
Awakening a Molecular Mummy: The Inter-and Intramolecular Photochemistry of Pyromellitic Diimides with Alkyl Carboxylates


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Solvents for Syntheses and Photoreactions
2.2. Synthesis of the Photochemical Starting Materials
2.3. Photoreactions
3. Results
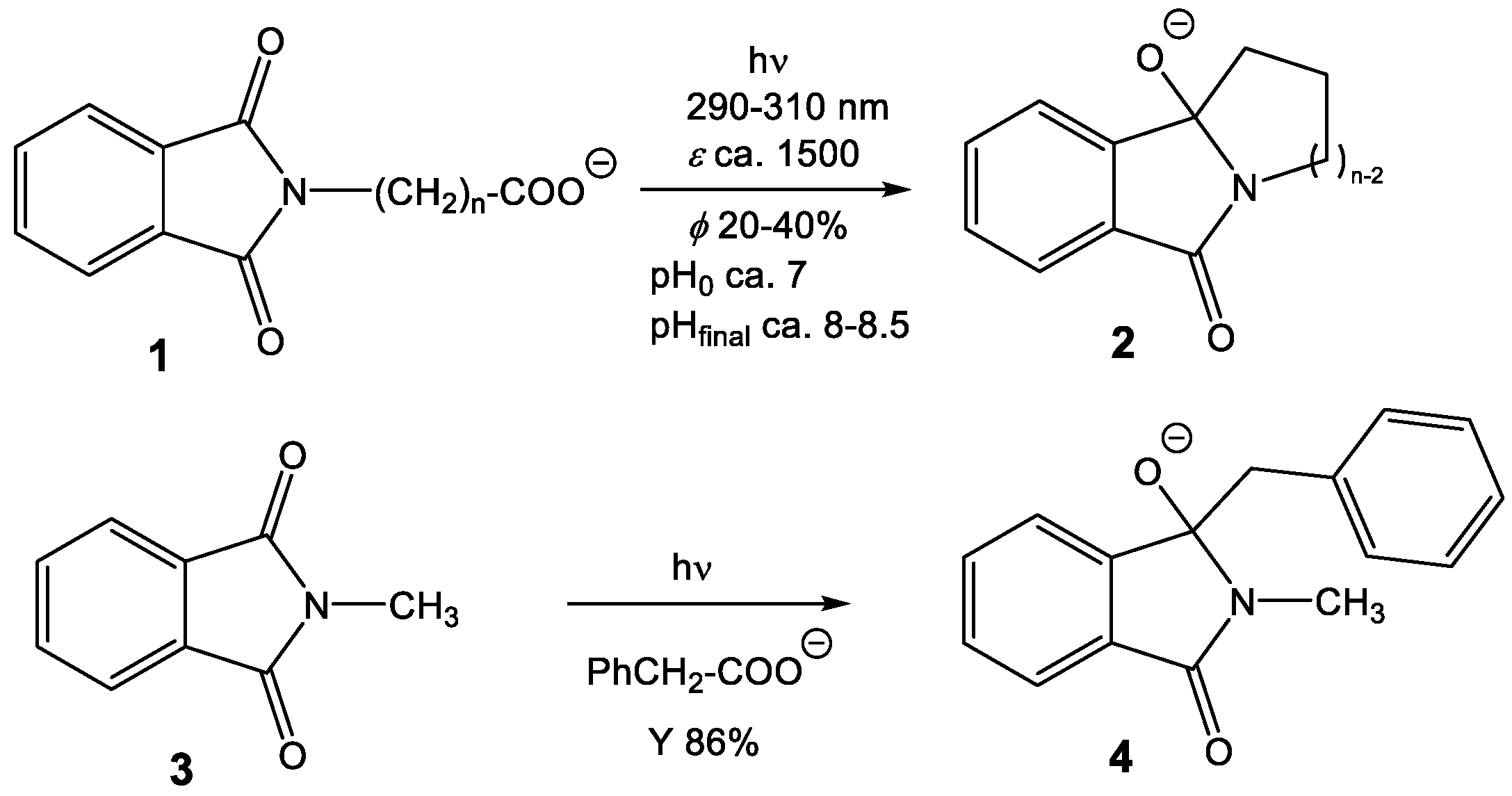
3.1. Intermolecular Reactivity
3.2. Intramolecular Reactivity
4. Discussion
4.1. Intermolecular Reactions
4.2. Intramolecular Reactions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Imahori, H. Porphyrin-fullerene linked systems as artificial photosynthetic mimics. Org. Biomol. Chem. 2004, 2, 1425–1433. [Google Scholar] [CrossRef] [PubMed]
- Langford, S.J.; Latter, M.J.; Woodward, C.P. Progress in charge transfer systems utilizing porphyrin donors and simple aromatic diimide acceptor units. Photochem. Photobiol. 2006, 82, 1530–1540. [Google Scholar] [CrossRef] [PubMed]
- Imahori, H.; Tamaki, K.; Araki, Y.; Hasobe, T.; Ito, O.; Shimomura, A.; Kundu, S.; Okada, T.; Sakata, Y.; Fukuzumi, S. Linkage dependent charge separation and charge recombination in porphyrin-pyromellitimide-fullerene triads. J. Phys. Chem. A 2022, 106, 2803–2814. [Google Scholar] [CrossRef]
- Okamoto, K.; Mori, Y.; Yamada, H.; Imahori, H.; Fukuzumi, S. Effects of metal ions on photoinduced electron transfer in zinc porphyrin-naphthalenediimide linked systems. Chem. Eur. J. 2004, 10, 474–483. [Google Scholar] [CrossRef]
- Balan, B.; Gopidas, K.B. An anthracene-appended beta-cyclodextrin-based dyad: Study of self-assembly and photoinduced electron-transfer processes. Chem. Eur. J. 2007, 13, 5173–5185. [Google Scholar] [CrossRef] [PubMed]
- Spenst, P.; Würthner, F. Photo- and redoxfunctional cyclophanes, macrocycles, and catenanes based on aromatic bisimides. J. Photochem. Photobiol. Rev. 2017, 31, 114–138. [Google Scholar] [CrossRef]
- Gust, D.; Moore, T.A. Photosynthetic model systems. Top. Curr. Chem. 1991, 159, 103–152. [Google Scholar]
- Redmore, N.P.; Rubtsov, I.V.; Therien, M.J. Synthesis, excited-state dynamics, and reactivity of a directly-linked pyromellitimide-(porphinato)zinc(II) complex. Inorg. Chem. 2002, 41, 566–570. [Google Scholar] [CrossRef]
- Giribabu, L.; Kumar, A.A.; Neeraja, V.; Maiya, B.G. Orientation dependence of energy transfer in an anthracene-porphyrin donor-acceptor system. Angew. Chem. Int. Ed. 2001, 40, 3621–3624. [Google Scholar] [CrossRef]
- Kalisch, W.W.; Senge, M.O.; Ruhlandt-Senge, K. Synthesis and crystal structures of cofacial bisoctaethylchlorins as structural models for the special pair. Photochem. Photobiol. 1998, 67, 312–323. [Google Scholar] [CrossRef]
- Hilczer, M.; Tachiya, M. Competitive electron transfers in model triad systems. MD simulations. J. Phys. Chem. 1996, 100, 8815–8826. [Google Scholar] [CrossRef]
- Mataga, N.; Shioyama, H.; Kanda, Y. Dynamics of charge recombination processes in the singlet electron-transfer state of pyrene-pyromellitic dianhydride systems in various solvents. Picosecond laser photolysis studies. J. Phys. Chem. 1987, 91, 314–317. [Google Scholar] [CrossRef]
- Miyauchi, M.; Ishida, Y.; Ogasawara, T.; Yokota, R. Highly soluble phenylethynyl-terminated imide oligomers based on KAPTON-type backbone structures for carbon fiber-reinforced composites with high heat resistance. Polym. J. 2013, 45, 594–600. [Google Scholar] [CrossRef]
- Gouzman, I.; Grossman, E.; Verker, R.; Atar, N.; Bolker, A.; Eliaz, N. Advances in polyimide-based materials for space applications. Adv. Mater. 2019, 31, 1807738. [Google Scholar] [CrossRef] [PubMed]
- Esther, A.C.M.; Dey, A.; Sridhara, N.; Yougandar, B.; Bera, P.; Anandan, C.; Rangappa, D.; Sharma, A.K. A study on degradation of germanium coating on Kapton used for spacecraft sunshield application. Surface Interface Anal. 2015, 47, 1155–1160. [Google Scholar] [CrossRef]
- Griesbeck, A.G.; Hoffmann, N.; Warzecha, K.-D. Photoinduced electron transfer chemistry: From studies on PET processes to applications in natural product synthesis. Acc. Chem. Res. 2007, 40, 128–140. [Google Scholar] [CrossRef]
- Griesbeck, A.G.; Henz, A.; Peters, K.; Peters, E.-M.; von Schnering, H.G. Photoelectron transfer-induced macrocyclization of N-phthaloyl-aminocarboxylic acids. Angew. Chem. Int. Ed. 1995, 34, 474–476. [Google Scholar] [CrossRef]
- Griesbeck, A.G.; Kramer, W.; Lex, J. Chirality memory in triplet 1,7-biradicals: Diastereo- and enantioselective synthesis of pyrrolo[1,4]benzodiazepines by decarboxylative photocyclization of anthranilic acid building blocks. Angew. Chem. Int. Ed. 2001, 40, 577–579. [Google Scholar] [CrossRef]
- Oelgemöller, M.; Griesbeck, A.G. Photoinduced electron transfer chemistry of phthalimides: An efficient tool for C-C-bond formation. J. Photochem. Photobiol. C Rev. 2002, 3, 109–127. [Google Scholar] [CrossRef]
- Griesbeck, A.G.; Heinrich, T.; Oelgemöller, M.; Molis, A.; Lex, J. A photochemical route to cyclopeptides with a minimum of activation and protection chemistry. J. Am. Chem. Soc. 2002, 124, 10972–10973. [Google Scholar] [CrossRef]
- Griesbeck, A.G.; Henz, A.; Kramer, W.; Lex, J.; Nerowski, F.; Oelgemöller, M.; Peters, K.; Peters, E.-M. Synthesis of medium- and large-ring compounds initiated by photochemical decarboxylation of ω-phthalimido alkylcarboxylates. Helv. Chim. Acta 1997, 80, 912–933. [Google Scholar] [CrossRef]
- Griesbeck, A.G.; Kramer, W.; Oelgemöller, M. Radical chemistry in water: Inter- and intramolecular photoinduced decarboxylations. Green Chem. 1999, 1, 205–207. [Google Scholar] [CrossRef]
- Horvat, M.; Mlinaric-Majerski, K.; Griesbeck, A.G.; Basaric, N. Photoinduced decarboxylation of 3-(N-phthalimido)adamantane-1-carboxylic acid and radical addition to electron deficient alkenes. Photochem. Photobiol. Sci. 2011, 10, 610–617. [Google Scholar] [CrossRef] [PubMed]
- Mandic, L.; Sohora, M.; Mihaljevic, B.; Biczok, L.; Basaric, N. The effect of the rate of photoinduced electron transfer on the photodecarboxylation efficiency in phthalimide photochemistry. J. Photochem. Photobiol. A Chem. 2021, 408, 113109. [Google Scholar] [CrossRef]
- Mandic, L.; Dzeba, I.; Jadresko, D.; Mihaljevic, B.; Biczok, L.; Basaric, N. Photophysical properties and electron transfer photochemical reactivity of substituted phthalimides. New J. Chem. 2020, 44, 17252–17266. [Google Scholar] [CrossRef]
- Griesbeck, A.G.; Nazarov, N.; Neudörfl, J.M.; Heffen, M. Intermolecular photodecarboxylation of electron-deficient substrates by phthalimides in water: Efficiency, selectivity and online monitoring. Green Chem. 2012, 14, 3004–3006. [Google Scholar] [CrossRef]
- Mumtaz, S.; Robertson, M.J.; Oelgemöller, M. Recent advances in photodecarboxylations involving phthalimides. Aust. J. Chem. 2018, 71, 634–648. [Google Scholar] [CrossRef]
- Griesbeck, A.G.; Neudörfl, J.; Goldfuss, B.; Molitor, S. Synthesis of 3-benzylated isoindolinones by photoredox decarboxylation of arylacetates in the presence of N-benzylphthalimide: Conductometry as a useful monitoring tool and the quest for the rate-determining step. ChemPhotoChem 2017, 1, 355–362. [Google Scholar] [CrossRef]
- Griesbeck, A.G.; Warzecha, K.-D.; Neudörfl, J.; Görner, H. PET-induced decarboxylative benzylation of N-alkylphthalimides: A concise route to the aristolactam skeleton. Synlett 2004, 2004, 2347–2350. [Google Scholar] [CrossRef]
- Hardcastle, I.R.; Ahmed, S.U.; Atkins, H.; Calvert, A.H.; Curtin, N.J.; Farnie, G.; Golding, B.T.; Griffin, R.J.; Guyenne, S.; Hutton, C.; et al. Isoindolinone-based inhibitors of the MDM2–p53 protein–protein interaction. Bioorg. Med. Chem. Lett. 2005, 15, 1515–1520. [Google Scholar] [CrossRef] [Green Version]
- Boulahjar, R.; Ouach, A.; Matteo, C.; Bourg, S.; Ravache, M.; le Guével, R.; Marionneau, S.; Oullier, T.; Lozach, O.; Meijer, L.; et al. Novel Tetrahydropyrido[1,2-a]isoindolone derivatives (valmerins): Potent cyclin-dependent kinase/glycogen synthase kinase 3 inhibitors with antiproliferative activities and antitumor effects in human tumor xenografts. J. Med. Chem. 2012, 55, 9589–9606. [Google Scholar] [CrossRef] [PubMed]
- Griesbeck, A.G.; Oelgemöller, M. Photodecarboxylative addition of carboxylates and α-keto carboxylates to phthalimides. Synlett 1999, 1999, 492–494. [Google Scholar] [CrossRef]
- Gosztola, D.D.; Niemzyk, M.P.; Svec, W.; Lucas, A.S.; Wasielewski, M.R. Excited doublet states of electrochemically generated aromatic imide and diimide radical anions. J. Phys Chem. A 2000, 104, 6545–6551. [Google Scholar] [CrossRef]
- Schindlbauer, H. Reaction of carboxylic anhydrides with formamide and N-methylformamide. Monatsh. Chem. 1973, 104, 848–853. [Google Scholar] [CrossRef]
- González, J.I.; Abajo, J.; Gonzáles-Babé, S.; Fontan, J. Polyanhydride-Imides. Angew. Makromol. Chem. 1976, 55, 85. [Google Scholar] [CrossRef]
- Hsiao, S.H.; Yang, C.P. Preparation of poly(amide-imide)s by direct polycondensation with triphenyl phosphite. IV. Aliphatic-aromatic poly(amide-imide)s based on N,N′-bis(o-carboxyalkyl)pyromellitimides. J. Polym. Sci. Polym. Chem. Ed. 1990, 28, 2169–2178. [Google Scholar] [CrossRef]
- Yang, C.-P.; Lin, J.-H.; Hsiao, S.-H. Synthesis and properties of aliphatic-aromatic poly(amide-imide)s from sulfonyldianilines and N,N′-bis(ω-carboxyalkyl)pyromellitimides. J. Polym. Sci. A Polym. Chem. 1991, 29, 1175–1182. [Google Scholar] [CrossRef]
- Penneau, J.-F.; Stallman, B.J.; Kasai, P.H.; Miller, L.L. An imide anion radical that dimerizes and assembles into π-stacks in solution. Chem. Mater. 1991, 3, 791–796. [Google Scholar] [CrossRef]
- Langhals, H.; Ritter-Faizade, U.; Stadler, P.; Havlicek, M.; Hofer, A.; Sariciftci, A.S. Persistent radical anions in the series of peri-arylenes: Broadband light absorption until far in the NIR and purely organic magnetism. Monatsh. Chem. Chem. Mon. 2019, 150, 885–900. [Google Scholar] [CrossRef] [Green Version]
- Griller, D.; Ingold, K.U. Persistent carbon-centered radicals. Acc. Chem. Res. 1976, 9, 13–19. [Google Scholar] [CrossRef]













Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kramer, W.H.; Razinoubakht, D.; Kaur, G.; Klein, A.; Garbe, S.; Neudörfl, J.; Molitor, S.; Zimmer, A.; Griesbeck, A.G. Awakening a Molecular Mummy: The Inter-and Intramolecular Photochemistry of Pyromellitic Diimides with Alkyl Carboxylates. Photochem 2022, 2, 717-732. https://doi.org/10.3390/photochem2030046
Kramer WH, Razinoubakht D, Kaur G, Klein A, Garbe S, Neudörfl J, Molitor S, Zimmer A, Griesbeck AG. Awakening a Molecular Mummy: The Inter-and Intramolecular Photochemistry of Pyromellitic Diimides with Alkyl Carboxylates. Photochem. 2022; 2(3):717-732. https://doi.org/10.3390/photochem2030046
Chicago/Turabian StyleKramer, Wolfgang H., Donya Razinoubakht, Gurjit Kaur, Axel Klein, Simon Garbe, Jörg Neudörfl, Sabrina Molitor, Anne Zimmer, and Axel G. Griesbeck. 2022. "Awakening a Molecular Mummy: The Inter-and Intramolecular Photochemistry of Pyromellitic Diimides with Alkyl Carboxylates" Photochem 2, no. 3: 717-732. https://doi.org/10.3390/photochem2030046
APA StyleKramer, W. H., Razinoubakht, D., Kaur, G., Klein, A., Garbe, S., Neudörfl, J., Molitor, S., Zimmer, A., & Griesbeck, A. G. (2022). Awakening a Molecular Mummy: The Inter-and Intramolecular Photochemistry of Pyromellitic Diimides with Alkyl Carboxylates. Photochem, 2(3), 717-732. https://doi.org/10.3390/photochem2030046