.; Gimby, B.; Gokhale, P.; Häner, T.; et al. OpenFermion: The Electronic Structure Package for Quantum Computers. arXiv 2017, arXiv:1710.07629. [Google Scholar]
- Virtanen, P.; Gommers, R.; Oliphant, T.E.; Haberland, M.; Reddy, T.; Cournapeau, D.; Burovski, E.; Peterson, P.; Weckesser, W.; Bright, J.; et al. SciPy 1.0: Fundamental Algorithms for Scientific Computing in Python. Nat. Methods 2020. [Google Scholar] [CrossRef] [Green Version]
- Truhlar, D.G.; Garrett, B.C.; Klippenstein, S.J. Current status of transition-state theory. J. Phys. Chem. 1996, 100, 12771–12800. [Google Scholar] [CrossRef]
- Gui, K.; Tomesh, T.; Gokhale, P.; Shi, Y.; Chong, F.T.; Martonosi, M.; Suchara, M. Term Grouping and Travelling Salesperson for Digital Quantum Simulation. arXiv 2020, arXiv:2001.05983. [Google Scholar]
- Qiskit team. Qiskit: An Open-source Framework for Quantum Computing. 2019. Available online: https://zenodo.org/record/2562111#.YBYhA3kRVPY (accessed on 15 October 2020).
- IBM-Q Team. IBM-Q QASM Backend, Specification v0.1.547. 2020. Available online: https://quantum-computing.ibm.com (accessed on 15 October 2020).
- Tilly, J.; Jones, G.; Chen, H.; Wossnig, L.; Grant, E. Computation of molecular excited states on IBMQ using a Discriminative Variational Quantum Eigensolver. arXiv 2020, arXiv:2001.04941. [Google Scholar]
- Santagati, R.; Wang, J.; Gentile, A.A.; Paesani, S.; Wiebe, N.; McClean, J.R.; Morley-Short, S.; Shadbolt, P.J.; Bonneau, D.; Silverstone, J.W.; et al. Witnessing eigenstates for quantum simulation of Hamiltonian spectra. Sci. Adv. 2018, 4, eaap9646. [Google Scholar] [CrossRef] [Green Version]
- McArdle, S.; Jones, T.; Endo, S.; Li, Y.; Benjamin, S.C.; Yuan, X. Variational ansatz-based quantum simulation of imaginary time evolution. Npj Quantum Inf. 2019, 5. [Google Scholar] [CrossRef] [Green Version]
- Somma, R.D. Quantum eigenvalue estimation via time series analysis. arXiv 2019, arXiv:1907.11748. [Google Scholar] [CrossRef]
- McClean, J.R.; Kimchi-Schwartz, M.E.; Carter, J.; de Jong, W.A. Hybrid quantum-classical hierarchy for mitigation of decoherence and determination of excited states. Phys. Rev. A 2017, 95. [Google Scholar] [CrossRef] [Green Version]
- Colless, J.I.; Ramasesh, V.V.; Dahlen, D.; Blok, M.S.; Kimchi-Schwartz, M.E.; McClean, J.R.; Carter, J.; de Jong, W.A.; Siddiqi, I. Computation of Molecular Spectra on a Quantum Processor with an Error-Resilient Algorithm. Phys. Rev. X 2018, 8, 011021. [Google Scholar] [CrossRef] [Green Version]
- Diker, F. Deterministic construction of arbitrary W states with quadratically increasing number of two-qubit gates. arXiv 2016, arXiv:1606.0929. [Google Scholar]
- Venturelli, D.; Do, M.; Rieffel, E.; Frank, J. Compiling quantum circuits to realistic hardware architectures using temporal planners. Quantum Sci. Technol. 2018, 3, 025004. [Google Scholar] [CrossRef] [Green Version]
- Nash, B.; Gheorghiu, V.; Mosca, M. Quantum circuit optimizations for NISQ architectures. Quantum Sci. Technol. 2020, 5, 025010. [Google Scholar] [CrossRef] [Green Version]
- Hazra, S.; Salunkhe, K.V.; Bhattacharjee, A.; Bothara, G.; Kundu, S.; Roy, T.; Patankar, M.P.; Vijay, R. Engineering cross resonance interaction in multi-modal quantum circuits. Appl. Phys. Lett. 2020, 116, 152601. [Google Scholar] [CrossRef]
- Zhang, Y.; Deng, H.; Li, Q.; Song, H.; Nie, L. Optimizing Quantum Programs against Decoherence: Delaying Qubits into Quantum Superposition. In Proceedings of the 2019 International Symposium on Theoretical Aspects of Software Engineering (TASE), Guilin, China, 29–31 July 2019; pp. 184–191. [Google Scholar]
- Holmes, A.; Jokar, M.R.; Pasandi, G.; Ding, Y.; Pedram, M.; Chong, F.T. NISQ+: Boosting quantum computing power by approximating quantum error correction. arXiv 2020, arXiv:2004.04794. [Google Scholar]
- Kay, A. Quantikz. 2019. Available online: https://royalholloway.figshare.com/articles/dataset/Quantikz/7000520/4 (accessed on 15 October 2020).
- Hunter, J.D. Matplotlib: A 2D graphics environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jouzdani, P.; Bringuier, S. Hybrid Quantum-Classical Eigensolver without Variation or Parametric Gates. Quantum Rep. 2021, 3, 137-152. https://doi.org/10.3390/quantum3010008
Jouzdani P, Bringuier S. Hybrid Quantum-Classical Eigensolver without Variation or Parametric Gates. Quantum Reports. 2021; 3(1):137-152. https://doi.org/10.3390/quantum3010008
Chicago/Turabian StyleJouzdani, Pejman, and Stefan Bringuier. 2021. "Hybrid Quantum-Classical Eigensolver without Variation or Parametric Gates" Quantum Reports 3, no. 1: 137-152. https://doi.org/10.3390/quantum3010008
APA StyleJouzdani, P., & Bringuier, S. (2021). Hybrid Quantum-Classical Eigensolver without Variation or Parametric Gates. Quantum Reports, 3(1), 137-152. https://doi.org/10.3390/quantum3010008