A Single Gene Expression Set Derived from Artificial Intelligence Predicted the Prognosis of Several Lymphoma Subtypes; and High Immunohistochemical Expression of TNFAIP8 Associated with Poor Prognosis in Diffuse Large B-Cell Lymphoma

 ,
, 
 ,
,
Abstract
:
1. Introduction
2. Materials and Methods
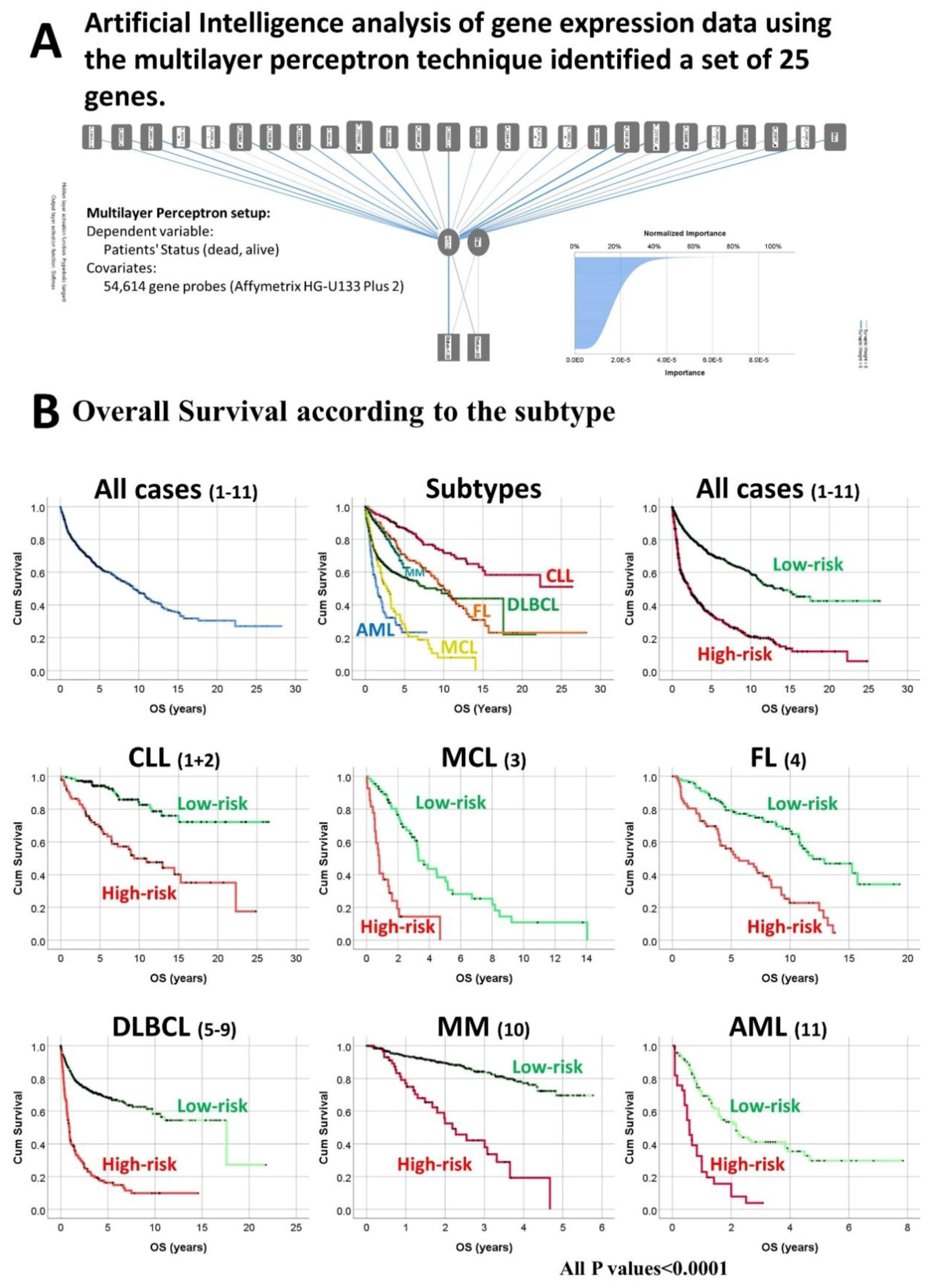
2.1. Subjects of Study and Set of Genes Derived from Artificial Intelligence Analysis
2.2. Gene Expression Analysis
2.3. Statistical Analysis
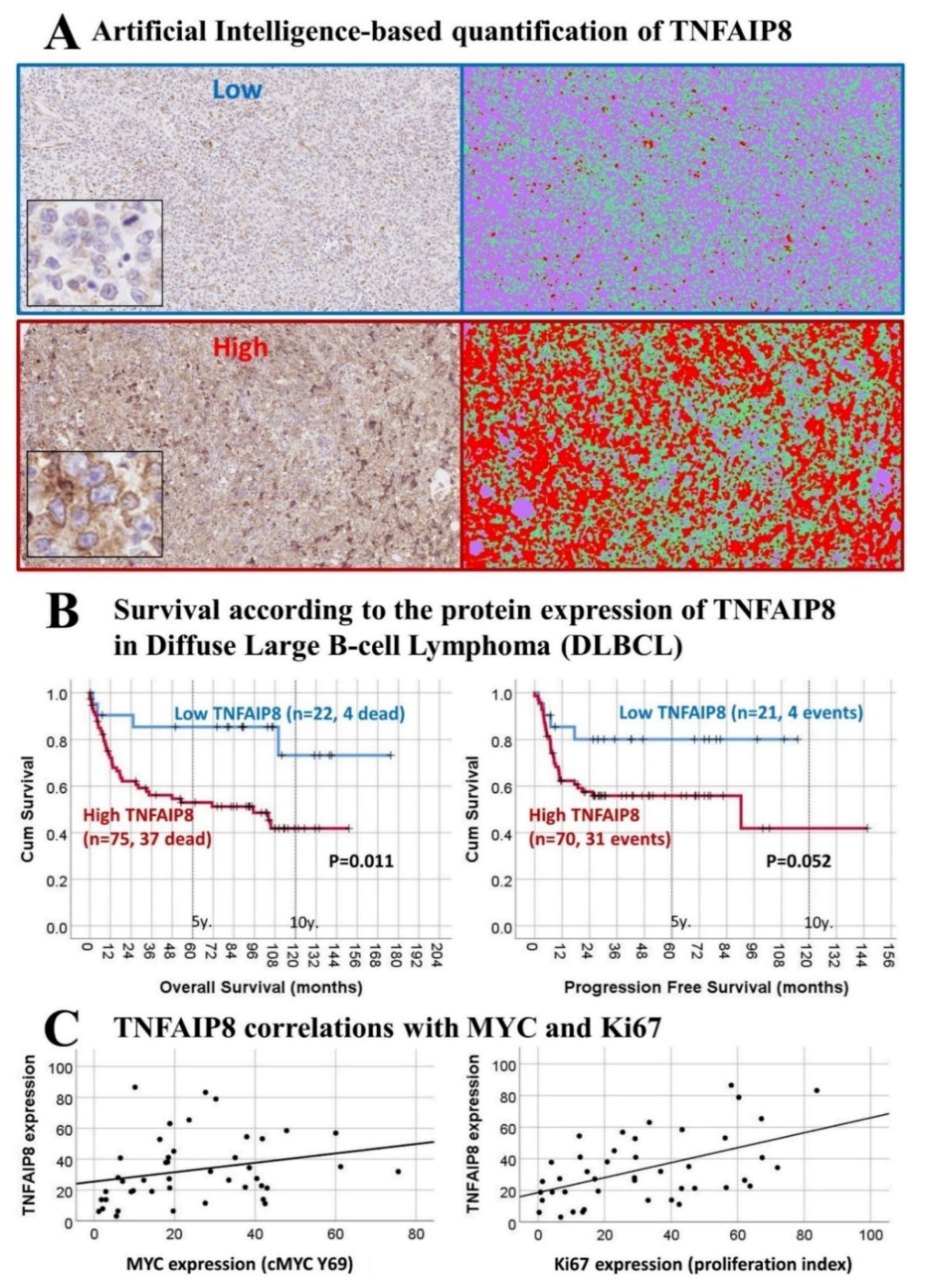
2.4. Validation of TNFAIP8 in an Independent Series of DLBCL
2.5. Immunohistochemistry of TNFAIP8 and Additional Markers
2.6. Conventional and Artificial Intelligence-Based Digital Image Analysis
3. Results
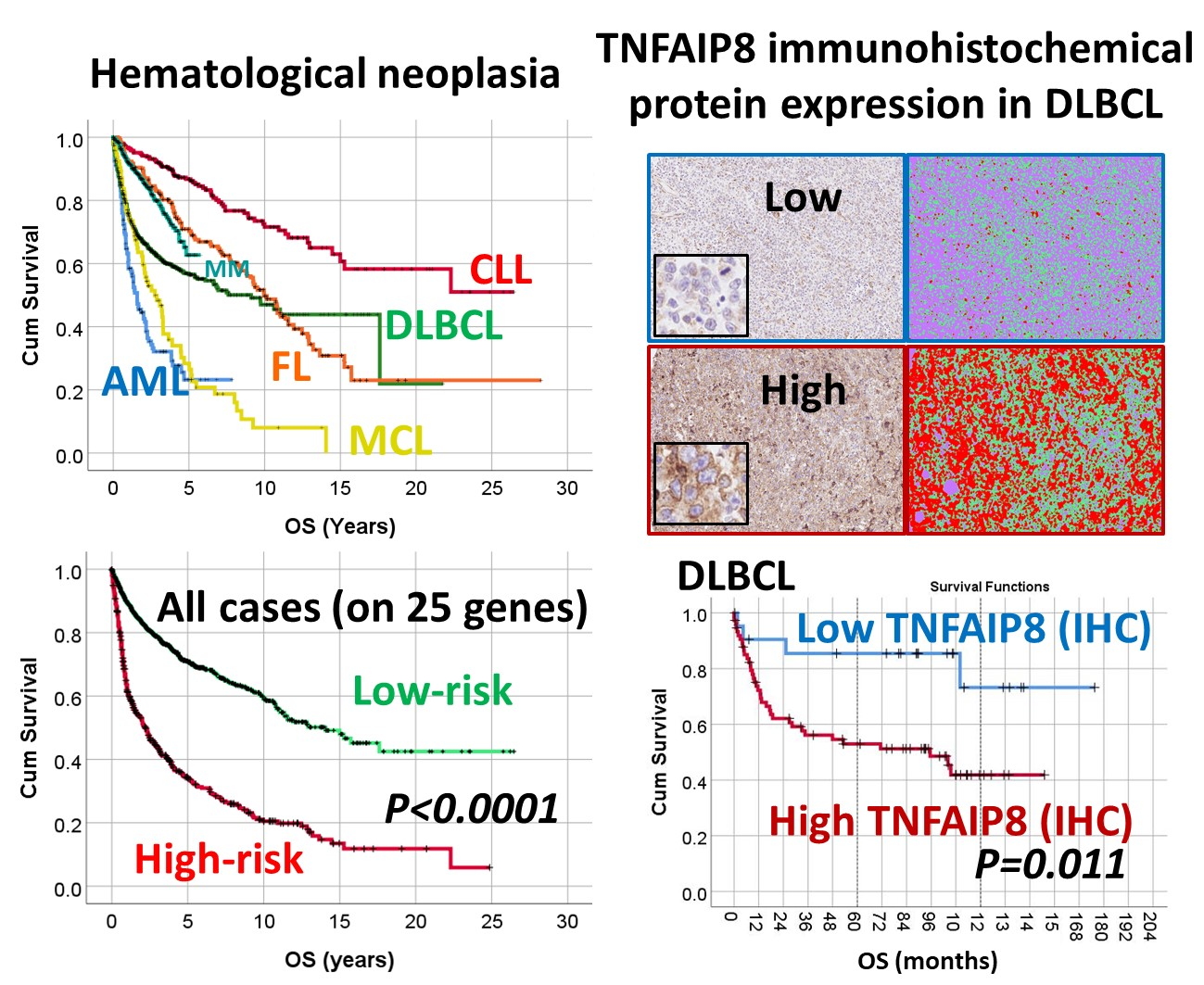
3.1. Overall Survival of the Lymphoma Subtypes and Acute Myeloid Leukemia.
3.2. Survival Analysis According to the Risk-Score Based on the Set of 25 Genes
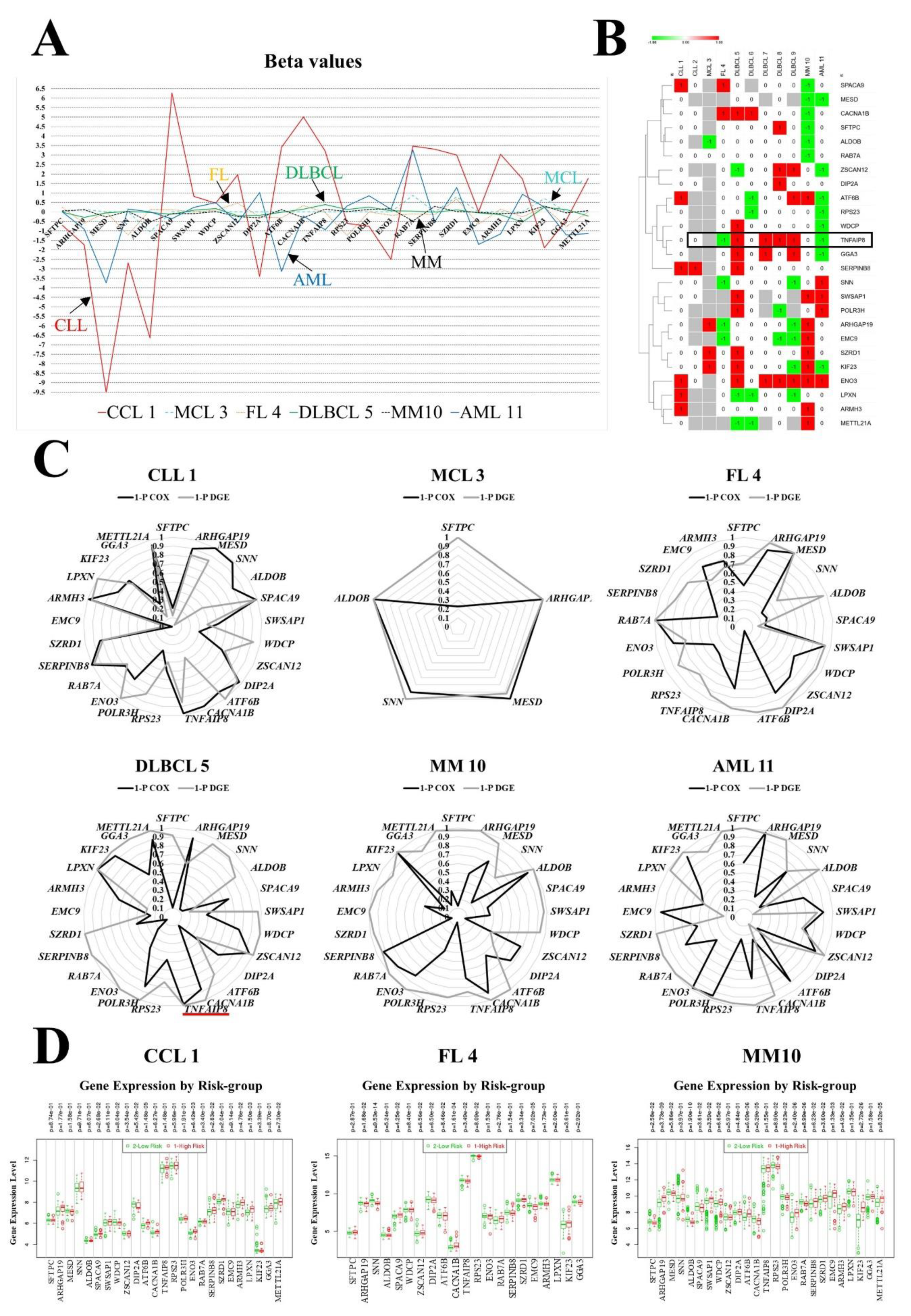
3.3. Gene Contribution to the Prognostic Model
3.4. Immunohistochemical Expression of TNFAIP8 in DLBCL
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Swerdlow, S.H.; Campo, E.; Harris, N.L. WHO Classification of Tumors of Haematopoietic and Lymphoid Tissues, 4th ed.; International Agency for Research on Cancer: Lyon, France, 2017. [Google Scholar]
- Li, S.; Young, K.H.; Medeiros, L.J. Diffuse large B-cell lymphoma. Pathology 2018, 50, 74–87. [Google Scholar] [CrossRef]
- Rosenwald, A.; Wright, G.; Leroy, K.; Yu, X.; Gaulard, P.; Gascoyne, R.D.; Chan, W.C.; Zhao, T.; Haioun, C.; Greiner, T.C.; et al. Molecular Diagnosis of Primary Mediastinal B Cell Lymphoma Identifies a Clinically Favorable Subgroup of Diffuse Large B Cell Lymphoma Related to Hodgkin Lymphoma. J. Exp. Med. 2003, 198, 851–862. [Google Scholar] [CrossRef]
- Lee, S.; Day, N.S.; Miles, R.R.; Perkins, S.L.; Lim, M.S.; Ayello, J.; Van De Ven, C.; Harrison, L.; El-Mallawany, N.K.; Goldman, S.; et al. Comparative genomic expression signatures of signal transduction pathways and targets in paediatric Burkitt lymphoma: A Children’s Oncology Group report. Br. J. Haematol. 2017, 177, 601–611. [Google Scholar] [CrossRef] [Green Version]
- Codony, C.; Crespo, M.; Abrisqueta, P.; Montserrat, E.; Bosch, F. Gene expression profiling in chronic lymphocytic leukaemia. Best Pract. Res. Clin. Haematol. 2009, 22, 211–222. [Google Scholar] [CrossRef]
- IBM Corp. IBM SPSS Neural Networks 25. IBM SPSS Statistics for Windows, Version 25.0; IBM Corp.: Armonk, NY, USA, 2017.
- Carreras, J.; Hamoudi, R.; Nakamura, N. Artificial Intelligence Analysis of Gene Expression Data Predicted the Prognosis of Patients with Diffuse Large B-Cell Lymphoma. Tokai J. Exp. Clin. Med. 2020, 45, 37–48. [Google Scholar] [PubMed]
- Herold, T.; Jurinovic, V.; Metzeler, K.H.; Boulesteix, A.-L.; Bergmann, M.; Seiler, T.; Mulaw, M.A.; Thoene, S.; Dufour, A.; Pasalic, Z.; et al. An eight-gene expression signature for the prediction of survival and time to treatment in chronic lymphocytic leukemia. Leukemia 2011, 25, 1639–1645. [Google Scholar] [CrossRef] [PubMed]
- International Cancer Genome Consortium. International network of cancer genome projects. Nature 2010, 464, 993–998. [Google Scholar] [CrossRef] [Green Version]
- Rosenwald, A.; Wright, G.; Wiestner, A.; Chan, W.C.; Connors, J.M.; Campo, E.; Gascoyne, R.D.; Grogan, T.M.; Muller-Hermelink, H.K.; Smeland, E.B.; et al. The proliferation gene expression signature is a quantitative integrator of oncogenic events that predicts survival in mantle cell lymphoma. Cancer Cell 2003, 3, 185–197. [Google Scholar] [CrossRef] [Green Version]
- Leich, E.; Salaverria, I.; Bea, S.; Zettl, A.; Wright, G.; Moreno, V.; Gascoyne, R.D.; Chan, W.-C.; Braziel, R.M.; Rimsza, L.M.; et al. Follicular lymphomas with and without translocation t(14;18) differ in gene expression profiles and genetic alterations. Blood 2009, 114, 826–834. [Google Scholar] [CrossRef] [PubMed]
- Lenz, G.; Wright, G.; Dave, S.S.; Xiao, W.; Powell, J.; Zhao, H.; Xu, W.; Tan, B.; Goldschmidt, N.; Iqbal, J.; et al. Lymphoma/Leukemia Molecular Profiling Project. Stromal gene signatures in large-B-cell lymphomas. N. Engl. J. Med. 2008, 359, 2313–2323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaknovich, R.; Geng, H.; Johnson, N.A.; Tsikitas, L.; Cerchietti, L.; Greally, J.M.; Gascoyne, R.D.; Elemento, O.; Melnick, A. DNA methylation signatures define molecular subtypes of diffuse large B-cell lymphoma. Blood 2010, 116, e81–e89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jais, J.P.; Haioun, C.; Molina, T.J.; Rickman, D.S.; De Reynies, A.; Berger, F.; Gisselbrecht, C.; Brière, J.; Reyes, F.; et al. The expression of 16 genes related to the cell of origin and immune response predicts survival in elderly patients with diffuse large B-cell lymphoma treated with CHOP and rituximab. Leukemia 2008, 22, 1917–1924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancer Genome Atlas Research Network; Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.M.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar]
- Hummel, M.; Bentink, S.; Berger, H. Molecular Mechanisms in Malignant Lymphomas Network Project of the Deutsche Krebshilfe. A biologic definition of Burkitt’s lymphoma from transcriptional and genomic profiling. N. Engl. J. Med. 2006, 354, 2419–2430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhan, F.; Huang, Y.; Colla, S.; Stewart, J.P.; Hanamura, I.; Gupta, S.; Epstein, J.; Yaccoby, S.; Sawyer, J.; Burington, B.; et al. The molecular classification of multiple myeloma. Blood 2006, 108, 2020–2028. [Google Scholar] [CrossRef] [Green Version]
- Tsuda, S.; Carreras, J.; Kikuti, Y.Y.; Nakae, H.; Dekiden-Monma, M.; Imai, J.; Tsuruya, K.; Nakamura, J.; Tsukune, Y.; Uchida, T.; et al. Prediction of steroid demand in the treatment of patients with ulcerative colitis by immunohistochemical analysis of the mucosal microenvironment and immune checkpoint: Role of macrophages and regulatory markers in disease severity. Pathol. Int. 2019, 69, 260–271. [Google Scholar] [CrossRef]
- Carreras, J.; Lopez-Guillermo, A.; Kikuti, Y.Y.; Itoh, J.; Masashi, M.; Ikoma, H.; Tomita, S.; Hiraiwa, S.; Hamoudi, R.; Rosenwald, A.; et al. High TNFRSF14 and low BTLA are associated with poor prognosis in Follicular Lymphoma and in Diffuse Large B-cell Lymphoma transformation. J. Clin. Exp. Hematop. 2019, 59, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Aguirre-Gamboa, R.; Gomez-Rueda, H.; Martínez-Ledesma, E.; Chacolla-Huaringa, R.; Rodriguez-Barrientos, A.; Tamez-Pena, J.; Treviño, V. SurvExpress: An online biomarker validation tool and database for cancer gene expression data using survival analysis. PLoS ONE 2013, 8, e74250. [Google Scholar]
- Neural Networks (Computer). NCBI, MeSH Unique ID: D016571. Year Introduced: 1992. Available online: https://www.ncbi.nlm.nih.gov/mesh/?term=%22Neural+Networks+(Computer)%22%5BMeSH+Terms%5D (accessed on 20 May 2020).
- The UniProt Consortium. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.K.; Zhang, R.Y.; Yong, Y.L.; Zhang, Z.Y.; Li, C.; Chen, Z.N.; Bian, H. Identification of crucial genes based on expression profiles of hepatocellular carcinomas by bioinformatics analysis. Peer J. 2019, 7, e7436. [Google Scholar] [CrossRef] [Green Version]
- Fabregat, A.; Jupe, S.; Matthews, L.; Sidiropoulos, K.; Gillespie, M.; Garapati, P.; Haw, R.; Jassal, B.; Korninger, F.; May, B.; et al. The Reactome Pathway Knowledgebase. Nucleic Acids Res. 2018, 46, D649–D655. [Google Scholar] [CrossRef] [PubMed]
- Li, X.L.; Ji, Y.M.; Song, R.; Li, X.N.; Guo, L.S. KIF23 Promotes Gastric Cancer by Stimulating Cell Proliferation. Dis. Markers 2019, 2019, 9751923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- David, M.D.; Petit, D.; Bertoglio, J. The RhoGAP ARHGAP19 controls cytokinesis and chromosome segregation in T lymphocytes. J. Cell Sci. 2014, 127, 400–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Wang, M.Y.; He, J.; Wang, J.-C.; Yang, Y.-J.; Jin, L.; Chen, Z.-Y.; Ma, X.-J.; Sun, M.-H.; Xia, K.-Q.; et al. Tumor necrosis factor-α induced protein 8 polymorphism and risk of non-Hodgkin’s lymphoma in a Chinese population: A case-control study. PLoS ONE 2012, 7, e37846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deeb, S.J.; Tyanova, S.; Hummel, M.; Schmidt-Supprian, M.; Cox, J.; Mann, M. Machine Learning-based Classification of Diffuse Large B-cell Lymphoma Patients by Their Protein Expression Profiles. Mol. Cell. Proteom. 2015, 14, 2947–2960. [Google Scholar] [CrossRef] [Green Version]
- You, Z.; Ouyang, H.; Lopatin, D.; Polver, J.P.; Wang, C.Y. Nuclear Factor-Kappa B-inducible Death Effector Domain-Containing Protein Suppresses Tumor Necrosis Factor-Mediated Apoptosis by Inhibiting caspase-8 Activity. J. Biol. Chem. 2001, 276, 26398–26404. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| N. | Series ID | Cases | Total Num. (%) | Log-Rank p Value | 5-y OS (±95% CI) | HR p Value | HR (95% CI) |
|---|---|---|---|---|---|---|---|
| Chronic lymphocytic leukemia (CLL) | |||||||
| 1 | GSE22762 GPL570 | 107 | 308 (15.2%) | Reference | 86.7% (84.9–88.5%) | 3.59 × 10−48 | reference |
| 2 | ICGC CLLE-ES v.2016 | 201 | |||||
| Mantle cell lymphoma (MCL) | |||||||
| 3 | LLMPP Rosenwald 2003 | 92 | 92 (4.5%) | 6.10 × 10−39 | 28.3% (26.7–29.9%) | 2.45 × 10−26 | 6.6 (4.6–9.3) |
| Follicular Lymphoma (FL) | |||||||
| 4 | GSE16131 GPL96 | 180 | 180 (8.9%) | 6.79 × 10−8 | 70.7% (68.2–73.2%) | 9.24 × 10−8 | 2.4 (1.7–3.2) |
| Diffuse Large B-cell Lymphoma (DLBCL) | |||||||
| 5 | GSE10846 | 414 | 741 (36.5%) | 1.03 × 10−17 | 56.5% (55.3–57.7%) | 1.10 × 10−19 | 3.5 (2.7–4.6) |
| 6 | GSE23501 | 69 | |||||
| 7 | E-TABM-346 | 52 | |||||
| 8 | TCGA DLBCL v.2016 | 47 | |||||
| 9 | GSE4475 | 159 | |||||
| Multiple Myeloma (MM) | |||||||
| 10 | GSE2658 | 559 | 559 (27.6%) | 2.47 × 10−8 | 62.7% (59.95–65.5%) | 7 × 10−5 | 1.9 (1.4–2.6) |
| Acute Myeloid Leukemia (AML) | |||||||
| 11 | TCGA-AML v.2016 | 149 | 149 (7.3%) | 1.47 × 10−46 | 23.2% (22.1–24.3%) | 6.11 × 10−37 | 8.3 (5.9–11.5) |
| All | Series 1–11 | 2029 | 2029 (100%) | 4.78 × 10−56 | 62.4% (61.6–63.2%) | - | - |
| Sub-Type | Series | Low-Risk/High-Risk | Log-Rank | 5-Year OS (95% CI) | HR | HR | |
|---|---|---|---|---|---|---|---|
| Num. (%) | p Value | Low-Risk | High-Risk | p Value | (95% CI) | ||
| CLL | 1–2 | 219 (71.1%)/89 (28.9%) | 3.07 × 10−10 | 94.2% (92.6–95.8%) | 69.2% (69.2–65.7%) | 7.63 × 10−09 | 4.3 (2.6–7.0) |
| MCL | 3 | 65 (70.7%)/27 (29.3%) | 1.46 × 10−09 | 38.4% (35.6–41.2%) | 0% (0–0%) | 3.22 × 10−08 | 5.2 (2.9–9.2) |
| FL | 4 | 113 (62.8%)/67 (37.2%) | 6.58 × 10−08 | 79.3% (76.2–83.4%) | 55.9% (52.4–59.4%) | 2.59 × 10−07 | 3.0 (1.9–4.6) |
| DLBCL | 5–9 | 587 (79.2%)/154 (20.8%) | 3.92 × 10−42 | 68.1% (66.5–69.7%) | 16.3% (15.7–16.9%) | 1.32 × 10−35 | 4.5 (3.5–5.7) |
| MM | 10 | 499 (89.3%)/60 (10.7%) | 1.84 × 10−16 | 69.6% (66.6–72.6%) | 0% (0–0%) | 1.63 × 10−13 | 5.3 (3.4–8.2) |
| AML | 11 | 116 (77.9%)/33 (22.1%) | 1.23 × 10−09 | 29.7% (27.9–31.5%) | 3.9% (3.8–4.0%) | 1.66 × 10−08 | 3.7 (2.4–5.9) |
| All | 1–11 | 1599 (78.8%)/430 (21.2%) | 9.26 × 10−59 | 70.9% (69.9–71.9%) | 34.4% (33.5–35.3%) | 9.53 × 10−53 | 3.2 (2.8–3.7) |
| High RNA of Gene | CLL 1 | CLL 2 | MCL 3 | FL 4 | DLBCL 5 | DLBCL 6 | DLBCL 7 | DLBCL 8 | DLBCL 9 | MM 10 | AML 11 | % |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SFTPC | NC | NC | - | NC | NC | NC | NC | HR | NC | LR | NC | 20 |
| ARHGAP19 | NC | - | HR | LR | NC | NC | NC | NC | LR | HR | NC | 40 |
| MESD | NC | - | - | - | NC | NC | - | NC | - | LR * | LR | 33 |
| SNN | NC | NC | - | LR | NC | NC | NC | NC | LR | NC | HR * | 30 |
| ALDOB | NC | - | LR | NC | NC | NC | NC | NC | NC | LR | NC | 20 |
| SPACA9 | HR | NC | - | HR | NC | - | NC | NC | NC | LR | NC | 33 |
| SWSAP1 | NC | NC | - | - | HR | NC | - | NC | - | HR | HR * | 43 |
| WDCP | NC | - | - | NC | HR | NC | NC | NC | NC | NC | LR | 22 |
| ZSCAN12 | NC | - | - | NC | LR | NC | NC | HR* | HR | NC | LR | 44 |
| DIP2A | NC | NC | - | NC | NC | NC | NC | HR | NC | NC | NC | 10 |
| ATF6B | HR | - | - | NC | NC | LR | NC | NC | HR | HR | LR | 56 |
| CACNA1B | NC | NC | - | HR | HR | HR | NC | NC | NC | LR | NC | 40 |
| TNFAIP8 | NC | NC | - | LR | HR | NC | HR | HR | HR | NC | LR | 60 |
| RPS23 | NC | - | - | NC | NC | LR | NC | NC | NC | NC | LR | 22 |
| POLR3H | NC | - | - | - | HR | NC | - | LR | - | NC | HR | 50 |
| ENO3 | HR | NC | - | NC | HR | NC | HR | HR* | HR | HR | HR | 70 |
| RAB7A | NC | NC | NC | NC | NC | NC | NC | NC | NC | LR | NC | 9 |
| SERPINB8 | HR | HR * | - | NC | HR | NC | NC | NC | NC | NC | NC | 30 |
| SZRD1 | NC | NC | HR | NC | HR | NC | NC | NC | NC | HR | NC | 27 |
| EMC9 | NC | - | - | LR | NC | NC | NC | LR | LR | HR | NC | 44 |
| ARMH3 | HR | - | - | NC | NC | NC | NC | NC | NC | HR | NC | 22 |
| LPXN | HR | - | - | NC | LR | LR | NC | NC | LR | NC | NC | 44 |
| KIF23 | NC | NC | HR | NC | HR | NC | NC | NC | LR | HR | LR | 45 |
| GGA3 | NC | - | - | NC | HR | NC | HR | NC | HR | NC | LR | 44 |
| METTL21A | NC | - | - | - | LR | LR | - | NC | - | HR | NC | 50 |
| Variable | Num. | % | p Value | Hazard Risk | 95.0% CI for HR | |
|---|---|---|---|---|---|---|
| Lower | Upper | |||||
| Sex Male | 54/97 | 55.7 | 0.714 | 1.124 | 0.603 | 2.095 |
| Age > 60 | 68/97 | 70.1 | 0.004 | 3.968 | 1.555 | 10.126 |
| Location | ||||||
| Nodal (+spleen) | 52/97 | 53.6 | Reference | - | - | - |
| Extranodal | ||||||
| Waldeyer’s ring | 9/97 | 9.3 | 0.238 | 0.238 | 0.032 | 1.774 |
| Gastrointestinal | 10/97 | 10.3 | 0.800 | 0.8 | 0.238 | 2.687 |
| Other extranodal | 26/97 | 26.8 | 1.625 | 1.625 | 0.847 | 3.12 |
| LDH High (>219) | 58/96 | 60.4 | 0.003 | 3.269 | 1.501 | 7.119 |
| Seric IL2RA High (>530) | 70/90 | 77.8 | 0.024 | 3.914 | 1.2 | 12.762 |
| ECOG Performance Status ≥2 | 13/77 | 16.9 | 3.90 × 10−4 | 4.019 | 1.863 | 8.668 |
| Clinical stage III or IV | 41/88 | 46.6 | 0.047 | 1.981 | 1.01 | 3.884 |
| Extranodal disease site >1 | 18/73 | 24.7 | 0.000381 | 3.784 | 1.816 | 7.884 |
| B symptoms | 18/79 | 22.8 | 0.311 | 1.491 | 0.689 | 3.226 |
| International Prognostic Index (IPI) | ||||||
| Low risk (L) | 30/80 | 37.5 | Reference | - | - | - |
| Low-intermediate risk (LI) | 25/80 | 31.3 | 0.011 | 3.523 | 1.334 | 9.304 |
| High-intermediate risk (HI) | 14/80 | 17.5 | 0.046 | 3.053 | 1.022 | 9.114 |
| High risk (H) | 11/80 | 13.8 | 0.005 | 5.035 | 1.615 | 15.701 |
| Cell-of-origin Subtype (Hans) | ||||||
| GCB | 31/94 | 33 | - | - | - | - |
| Non-GCB | 63/94 | 67 | 0.011 | 2.906 | 1.283 | 6.583 |
| High RGS1 expression | 52/96 | 54.2 | 0.032 | 2.147 | 1.066 | 4.323 |
| Positive BCL2 expression | 73/92 | 79.3 | 0.024 | 3.887 | 1.195 | 12.639 |
| Epstein–Barr virus, EBER+ | 17/95 | 17.9 | 0.005 | 2.822 | 1.371 | 5.809 |
| Treatment | ||||||
| RCHOP | 64/89 | 71.9 | Reference | - | - | - |
| RCHOP-like | 20/89 | 22.5 | 0.148 | 1.715 | 0.826 | 3.561 |
| Others | 5/89 | 5.6 | 0.394 | 1.881 | 0.44 | 8.037 |
| Response to treatment | ||||||
| CR | 63/85 | 74.1 | Reference | - | - | - |
| PR+PD+SD+NC | 22/85 | 25.9 | 2.06 × 10−12 | 16.044 | 7.401 | 34.779 |
| Overall survival (outcome) | ||||||
| Dead | 41/97 | 42.3 | - | - | - | - |
| Alive | 56/97 | 57.7 | - | - | - | - |
| Predictors for High TNFAIP8 | p Value | Odds Ratio | 95% C.I. for OR | |
|---|---|---|---|---|
| Lower | Upper | |||
| Sex Male | 0.394 | 0.653 | 0.245 | 1.74 |
| Age >60 | 0.022 | 3.167 | 1.177 | 8.519 |
| Location | ||||
| Nodal (+spleen) | Reference | - | - | - |
| Extranodal | ||||
| Waldeyer’s ring | 0.357 | 0.507 | 0.119 | 2.15 |
| Gastrointestinal | 0.941 | 0.946 | 0.215 | 4.154 |
| Other extranodal | 0.998 | 6.51 × 10+8 | 0 | . |
| LDH High (>219) | 0.522 | 1.369 | 0.523 | 3.582 |
| Serum soluble IL2RA High (>530) | 0.001 | 6 | 1.991 | 18.078 |
| ECOG Performance Status ≥2 | 0.823 | 0.85 | 0.204 | 3.539 |
| Clinical stage III or IV | 0.373 | 1.577 | 0.579 | 4.298 |
| Extranodal disease site >1 | 0.149 | 4.744 | 0.572 | 39.361 |
| B symptoms | 0.194 | 2.844 | 0.588 | 13.765 |
| High-intermediate+high IPI | 0.352 | 1.793 | 0.524 | 6.129 |
| Non-GCB Subtype (Hans’ algorithm) | 0.000474 | 6.588 | 2.289 | 18.963 |
| High RGS1 protein expression | 0.159 | 2.004 | 0.762 | 5.271 |
| Positive BCL2 protein expression (>30%) | 0.109 | 2.458 | 0.819 | 7.38 |
| High CD163+ tumor-associated macrophages (TAMs) | 0.017 | 3.3 | 1.235 | 8.819 |
| Epstein–Barr virus, EBER+ | 0.968 | 0.975 | 0.283 | 3.363 |
| Absence of clinical response to treatment | 0.213 | 2.341 | 0.614 | 8.927 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carreras, J.; Kikuti, Y.Y.; Miyaoka, M.; Hiraiwa, S.; Tomita, S.; Ikoma, H.; Kondo, Y.; Ito, A.; Shiraiwa, S.; Hamoudi, R.; et al. A Single Gene Expression Set Derived from Artificial Intelligence Predicted the Prognosis of Several Lymphoma Subtypes; and High Immunohistochemical Expression of TNFAIP8 Associated with Poor Prognosis in Diffuse Large B-Cell Lymphoma. AI 2020, 1, 342-360. https://doi.org/10.3390/ai1030023
Carreras J, Kikuti YY, Miyaoka M, Hiraiwa S, Tomita S, Ikoma H, Kondo Y, Ito A, Shiraiwa S, Hamoudi R, et al. A Single Gene Expression Set Derived from Artificial Intelligence Predicted the Prognosis of Several Lymphoma Subtypes; and High Immunohistochemical Expression of TNFAIP8 Associated with Poor Prognosis in Diffuse Large B-Cell Lymphoma. AI. 2020; 1(3):342-360. https://doi.org/10.3390/ai1030023
Chicago/Turabian StyleCarreras, Joaquim, Yara Y. Kikuti, Masashi Miyaoka, Shinichiro Hiraiwa, Sakura Tomita, Haruka Ikoma, Yusuke Kondo, Atsushi Ito, Sawako Shiraiwa, Rifat Hamoudi, and et al. 2020. "A Single Gene Expression Set Derived from Artificial Intelligence Predicted the Prognosis of Several Lymphoma Subtypes; and High Immunohistochemical Expression of TNFAIP8 Associated with Poor Prognosis in Diffuse Large B-Cell Lymphoma" AI 1, no. 3: 342-360. https://doi.org/10.3390/ai1030023
APA StyleCarreras, J., Kikuti, Y. Y., Miyaoka, M., Hiraiwa, S., Tomita, S., Ikoma, H., Kondo, Y., Ito, A., Shiraiwa, S., Hamoudi, R., Ando, K., & Nakamura, N. (2020). A Single Gene Expression Set Derived from Artificial Intelligence Predicted the Prognosis of Several Lymphoma Subtypes; and High Immunohistochemical Expression of TNFAIP8 Associated with Poor Prognosis in Diffuse Large B-Cell Lymphoma. AI, 1(3), 342-360. https://doi.org/10.3390/ai1030023