


Chronic Rhinosinusitis with Nasal Polyposis in People with Cystic Fibrosis


Abstract
:1. Cystic Fibrosis: Background and Overview
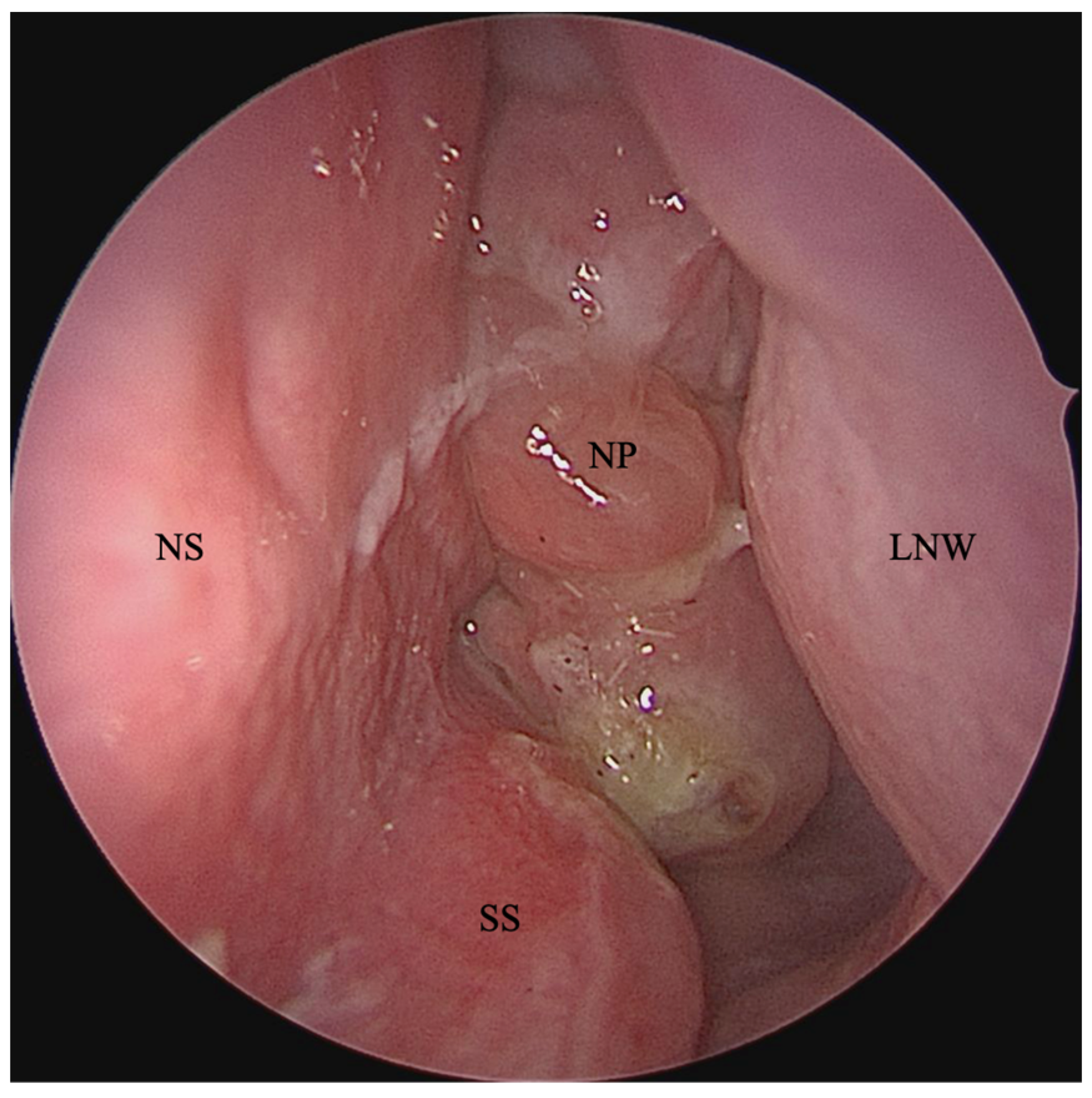
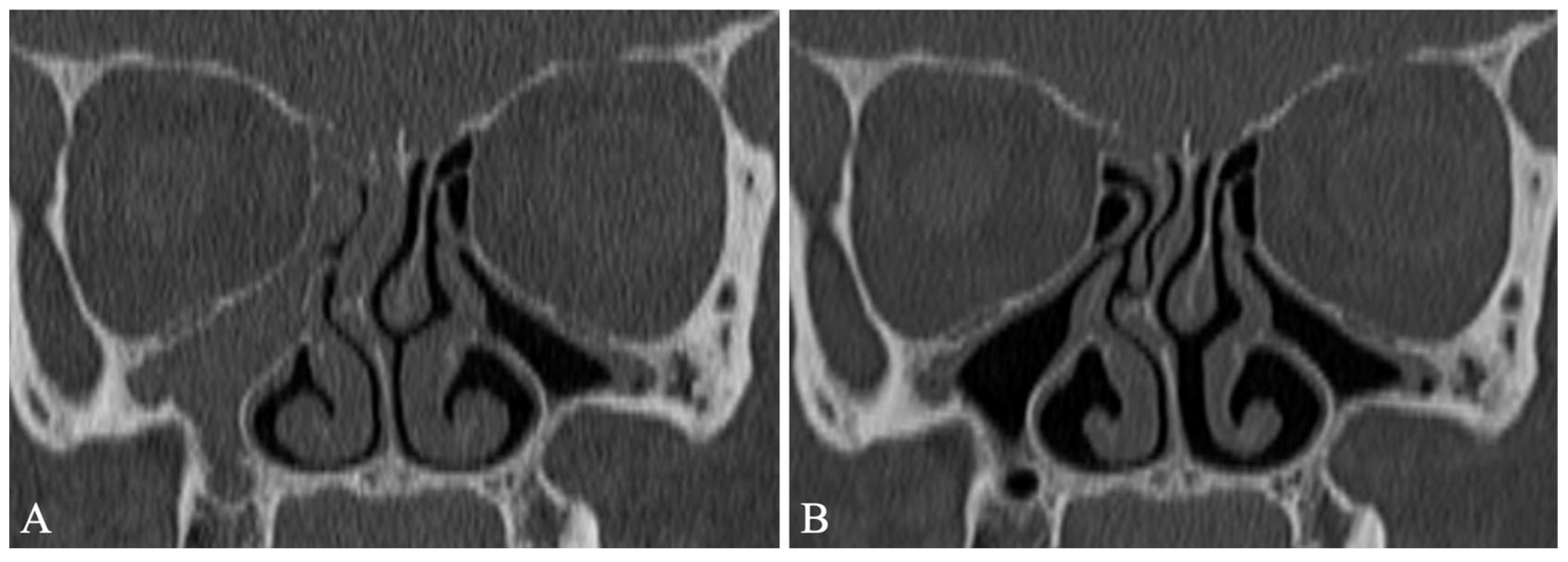
2. Chronic Rhinosinusitis with Nasal Polyposis in People with Cystic Fibrosis
3. Management of Nasal Polyps in People with Cystic Fibrosis
4. The Impact of Highly Effective Modulator Therapy on Sinusitis and Nasal Polyps
5. Unique Considerations for Managing Chronic Rhinosinusitis in the Pediatric Population
6. Future Directions in the Management of Cystic Fibrosis-Related Chronic Rhinosinusitis with Nasal Polyposis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Walters, S.; Mehta, A. Chapter 2: Epidemiology of cystic fibrosis. In Cystic Fibrosis, 3rd ed.; Hodson, M., Ed.; Taylor & Francis Group: London, UK, 2007; pp. 21–45. [Google Scholar]
- O’Sullivan, B.P.; Freedman, S.D. Cystic fibrosis. Lancet 2009, 373, 1891–1904. [Google Scholar] [CrossRef] [PubMed]
- Yung, M.W.; Gould, J.; Upton, G.J.G. Nasal polyposis in children with cystic fibrosis: A long-term follow-up study. Ann. Otol. Rhinol. Laryngol. 2002, 111 Pt 1, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- Di Lullo, A.M.; Iacotucci, P.; Comegna, M.; Amato, F.; Dolce, P.; Castaldo, G.; Cantone, E.; Carnovale, V. Cystic Fibrosis: The Sense of Smell. Am. J. Rhinol. Allergy 2020, 34, 35–42. [Google Scholar] [CrossRef]
- Mall, M.A.; Galietta, L.J.V. Targeting ion channels in cystic fibrosis. J. Cyst. Fibros. 2015, 14, 561–570. [Google Scholar] [CrossRef] [PubMed]
- Bock, J.M.; Schien, M.; Fischer, C.; Naehrlich, L.; Kaeding, M.; Guntinas-Lichius, O.; Gerber, A.; Arnold, C.; Mainz, J.G. Importance to question sinonasal symptoms and to perform rhinoscopy and rhinomanometry in cystic fibrosis patients. Pediatr. Pulmonol. 2017, 52, 167–174. [Google Scholar] [CrossRef]
- Cystic Fibrosis Foundation Patient Registry, 2021 Annual Data Report; Cystic Fibrosis Foundation: Bethesda, MD, USA, 2022.
- Tipirneni, K.E.; Woodworth, B.A. Medical and Surgical Advancements in the Management of Cystic Fibrosis Chronic Rhinosinusitis. Curr. Otorhinolaryngol. Rep. 2017, 5, 24–34. [Google Scholar] [CrossRef]
- Johnson, B.J.; Choby, G.W.; O’Brien, E.K. Chronic rhinosinusitis in patients with cystic fibrosis—Current management and new treatments. Laryngoscope Investig. Otolaryngol. 2020, 5, 368–374. [Google Scholar] [CrossRef] [PubMed]
- Illing, E.A.; Woodworth, B.A. Management of the upper airway in cystic fibrosis. Curr. Opin. Pulm. Med. 2014, 20, 623–631. [Google Scholar] [CrossRef]
- Alanin, M.C.; Aanaes, K.; Høiby, N.; Pressler, T.; Skov, M.; Nielsen, K.G.; Taylor-Robinson, D.; Waldmann, E.; Krogh Johansen, H.; von Buchwald, C. Sinus surgery postpones chronic Gram-negative lung infection: Cohort study of 106 patients with cystic fibrosis. Rhinology 2016, 54, 206–213. [Google Scholar] [CrossRef]
- Johansen, H.K.; Aanaes, K.; Pressler, T.; Nielsen, K.G.; Fisker, J.; Skov, M.; Høiby, N.; von Buchwald, C. Colonisation and infection of the paranasal sinuses in cystic fibrosis patients is accompanied by a reduced PMN response. J. Cyst. Fibros. 2012, 11, 525–531. [Google Scholar] [CrossRef]
- Safi, C.; Zheng, Z.; Dimango, E.; Keating, C.; Gudis, D.A. Chronic Rhinosinusitis in Cystic Fibrosis: Diagnosis and Medical Management. Med. Sci. 2019, 7, 32. [Google Scholar] [CrossRef] [PubMed]
- Doull, I.J. Recent advances in cystic fibrosis. Arch. Dis. Child. 2001, 85, 62–66. [Google Scholar] [CrossRef]
- Liang, J.; Higgins, T.; Ishman, S.L.; Boss, E.F.; Benke, J.R.; Lin, S.Y. Medical management of chronic rhinosinusitis in cystic fibrosis: A systematic review: Medical Management of CRS in CF. Laryngoscope 2014, 124, 1308–1313. [Google Scholar] [CrossRef]
- Suy, P.; Coudert, A.; Vrielynck, S.; Truy, E.; Hermann, R.; Ayari-Khalfallah, S. Evolution of sinonasal clinical features in children with cystic fibrosis. Int. J. Pediatr. Otorhinolaryngol. 2019, 124, 47–53. [Google Scholar] [CrossRef]
- Hulka, G.F. Head and Neck Manifestations of Cystic Fibrosis and Ciliary Dyskinesia. Otolaryngol. Clin. N. Am. 2000, 33, 1333–1341. [Google Scholar] [CrossRef] [PubMed]
- Kimple, A.J.; Senior, B.A.; Naureckas, E.T.; Gudis, D.A.; Meyer, T.; Hempstead, S.E.; Resnick, H.E.; Albon, D.; Barfield, W.; Benoit, M.M.; et al. Cystic Fibrosis Foundation otolaryngology care multidisciplinary consensus recommendations. Int. Forum Allergy Rhinol. 2022, 12, 1089–1103. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, B.; Richardson, M. Impact of sinusitis in cystic fibrosis. J. Allergy Clin. Immunol. 1992, 90, 547–552. [Google Scholar] [CrossRef]
- Le, C.; McCrary, H.C.; Chang, E. Cystic Fibrosis Sinusitis. Adv. Otorhinolaryngol. 2016, 79, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Spielman, D.B.; Beswick, D.M.; Kimple, A.J.; Senior, B.A.; Aanaes, K.; Woodworth, B.A.; Schlosser, R.J.; Lee, S.; Cho, D.; Adappa, N.D.; et al. The management of cystic fibrosis chronic rhinosinusitis: An evidenced-based review with recommendations. Int. Forum Allergy Rhinol. 2022, 12, 1148–1183. [Google Scholar] [CrossRef]
- Beswick, D.M.; Schlosser, R.J. Chronic rhinosinusitis in people with Cystic Fibrosis: Expanding evidence and future directions. J. Cyst. Fibros. 2022, 21, 737–738. [Google Scholar] [CrossRef]
- Rosenfeld, R.M.; Piccirillo, J.F.; Chandrasekhar, S.S.; Brook, I.; Kumar, K.A.; Kramper, M.; Orlandi, R.R.; Palmer, J.N.; Patel, Z.M.; Peters, A.; et al. Clinical Practice Guideline (Update): Adult Sinusitis. Otolaryngol. Head. Neck Surg. 2015, 152 (Suppl. S2), S1–S39. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, O.G.; Rowan, N.R. Olfactory Dysfunction and Chronic Rhinosinusitis. Immunol. Allergy Clin. N. Am. 2020, 40, 223–232. [Google Scholar] [CrossRef]
- Lindig, J.; Steger, C.; Beiersdorf, N.; Michl, R.; Beck, J.F.; Hummel, T.; Mainz, J.G. Smell in cystic fibrosis. Eur. Arch. Otorhinolaryngol. 2013, 270, 915–921. [Google Scholar] [CrossRef] [PubMed]
- Beswick, D.M.; Humphries, S.M.; Balkissoon, C.D.; Strand, M.; Miller, J.E.; Khatiwada, A.; Vladar, E.K.; Lynch, D.A.; Taylor-Cousar, J.L. Olfactory dysfunction in people with cystic fibrosis with at least one copy of F508del. Int. Forum Allergy Rhinol. 2022, 12, 963–966. [Google Scholar] [CrossRef] [PubMed]
- Beswick, D.M.; Humphries, S.M.; Balkissoon, C.D.; Strand, M.; Vladar, E.K.; Ramakrishnan, V.R.; Taylor-Cousar, J.L. Olfactory dysfunction in cystic fibrosis: Impact of CFTR modulator therapy. J. Cyst. Fibros. 2022, 21, e141–e147. [Google Scholar] [CrossRef]
- Schwartz, C.E.; Andresen, E.M.; Nosek, M.A.; Krahn, G.L. RRTC Expert Panel on Health Status Measurement. Response shift theory: Important implications for measuring quality of life in people with disability. Arch. Phys. Med. Rehabil. 2007, 88, 529–536. [Google Scholar] [CrossRef]
- Stevens, W.W.; Schleimer, R.P.; Kern, R.C. Chronic Rhinosinusitis with Nasal Polyps. J. Allergy Clin. Immunol. Pract. 2016, 4, 565–572. [Google Scholar] [CrossRef]
- Fokkens, W.J.; Lund, V.J.; Hopkins, C.; Hellings, P.W.; Kern, R.; Reitsma, S.; Toppila-Salmi, S.; Bernal-Sprekelsen, M.; Mullol, J.; Alobid, I.; et al. European Position Paper on Rhinosinusitis and Nasal Polyps 2020. Rhinology 2020, 58 (Suppl. S29), 1–464. [Google Scholar] [CrossRef]
- Hadfield, P.J.; Rowe-Jones, J.M.; Mackay, I.S. The prevalence of nasal polyps in adults with cystic fibrosis. Clin. Otolaryngol. 2000, 25, 19–22. [Google Scholar] [CrossRef]
- Cimmino, M.; Cavaliere, M.; Nardone, M.; Plantulli, A.; Orefice, A.; Esposito, V.; Raia, V. Clinical characteristics and genotype analysis of patients with cystic fibrosis and nasal polyposis. Clin. Otolaryngol Allied Sci. 2003, 28, 125–132. [Google Scholar] [CrossRef]
- Kerrebijn, J.D.; Poublon, R.M.; Overbeek, S.E. Nasal and paranasal disease in adult cystic fibrosis patients. Eur. Respir. J. 1992, 5, 1239–1242. [Google Scholar]
- De Gaudemar, I.; Contencin, P.; Van den Abbeele, T.; Munck, A.; Navarro, J.; Narcy, P. Is nasal polyposis in cystic fibrosis a direct manifestation of genetic mutation or a complication of chronic infection? Rhinology 1996, 34, 194–197. [Google Scholar]
- Triglia, J.M.; Nicollas, R. Nasal and sinus polyposis in children. Laryngoscope 1997, 107, 963–966. [Google Scholar] [CrossRef]
- Slieker, M.G.; Schilder, A.G.M.; Uiterwaal, C.S.P.M.; van der Ent, C.K. Children with cystic fibrosis: Who should visit the otorhinolaryngologist? Arch. Otolaryngol. Head Neck Surg. 2002, 128, 1245–1248. [Google Scholar] [CrossRef] [PubMed]
- Schraven, S.P.; Wehrmann, M.; Wagner, W.; Blumenstock, G.; Koitschev, A. Prevalence and histopathology of chronic polypoid sinusitis in pediatric patients with cystic fibrosis. J. Cyst. Fibros. 2011, 10, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Akdis, C.A.; Bachert, C.; Cingi, C.; Dykewicz, M.S.; Hellings, P.W.; Naclerio, R.M.; Schleimer, R.P.; Ledford, D. Endotypes and phenotypes of chronic rhinosinusitis: A PRACTALL document of the European Academy of Allergy and Clinical Immunology and the American Academy of Allergy, Asthma & Immunology. J. Allergy Clin. Immunol. 2013, 131, 1479–1490. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Van Zele, T.; Perez-Novo, C.; Van Bruaene, N.; Holtappels, G.; DeRuyck, N.; Van Cauwenberge, P.; Bachert, C. Different types of T-effector cells orchestrate mucosal inflammation in chronic sinus disease. J. Allergy Clin. Immunol. 2008, 122, 961–968. [Google Scholar] [CrossRef]
- Mahdavinia, M.; Suh, L.A.; Carter, R.G.; Stevens, W.W.; Norton, J.E.; Kato, A.; Tan, B.K.; Kern, R.C.; Conley, D.B.; Chandra, R.; et al. Increased noneosinophilic nasal polyps in chronic rhinosinusitis in US second-generation Asians suggest genetic regulation of eosinophilia. J. Allergy Clin. Immunol. 2015, 135, 576–579. [Google Scholar] [CrossRef]
- Bachert, C.; Zhang, N.; Cavaliere, C.; Weiping, W.; Gevaert, E.; Krysko, O. Biologics for chronic rhinosinusitis with nasal polyps. J. Allergy Clin. Immunol. 2020, 145, 725–739. [Google Scholar] [CrossRef]
- Hamilos, D.L. Chronic Rhinosinusitis in Patients with Cystic Fibrosis. J. Allergy Clin. Immunol. Pract. 2016, 4, 605–612. [Google Scholar] [CrossRef]
- Van Zele, T.; Claeys, S.; Gevaert, P.; Van Maele, G.; Holtappels, G.; Van Cauwenberge, P.; Bachert, C. Differentiation of chronic sinus diseases by measurement of inflammatory mediators. Allergy 2006, 61, 1280–1289. [Google Scholar] [CrossRef]
- Feuillet-Fieux, M.N.; Lenoir, G.; Sermet, I.; Elie, C.; Djadi-Prat, J.; Ferrec, M.; Magen, M.; Couloigner, V.; Manach, Y.; Lacour, B.; et al. Nasal polyposis and cystic fibrosis(CF): Review of the literature. Rhinology 2011, 49, 347–355. [Google Scholar] [CrossRef]
- Henriksson, G.; Westrin, K.M.; Karpati, F.; Wikstroïm, A.C.; Stierna, P.; Hjelte, L. Nasal Polyps in Cystic Fibrosis. Chest 2002, 121, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Clancy, J.P.; Jain, M. Personalized medicine in cystic fibrosis: Dawning of a new era. Am. J. Respir. Crit. Care Med. 2012, 186, 593–597. [Google Scholar] [CrossRef]
- Koch, C.; Høiby, N. Pathogenesis of cystic fibrosis. Lancet 1993, 341, 1065–1069. [Google Scholar] [CrossRef]
- Cimmino, M.; Nardone, M.; Cavaliere, M.; Plantulli, A.; Sepe, A.; Esposito, V.; Mazzarella, G.; Raia, V. Dornase Alfa as Postoperative Therapy in Cystic Fibrosis Sinonasal Disease. Arch. Otolaryngol. Head Neck Surg. 2005, 131, 1097. [Google Scholar] [CrossRef]
- Yang, C.; Montgomery, M. Dornase alfa for cystic fibrosis. Cochrane Database Syst. Rev. 2021, 3, CD001127. [Google Scholar] [CrossRef] [PubMed]
- Mainz, J.G.; Schien, C.; Schiller, I.; Schädlich, K.; Koitschev, A.; Koitschev, C.; Riethmüller, J.; Graepler-Mainka, U.; Wiedemann, B.; Beck, J.F. Sinonasal inhalation of dornase alfa administered by vibrating aerosol to cystic fibrosis patients: A double-blind placebo-controlled cross-over trial. J. Cyst. Fibros. 2014, 13, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Shah, G.B.; De Keyzer, L.; Russell, J.A.; Halderman, A. Treatment of chronic rhinosinusitis with dornase alfa in patients with cystic fibrosis: A systematic review: Dornase alfa for CRS CF patients. Int. Forum Allergy Rhinol. 2018, 8, 729–736. [Google Scholar] [CrossRef]
- Mainz, J.G.; Schumacher, U.; Schädlich, K.; Hentschel, J.; Koitschev, C.; Koitschev, A.; Riethmüller, J.; Prenzel, F.; Sommerburg, O.; Wiedemann, B.; et al. Sino nasal inhalation of isotonic versus hypertonic saline (6.0%) in CF patients with chronic rhinosinusitis—Results of a multicenter, prospective, randomized, double-blind, controlled trial. J. Cyst. Fibros. 2016, 15, e57–e66. [Google Scholar] [CrossRef]
- Mainz, J.G.; Koitschev, A. Pathogenesis and management of nasal polyposis in cystic fibrosis. Curr. Allergy Asthma Rep. 2012, 12, 163–174. [Google Scholar] [CrossRef]
- Hadfield, P.J.; Rowe-Jones, J.M.; Mackay, I.S. A prospective treatment trial of nasal polyps in adults with cystic fibrosis. Rhinology 2000, 38, 63–65. [Google Scholar]
- Donaldson, J.D.; Gillespie, C.T. Observations on the efficacy of intranasal beclomethasone dipropionate in cystic fibrosis patients. J. Otolaryngol. 1988, 17, 43–45. [Google Scholar] [PubMed]
- Mainz, J.G.; Schädlich, K.; Schien, C.; Michl, R.; Schelhorn-Neise, P.; Koitschev, A.; Koitschev, C.; Keller, P.M.; Riethmüller, J.; Wiedemann, B.; et al. Sinonasal inhalation of tobramycin vibrating aerosol in cystic fibrosis patients with upper airway Pseudomonas aeruginosa colonization: Results of a randomized, double-blind, placebo-controlled pilot study. Drug Des. Dev. Ther. 2014, 8, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Moss, R.B.; King, V.V. Management of sinusitis in cystic fibrosis by endoscopic surgery and serial antimicrobial lavage. Reduction in recurrence requiring surgery. Arch. Otolaryngol. Head Neck Surg. 1995, 121, 566–572. [Google Scholar] [CrossRef]
- Di Cicco, M.; Alicandro, G.; Claut, L.; Cariani, L.; Luca, N.; Defilippi, G.; Costantini, D.; Colombo, C. Efficacy and tolerability of a new nasal spray formulation containing hyaluronate and tobramycin in cystic fibrosis patients with bacterial rhinosinusitis. J. Cyst. Fibros. 2014, 13, 455–460. [Google Scholar] [CrossRef]
- Lazio, M.S.; Luparello, P.; Mannelli, G.; Santoro, G.P.; Bresci, S.; Braggion, C.; Gallo, O.; Maggiore, G. Quality of Life and Impact of Endoscopic Sinus Surgery in Adult Patients With Cystic Fibrosis. Am. J. Rhinol. Allergy. 2019, 33, 413–419. [Google Scholar] [CrossRef] [PubMed]
- Khalid, A.N.; Mace, J.; Smith, T.L. Outcomes of sinus surgery in adults with cystic fibrosis. Otolaryngol. Head Neck Surg. 2009, 141, 358–363. [Google Scholar] [CrossRef]
- Taylor, R.J.; Miller, J.D.; Rose, A.S.; Drake, A.F.; Zdanski, C.J.; Senior, B.A.; Ebert, C.S.; Zanation, A.M. Comprehensive quality of life outcomes for pediatric patients undergoing endoscopic sinus surgery. Rhinology 2014, 52, 327–333. [Google Scholar] [CrossRef]
- Ayoub, N.; Thamboo, A.; Habib, A.R.; Nayak, J.V.; Hwang, P.H. Determinants and outcomes of upfront surgery versus medical therapy for chronic rhinosinusitis in cystic fibrosis: Chronic rhinosinusitis in cystic fibrosis. Int. Forum Allergy Rhinol. 2017, 7, 450–458. [Google Scholar] [CrossRef]
- Kerem, B.; Rommens, J.M.; Buchanan, J.A.; Markiewicz, D.; Cox, T.K.; Chakravarti, A.; Buchwald, M.; Tsui, L.C. Identification of the cystic fibrosis gene: Genetic analysis. Science 1989, 245, 1073–1080. [Google Scholar] [CrossRef]
- Middleton, P.G.; Taylor-Cousar, J.L. Development of elexacaftor–tezacaftor–ivacaftor: Highly effective CFTR modulation for the majority of people with Cystic Fibrosis. Expert. Rev. Respir. Med. 2021, 15, 723–735. [Google Scholar] [CrossRef] [PubMed]
- Middleton, P.G.; Mall, M.A.; Dřevínek, P.; Lands, L.C.; McKone, E.F.; Polineni, D.; Ramsey, B.W.; Taylor-Cousar, J.L.; Tullis, E.; Vermeulen, F.; et al. Elexacaftor-Tezacaftor-Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele. N. Engl. J. Med. 2019, 381, 1809–1819. [Google Scholar] [CrossRef] [PubMed]
- Boyle, M.P.; De Boeck, K. A new era in the treatment of cystic fibrosis: Correction of the underlying CFTR defect. Lancet Respir. Med. 2013, 1, 158–163. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, B.W.; Davies, J.; McElvaney, N.G.; Tullis, E.; Bell, S.C.; Dřevínek, P.; Griese, M.; McKone, E.F.; Wainwright, C.E.; Konstan, M.W.; et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N. Engl. J. Med. 2011, 365, 1663–1672. [Google Scholar] [CrossRef]
- Heijerman, H.G.M.; McKone, E.F.; Downey, D.G.; Van Braeckel, E.; Rowe, S.M.; Tullis, E.; Mall, M.A.; Welter, J.J.; Ramsey, B.W.; McKee, C.M.; et al. Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: A double-blind, randomised, phase 3 trial. Lancet 2019, 394, 1940–1948. [Google Scholar] [CrossRef]
- Keating, D.; Marigowda, G.; Burr, L.; Daines, C.; Mall, M.A.; McKone, E.F.; Ramsey, B.W.; Rowe, S.M.; Sass, L.A.; Tullis, E.; et al. VX-445-Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis and One or Two Phe508del Alleles. N. Engl. J. Med. 2018, 379, 1612–1620. [Google Scholar] [CrossRef] [PubMed]
- Trikafta United States Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/212273s004lbl.pdf (accessed on 10 May 2023).
- Stapleton, A.L.; Kimple, A.J.; Goralski, J.L.; Nouraie, S.M.; Branstetter, B.F.; Shaffer, A.D.; Pilewski, J.M.; Senior, B.A.; Lee, S.E.; Zemke, A.C. Elexacaftor-Tezacaftor- Ivacaftor improves sinonasal outcomes in cystic fibrosis. J. Cyst. Fibros. 2022, 21, 792–799. [Google Scholar] [CrossRef]
- DiMango, E.; Overdevest, J.; Keating, C.; Francis, S.F.; Dansky, D.; Gudis, D. Effect of highly effective modulator treatment on sinonasal symptoms in cystic fibrosis. J. Cyst. Fibros. 2021, 20, 460–463. [Google Scholar] [CrossRef]
- Beswick, D.M.; Humphries, S.M.; Balkissoon, C.D.; Strand, M.; Vladar, E.K.; Lynch, D.A.; Taylor-Cousar, J.L. Impact of Cystic Fibrosis Transmembrane Conductance Regulator Therapy on Chronic Rhinosinusitis and Health Status: Deep Learning CT Analysis and Patient-reported Outcomes. Ann. Am. Thorac. Soc. 2022, 19, 12–19. [Google Scholar] [CrossRef]
- Benninger, L.A.; Trillo, C.; Lascano, J. CFTR modulator use in post lung transplant recipients. J. Heart Lung Transplant. 2021, 40, 1498–1501. [Google Scholar] [CrossRef]
- Hayes, D.; Darland, L.K.; Hjelm, M.A.; Mansour, H.M.; Wikenheiser-Brokamp, K.A. To treat or not to treat: CFTR modulators after lung transplantation. Pediatr. Transplant. 2021, 25, e14007. [Google Scholar] [CrossRef]
- Ramos, K.J.; Guimbellot, J.S.; Valapour, M.; Bartlett, L.; Wai, T.H.; Goss, C.H.; Pilewski, J.M.; Faro, A.; Diamond, J.M. Use of elexacaftor/tezacaftor/ivacaftor among cystic fibrosis lung transplant recipients. J. Cyst. Fibros. 2022, 21, 745–752. [Google Scholar] [CrossRef]
- Gallant, J.N.; Mitchell, M.B.; Virgin, F.W. Update on sinus disease in children with cystic fibrosis: Advances in treatment modalities, microbiology, and health-related quality-of-life instruments. Curr. Opin. Otolaryngol. Head Neck Surg. 2018, 26, 417–420. [Google Scholar] [CrossRef]
- Gentile, V.G.; Isaacson, G. Patterns of sinusitis in cystic fibrosis. Laryngoscope 1996, 106, 1005–1009. [Google Scholar] [CrossRef] [PubMed]
- Gergin, O.; Kawai, K.; MacDougall, R.D.; Robson, C.D.; Moritz, E.; Cunningham, M.; Adil, E. Sinus Computed Tomography Imaging in Pediatric Cystic Fibrosis: Added Value? Otolaryngol. Head Neck Surg. 2016, 155, 160–165. [Google Scholar] [CrossRef] [PubMed]
- Sommerburg, O.; Wielpütz, M.O.; Trame, J.P.; Wuennenmann, F.; Opdazaite, E.; Stahl, M.; Puderbach, M.U.; Kopp-Schneider, A.; Fritzsching, E.; Kauczor, H.; et al. Magnetic Resonance Imaging Detects Chronic Rhinosinusitis in Infants and Preschool Children with Cystic Fibrosis. Ann. Am. Thorac. Soc. 2020, 17, 714–723. [Google Scholar] [CrossRef] [PubMed]
- Goralski, J.L.; Hoppe, J.E.; Mall, M.A.; McColley, S.A.; McKone, E.; Ramsey, B.; Rayment, J.H.; Robinson, P.; Stehling, F.; Taylor-Cousar, J.L.; et al. Phase 3 Open-Label Clinical Trial of Elexacaftor/Tezacaftor/Ivacaftor in Children Aged 2 Through 5 Years with Cystic Fibrosis and at Least One F508del Allele. Am. J. Respir. Crit. Care Med. 2023, 208, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Kalydeco United States Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/203188s022l_207925s003lbl.pdf (accessed on 10 May 2023).
- Macdonald, K.I.; Gipsman, A.; Magit, A.; Fandino, M.; Massoud, E.; Witterick, I.J.; Hong, P. Endoscopic sinus surgery in patients with cystic fibrosis: A systematic review and meta-analysis of pulmonary function. Rhinology. 2012, 50, 360–369. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Higgins, T.S.; Ishman, S.L.; Boss, E.F.; Benke, J.R.; Lin, S.Y. Surgical management of chronic rhinosinusitis in cystic fibrosis: A systematic review: Surgical management of CRS in CF. Int. Forum Allergy Rhinol. 2013, 3, 814–822. [Google Scholar] [CrossRef] [PubMed]
- Chaaban, M.R.; Kejner, A.; Rowe, S.M.; Woodworth, B.A. Cystic Fibrosis Chronic Rhinosinusitis: A Comprehensive Review. Am. J. Rhinol. Allergy 2013, 27, 387–395. [Google Scholar] [CrossRef]
- Pirsig, W. Growth of the deviated septum and its influence on midfacial development. Facial Plast. Surg. 1992, 8, 224–232. [Google Scholar] [CrossRef] [PubMed]
- Verwoerd-Verhoef, H.L.; Verwoerd, C.D.A. Surgery of the lateral nasal wall and ethmoid: Effects on sinonasal growth. Int. J. Pediatr. Otorhinolaryngol. 2003, 67, 263–269. [Google Scholar] [CrossRef]
- Bothwell, M.R.; Piccirillo, J.F.; Lusk, R.P.; Ridenour, B.D. Long-Term Outcome of Facial Growth after Functional Endoscopic Sinus Surgery. Otolaryngol. Head Neck Surg. 2002, 126, 628–634. [Google Scholar] [CrossRef]
- Van Peteghem, A.; Clement, P.A.R. Influence of extensive functional endoscopic sinus surgery (FESS) on facial growth in children with cystic fibrosis. Int. J. Pediatr. Otorhinolaryngol. 2006, 70, 1407–1413. [Google Scholar] [CrossRef]
- Helmen, Z.M.; Little, R.E.; Robey, T. Utility of Second-Look Endoscopy with Debridement After Pediatric Functional Endoscopic Sinus Surgery in Patients with Cystic Fibrosis. Ann. Otol. Rhinol. Laryngol. 2020, 129, 1153–1162. [Google Scholar] [CrossRef]
- Steinke, J.W.; Payne, S.C.; Chen, P.G.; Negri, J.; Stelow, E.B.; Borish, L. Etiology of Nasal Polyps in Cystic Fibrosis: Not a Unimodal Disease. Ann. Otol. Rhinol. Laryngol. 2012, 121, 579–586. [Google Scholar] [CrossRef]
- Beswick, D.M.; Humphries, S.M.; Miller, J.E.; Balkissoon, C.D.; Khatiwada, A.; Vladar, E.K.; Ramakrishnan, V.R.; Lynch, D.A.; Taylor-Cousar, J.L. Objective and patient-based measures of chronic rhinosinusitis in people with cystic fibrosis treated with highly effective modulator therapy. Int. Forum Allergy Rhinol. 2022, 12, 1435–1438. [Google Scholar] [CrossRef]
- Eischen, E.; Gliksman, M.F.; Segarra, D.; Murtagh, R.D.; Ryan, L.E.; Parasher, A.K.; Tabor, M.H. Correlation between CT imaging and symptom scores in cystic fibrosis associated chronic rhinosinusitis. Am. J. Otolaryngol. 2023, 44, 103858. [Google Scholar] [CrossRef]
- Sawicki, G.S.; Sellers, D.E.; Robinson, W.M. High Treatment Burden in Adults with Cystic Fibrosis: Challenges to Disease Self-Management. J. Cyst. Fibros. 2009, 8, 91–96. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Treatment for Cystic Fibrosis-Related Chronic Rhinosinusitis | Mechanism | Effect on Nasal Polyps | |
|---|---|---|---|
| Local Therapies | Dornase-alpha | Cleaves extracellular DNA, resulting in decreased secretion viscosity and improved mucociliary clearance. | None |
| Nasal saline irrigations | Clears retained mucus and pathogens from the paranasal sinuses. | None | |
| Topical corticosteroids | Decreases inflammation of the sinonasal mucosa. | Reduction in polyps | |
| Topical antibiotics | Treats bacterial infections, which results in decreased inflammation of the paranasal sinuses. | Unknown | |
| Endoscopic sinus surgery | Improves ventilation of the paranasal sinuses, removes polyps, and allows for improved delivery of topical medications postoperatively. | Reduction in polyps | |
| Systemic Therapies | Oral antibiotics | Treats bacterial infections of the paranasal sinuses. | Unknown |
| Oral corticosteroids | Decreases inflammation of the sinonasal mucosa. | Possible reduction in polyps, although evidence is limited | |
| Highly effective modulator therapy | Significantly improves CFTR dysfunction. | Reduction in polyps |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miller, J.E.; Taylor-Cousar, J.L.; Beswick, D.M. Chronic Rhinosinusitis with Nasal Polyposis in People with Cystic Fibrosis. Sinusitis 2023, 7, 27-37. https://doi.org/10.3390/sinusitis7020004
Miller JE, Taylor-Cousar JL, Beswick DM. Chronic Rhinosinusitis with Nasal Polyposis in People with Cystic Fibrosis. Sinusitis. 2023; 7(2):27-37. https://doi.org/10.3390/sinusitis7020004
Chicago/Turabian StyleMiller, Jessa E., Jennifer L. Taylor-Cousar, and Daniel M. Beswick. 2023. "Chronic Rhinosinusitis with Nasal Polyposis in People with Cystic Fibrosis" Sinusitis 7, no. 2: 27-37. https://doi.org/10.3390/sinusitis7020004
APA StyleMiller, J. E., Taylor-Cousar, J. L., & Beswick, D. M. (2023). Chronic Rhinosinusitis with Nasal Polyposis in People with Cystic Fibrosis. Sinusitis, 7(2), 27-37. https://doi.org/10.3390/sinusitis7020004