Protein Intrinsic Disorder: Role in Signaling, Regulation and Membrane-Less Organelle Formation
(Closed)
Share This Topical Collection
Editors
 Dr. Nathalie Sibille
Dr. Nathalie Sibille
Dr. Nathalie Sibille
E-Mail
Website
Collection Editor
Centre de Biochimie Structurale (CBS), CNRS, INSERM, University of Montpellier, Montpellier, France
Interests: intrinsically disordered proteins (IDPs); post-translational modifications (PTMs); signaling; nuclear magnetic resonance (NMR); small-angle X-ray scattering (SAXS)
Special Issues, Collections and Topics in MDPI journals
 Dr. Sonia Longhi
Dr. Sonia Longhi
Dr. Sonia Longhi
E-Mail
Website
Collection Editor
Architecture and Function of Biological Macromolecules (AFMB), UMR 7257 CNRS & Aix-Marseille University, 13288 Marseille, France
Interests: intrinsically disordered proteins; folding copuled to binding; protein-protein interactions; structural transitions; paramyxoviruses
Special Issues, Collections and Topics in MDPI journals
 Dr. Carine Van Heijenoort
Dr. Carine Van Heijenoort
Dr. Carine Van Heijenoort
E-Mail
Website
Collection Editor
ICSN-CNRS, Université Paris-Saclay, Gif-sur-Yvette, France
Interests: structural biology; NMR; intrinsically disordered proteins; relation structure; dynamics and function of proteins; protein-protein interactions; protein-ligand interaction
Topical Collection Information
Dear Colleagues,
Intrinsically disordered proteins (IDPs) are fascinating multifaceted proteins. Their discovery dates back to the mid-1990s, and it is now well established that IDPs are very common in the protein realm. They are particularly abundant in the proteome of eukaryotes, with this prevalence being tied to the higher complexity of the latter compared to prokaryotes. The lack of stable secondary and tertiary structure that typifies these proteins and that is encoded by their amino acid sequence makes these proteins very elusive; thus, IDPs and their complexes are very difficult to characterize at the structural level due to their inherent flexibility. Although IDPs lack a fixed 3D structure, they can feature transiently populated secondary structure elements, which are often conserved and linked to function. In particular, these elements often serve as binding sites for partners and attenuate the entropic cost of the disorder-to-order transition triggered by binding to the partner. Many IDPs conserve a considerable extent of residual disorder upon binding to a partner, leading to so-called “fuzzy complexes”. By tuning the extent of preconfiguration and/or of fuzziness, IDPs can finely tune their affinity towards multiple partners. They can establish interactions ranging from low to high affinity, while conserving specificity in all cases. All these features are also modified by frequent post-translational modifications due to their high flexibility and accessibility. As such, they are the target of more than 300 PTMs that ultimately modulate their structural features and their ability to interact with partners. Thanks to these peculiar biophysical and biochemical features, IDPs are widely involved in signaling and regulation. Their action is also highly dependent on their expression level and local concentration in the cell. In the last five years, a growing number of studies has thus addressed the ability of IDPs to undergo liquid–liquid phase separation (LLPS), a phenomenon that underlies the formation of membrane-less organelles (MLOs). As a consequence, the deregulation of processes implying IDPs is at the origin of a multitude of diseases such as cancer, neurodegeneration, cardiovascular diseases, and diabetes. In order to better understand the mechanisms behind these diseases, it is necessary to decipher their structural bases. This Topical Collection on “Protein Intrinsic Disorder: Role in Signaling, Regulation and Membrane-Less Organelle Formation” aims at further deepening our understanding of how the physicochemical properties of IDPs enable them to play a crucial role in the regulation of many critical biological processes.
Dr. Nathalie Sibille
Dr. Sonia Longhi
Dr. Carine Van Heijenoort
Collection Editors
Manuscript Submission Information
Manuscripts should be submitted online at www.mdpi.com by registering and logging in to this website. Once you are registered, click here to go to the submission form. Manuscripts can be submitted until the deadline. All submissions that pass pre-check are peer-reviewed. Accepted papers will be published continuously in the journal (as soon as accepted) and will be listed together on the collection website. Research articles, review articles as well as short communications are invited. For planned papers, a title and short abstract (about 100 words) can be sent to the Editorial Office for announcement on this website.
Submitted manuscripts should not have been published previously, nor be under consideration for publication elsewhere (except conference proceedings papers). All manuscripts are thoroughly refereed through a single-blind peer-review process. A guide for authors and other relevant information for submission of manuscripts is available on the Instructions for Authors page. Biomolecules is an international peer-reviewed open access monthly journal published by MDPI.
Please visit the Instructions for Authors page before submitting a manuscript.
The Article Processing Charge (APC) for publication in this open access journal is 2700 CHF (Swiss Francs).
Submitted papers should be well formatted and use good English. Authors may use MDPI's
English editing service prior to publication or during author revisions.
Keywords
- intrinsically disordered proteins (IDPs)
- signaling
- regulation
- post-translational modification (PTM)
- membrane-less organelle formation (MLOs)
- liquid–liquid phase separation (LLPS)
- nuclear magnetic resonance (NMR)
- integrative structural biology
Published Papers (12 papers)
Open AccessEditor’s ChoiceArticle
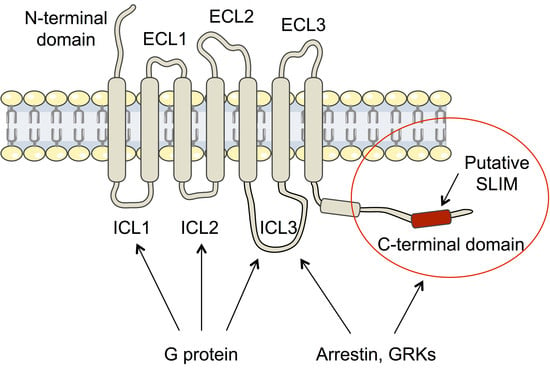
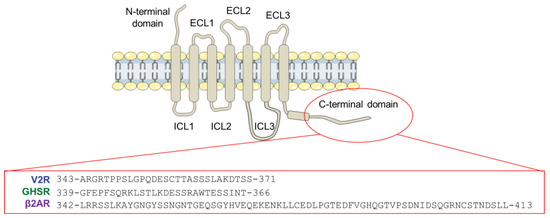
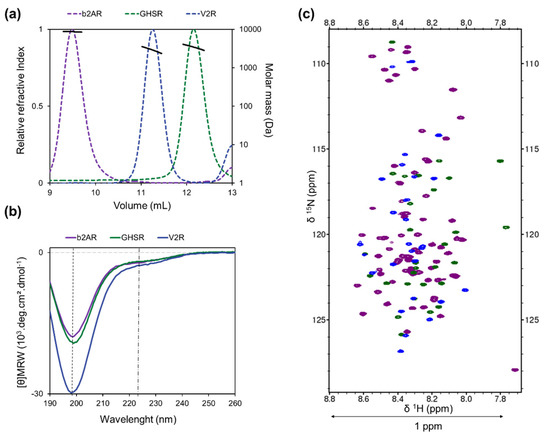
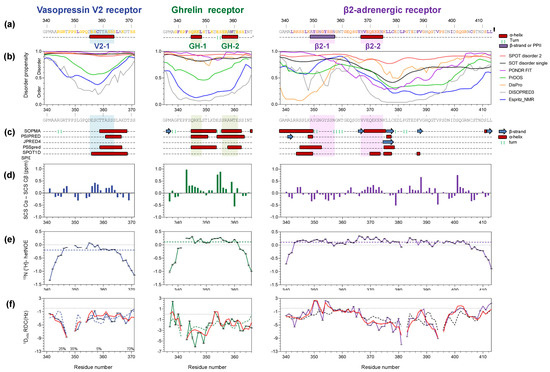
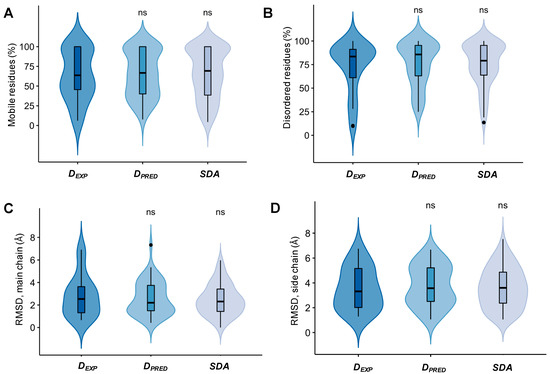
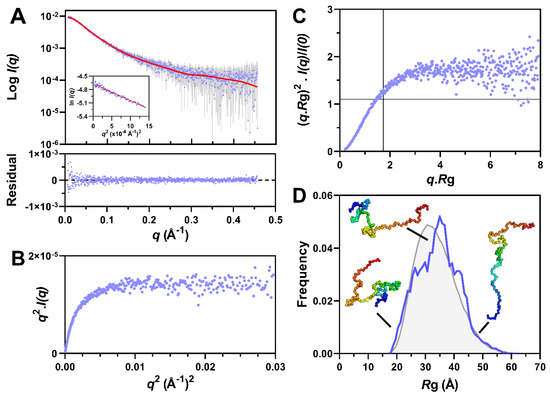
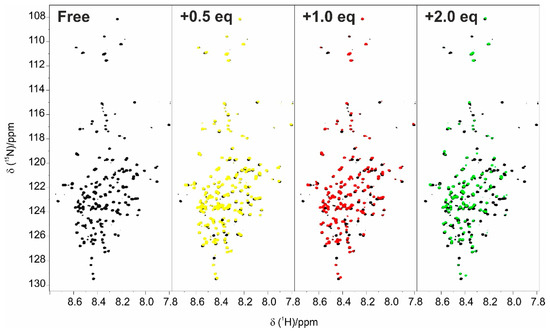
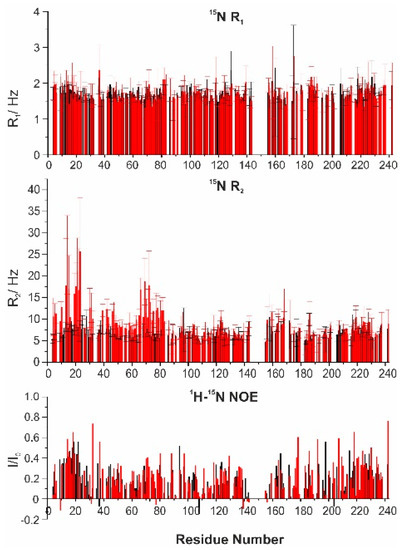
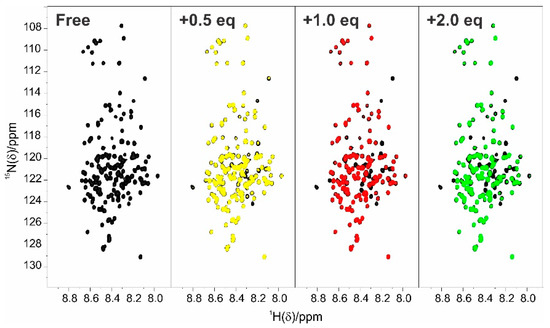
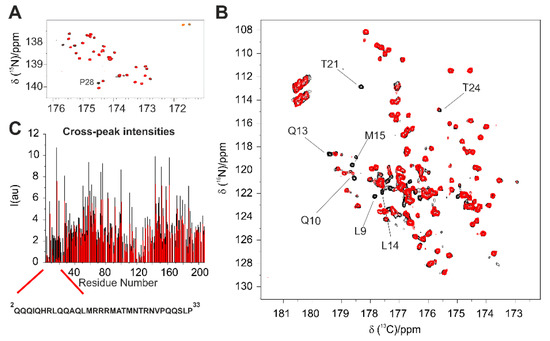
Structural Insights into the Intrinsically Disordered GPCR C-Terminal Region, Major Actor in Arrestin-GPCR Interaction
by
Myriam Guillien, Assia Mouhand, Aurélie Fournet, Amandine Gontier, Aleix Martí Navia, Tiago N. Cordeiro, Frédéric Allemand, Aurélien Thureau, Jean-Louis Banères, Pau Bernadó and Nathalie Sibille
Cited by 6 | Viewed by 4747
Abstract
Arrestin-dependent pathways are a central component of G protein-coupled receptor (GPCRs) signaling. However, the molecular processes regulating arrestin binding are to be further illuminated, in particular with regard to the structural impact of GPCR C-terminal disordered regions. Here, we used an integrated biophysical
[...] Read more.
Arrestin-dependent pathways are a central component of G protein-coupled receptor (GPCRs) signaling. However, the molecular processes regulating arrestin binding are to be further illuminated, in particular with regard to the structural impact of GPCR C-terminal disordered regions. Here, we used an integrated biophysical strategy to describe the basal conformations of the C-terminal domains of three class A GPCRs, the vasopressin V2 receptor (V2R), the growth hormone secretagogue or ghrelin receptor type 1a (GHSR) and the β2-adernergic receptor (β2AR). By doing so, we revealed the presence of transient secondary structures in these regions that are potentially involved in the interaction with arrestin. These secondary structure elements differ from those described in the literature in interaction with arrestin. This suggests a mechanism where the secondary structure conformational preferences in the C-terminal regions of GPCRs could be a central feature for optimizing arrestins recognition.
Full article
►▼
Show Figures
Open AccessFeature PaperArticle
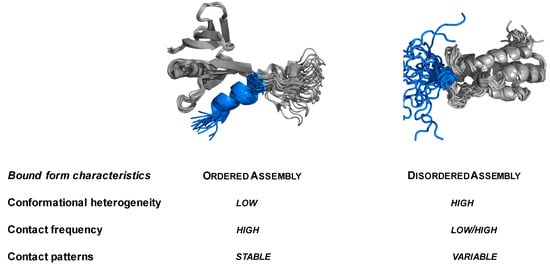
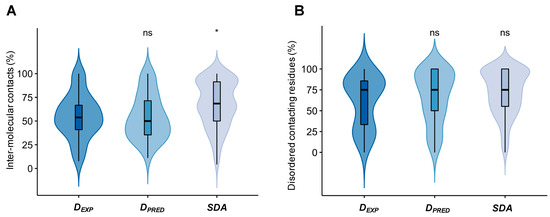
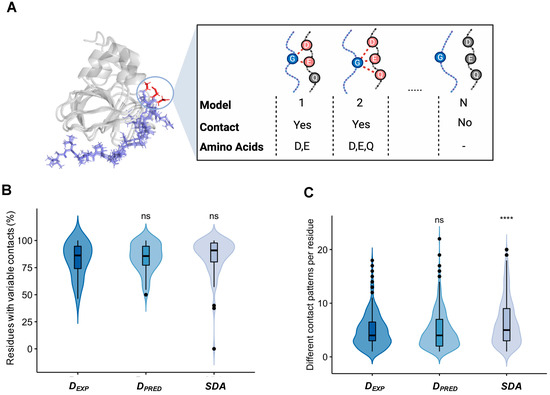
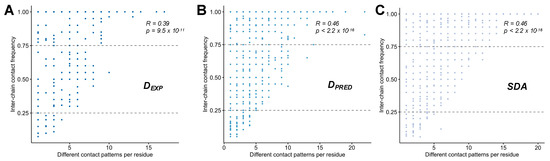
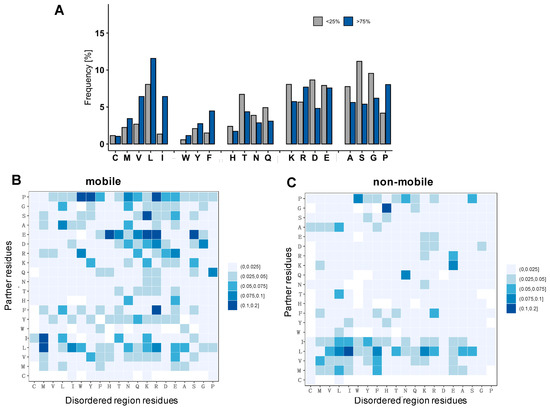
Molecular Determinants of Selectivity in Disordered Complexes May Shed Light on Specificity in Protein Condensates
by
Alexander Miguel Monzon, Damiano Piovesan and Monika Fuxreiter
Cited by 4 | Viewed by 2032
Abstract
Biomolecular condensates challenge the classical concepts of molecular recognition. The variable composition and heterogeneous conformations of liquid-like protein droplets are bottlenecks for high-resolution structural studies. To obtain atomistic insights into the organization of these assemblies, here we have characterized the conformational ensembles of
[...] Read more.
Biomolecular condensates challenge the classical concepts of molecular recognition. The variable composition and heterogeneous conformations of liquid-like protein droplets are bottlenecks for high-resolution structural studies. To obtain atomistic insights into the organization of these assemblies, here we have characterized the conformational ensembles of specific disordered complexes, including those of droplet-driving proteins. First, we found that these specific complexes exhibit a high degree of conformational heterogeneity. Second, we found that residues forming contacts at the interface also sample many conformations. Third, we found that different patterns of contacting residues form the specific interface. In addition, we observed a wide range of sequence motifs mediating disordered interactions, including charged, hydrophobic and polar contacts. These results demonstrate that selective recognition can be realized by variable patterns of weakly defined interaction motifs in many different binding configurations. We propose that these principles also play roles in determining the selectivity of biomolecular condensates.
Full article
►▼
Show Figures
Open AccessReview
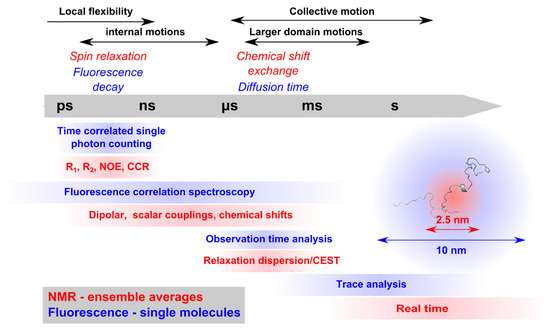
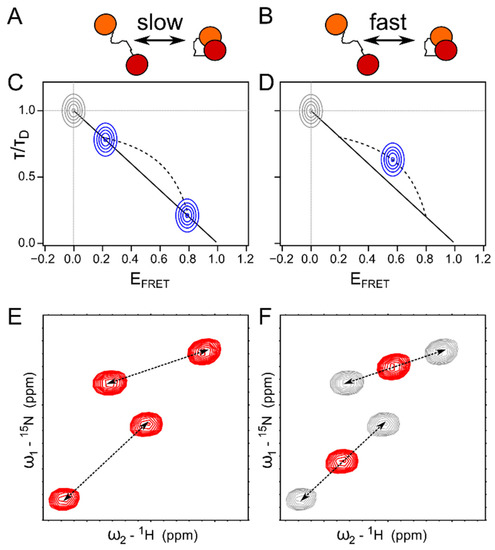
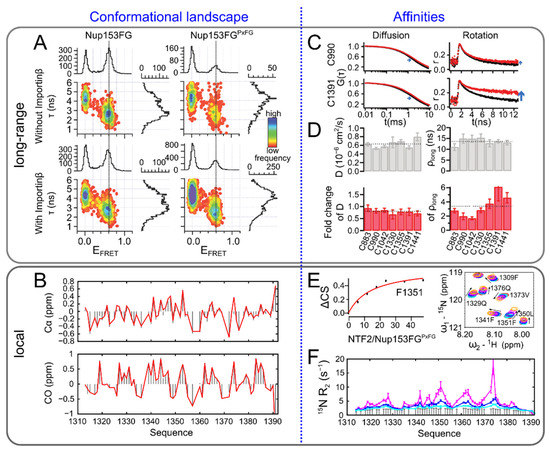
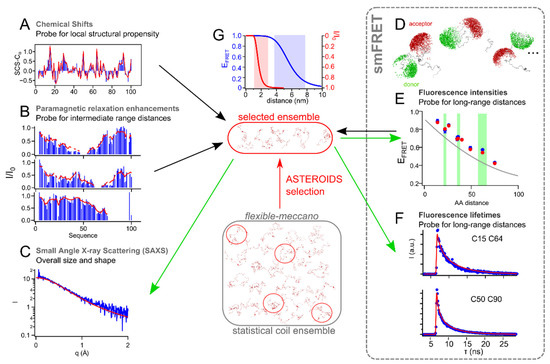
Synergies of Single Molecule Fluorescence and NMR for the Study of Intrinsically Disordered Proteins
by
Samuel Naudi-Fabra, Martin Blackledge and Sigrid Milles
Cited by 10 | Viewed by 4352
Abstract
Single molecule fluorescence and nuclear magnetic resonance spectroscopy (NMR) are two very powerful techniques for the analysis of intrinsically disordered proteins (IDPs). Both techniques have individually made major contributions to deciphering the complex properties of IDPs and their interactions, and it has become
[...] Read more.
Single molecule fluorescence and nuclear magnetic resonance spectroscopy (NMR) are two very powerful techniques for the analysis of intrinsically disordered proteins (IDPs). Both techniques have individually made major contributions to deciphering the complex properties of IDPs and their interactions, and it has become evident that they can provide very complementary views on the distance-dynamics relationships of IDP systems. We now review the first approaches using both NMR and single molecule fluorescence to decipher the molecular properties of IDPs and their interactions. We shed light on how these two techniques were employed synergistically for multidomain proteins harboring intrinsically disordered linkers, for veritable IDPs, but also for liquid–liquid phase separated systems. Additionally, we provide insights into the first approaches to use single molecule Förster resonance energy transfer (FRET) and NMR for the description of multiconformational models of IDPs.
Full article
►▼
Show Figures
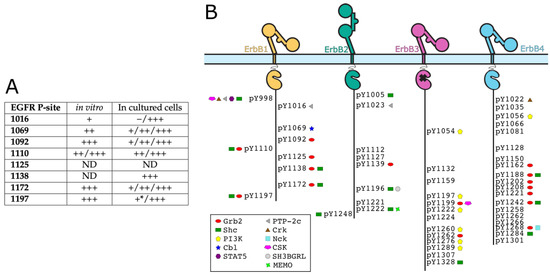
Open AccessEditor’s ChoiceReview
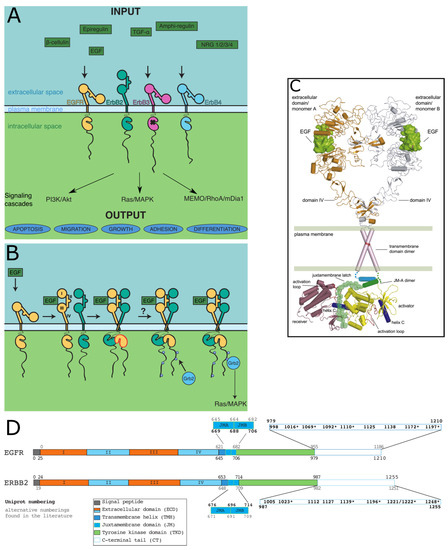
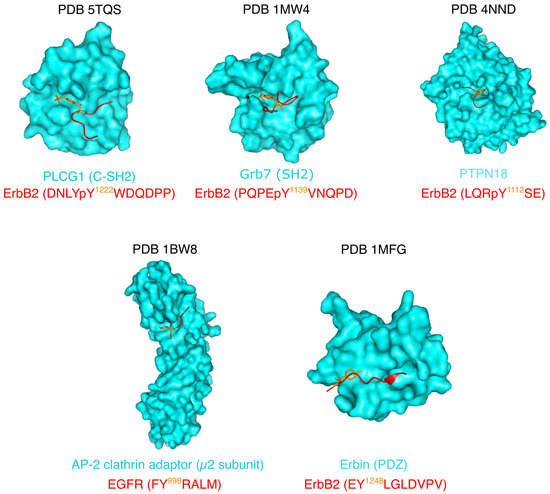
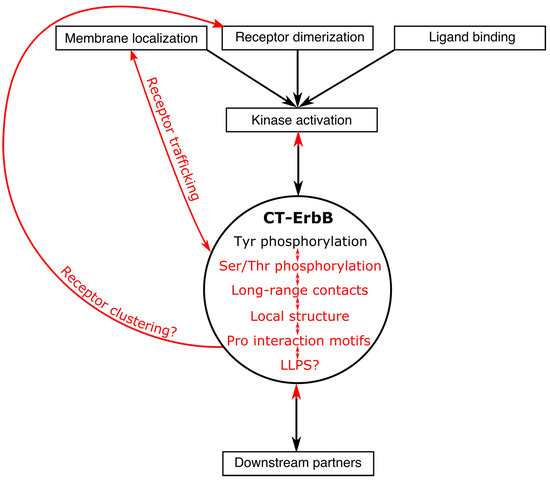
Expanding the Disorder-Function Paradigm in the C-Terminal Tails of Erbbs
by
Louise Pinet, Nadine Assrir and Carine van Heijenoort
Cited by 3 | Viewed by 2832
Abstract
ErbBs are receptor tyrosine kinases involved not only in development, but also in a wide variety of diseases, particularly cancer. Their extracellular, transmembrane, juxtamembrane, and kinase folded domains were described extensively over the past 20 years, structurally and functionally. However, their whole C-terminal
[...] Read more.
ErbBs are receptor tyrosine kinases involved not only in development, but also in a wide variety of diseases, particularly cancer. Their extracellular, transmembrane, juxtamembrane, and kinase folded domains were described extensively over the past 20 years, structurally and functionally. However, their whole C-terminal tails (CTs) following the kinase domain were only described at atomic resolution in the last 4 years. They were shown to be intrinsically disordered. The CTs are known to be tyrosine-phosphorylated when the activated homo- or hetero-dimers of ErbBs are formed. Their phosphorylation triggers interaction with phosphotyrosine binding (PTB) or Src Homology 2 (SH2) domains and activates several signaling pathways controling cellular motility, proliferation, adhesion, and apoptosis. Beyond this passive role of phosphorylated domain and site display for partners, recent structural and function studies unveiled active roles in regulation of phosphorylation and interaction: the CT regulates activity of the kinase domain; different phosphorylation states have different compaction levels, potentially modulating the succession of phosphorylation events; and prolines have an important role in structure, dynamics, and possibly regulatory interactions. Here, we review both the canonical role of the disordered CT domains of ErbBs as phosphotyrosine display domains and the recent findings that expand the known range of their regulation functions linked to specific structural and dynamic features.
Full article
►▼
Show Figures
Open AccessArticle
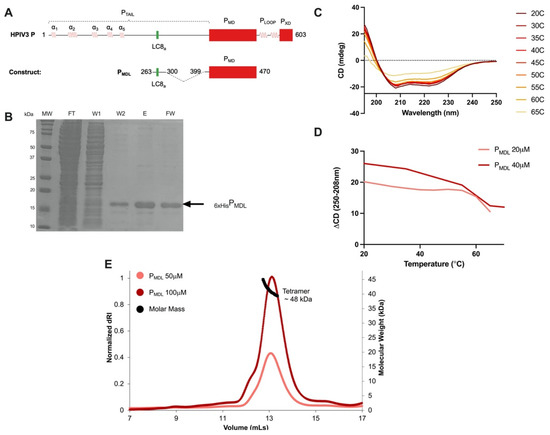
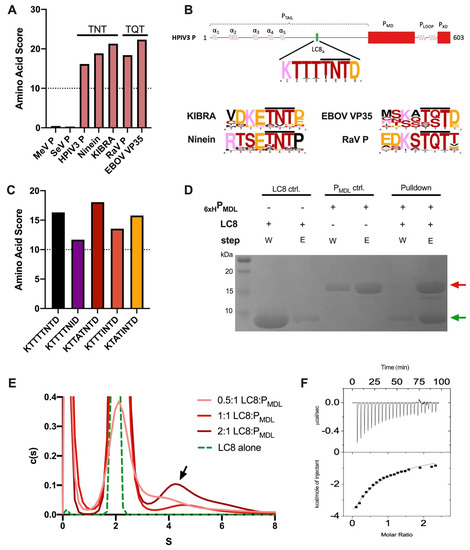
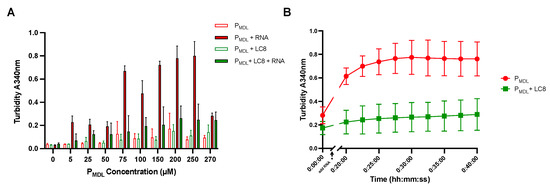
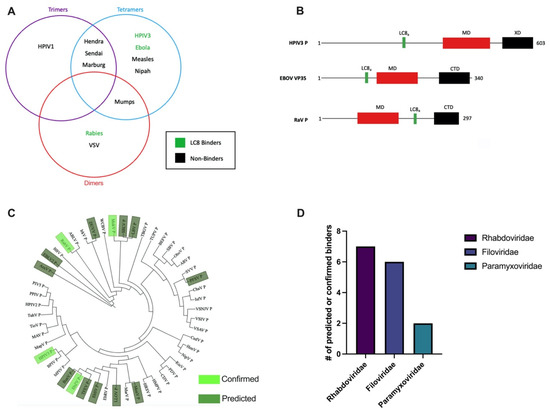
Human Parainfluenza Virus 3 Phosphoprotein Is a Tetramer and Shares Structural and Interaction Features with Ebola Phosphoprotein VP35
by
Joaquin Rodriguez Galvan, Brianna Donner, Cat Hoang Veseley, Patrick Reardon, Heather M. Forsythe, Jesse Howe, Gretchen Fujimura and Elisar Barbar
Cited by 6 | Viewed by 2781
Abstract
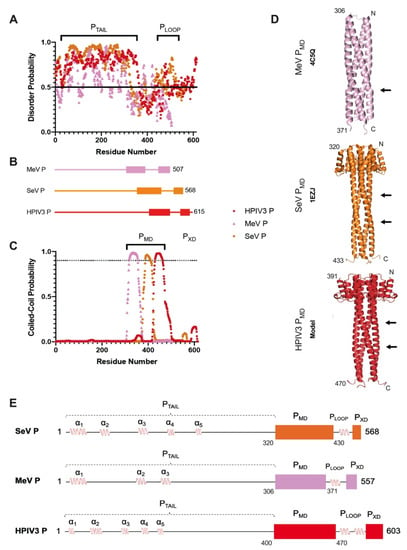
The human parainfluenza virus 3 (HPIV3) poses a risk for pneumonia development in young children and immunocompromised patients. To investigate mechanisms of HPIV3 pathogenesis, we characterized the association state and host protein interactions of HPIV3 phosphoprotein (HPIV3 P), an indispensable viral polymerase cofactor.
[...] Read more.
The human parainfluenza virus 3 (HPIV3) poses a risk for pneumonia development in young children and immunocompromised patients. To investigate mechanisms of HPIV3 pathogenesis, we characterized the association state and host protein interactions of HPIV3 phosphoprotein (HPIV3 P), an indispensable viral polymerase cofactor. Sequence analysis and homology modeling predict that HPIV3 P possesses a long, disordered N-terminal tail (P
TAIL) a coiled-coil multimerization domain (P
MD), similar to the well-characterized paramyxovirus phosphoproteins from measles and Sendai viruses. Using a recombinantly expressed and purified construct of P
MD and P
TAIL, we show that HPIV3 P in solution is primarily an alpha-helical tetramer that is stable up to 60 °C. Pulldown and isothermal titration calorimetry experiments revealed that HPIV3 P binds the host hub protein LC8, and turbidity experiments demonstrated a new role for LC8 in increasing the solubility of HPIV3 P in the presence of crowding agents such as RNA. For comparison, we show that the multimerization domain of the Zaire Ebola virus phosphoprotein VP35 is also a tetramer and binds LC8 but with significantly higher affinity. Comparative analysis of the domain architecture of various virus phosphoproteins in the order Mononegavirales show multiple predicted and verified LC8 binding motifs, suggesting its prevalence and importance in regulating viral phosphoprotein structures. Our work provides evidence for LC8 binding to phosphoproteins with multiple association states, either tetrameric, as in the HPIV3 and Ebola phosphoproteins shown here, or dimeric as in rabies virus phosphoprotein. Taken together the data suggest that the association states of a virus-specific phosphoprotein and the complex formed by binding of the phosphoprotein to host LC8 are important regulators of viral function.
Full article
►▼
Show Figures
Open AccessReview
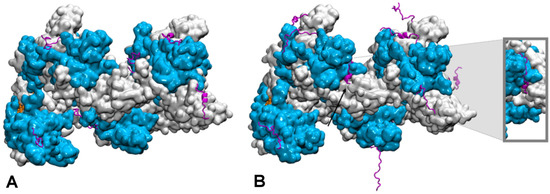
When Order Meets Disorder: Modeling and Function of the Protein Interface in Fuzzy Complexes
by
Sophie Sacquin-Mora and Chantal Prévost
Cited by 5 | Viewed by 3403
Abstract
The degree of proteins structural organization ranges from highly structured, compact folding to intrinsic disorder, where each degree of self-organization corresponds to specific functions: well-organized structural motifs in enzymes offer a proper environment for precisely positioned functional groups to participate in catalytic reactions;
[...] Read more.
The degree of proteins structural organization ranges from highly structured, compact folding to intrinsic disorder, where each degree of self-organization corresponds to specific functions: well-organized structural motifs in enzymes offer a proper environment for precisely positioned functional groups to participate in catalytic reactions; at the other end of the self-organization spectrum, intrinsically disordered proteins act as binding hubs via the formation of multiple, transient and often non-specific interactions. This review focusses on cases where structurally organized proteins or domains associate with highly disordered protein chains, leading to the formation of interfaces with varying degrees of fuzziness. We present a review of the computational methods developed to provide us with information on such fuzzy interfaces, and how they integrate experimental information. The discussion focusses on two specific cases, microtubules and homologous recombination nucleoprotein filaments, where a network of intrinsically disordered tails exerts regulatory function in recruiting partner macromolecules, proteins or DNA and tuning the atomic level association. Notably, we show how computational approaches such as molecular dynamics simulations can bring new knowledge to help bridging the gap between experimental analysis, that mostly concerns ensemble properties, and the behavior of individual disordered protein chains that contribute to regulation functions.
Full article
►▼
Show Figures
Open AccessEditor’s ChoiceArticle
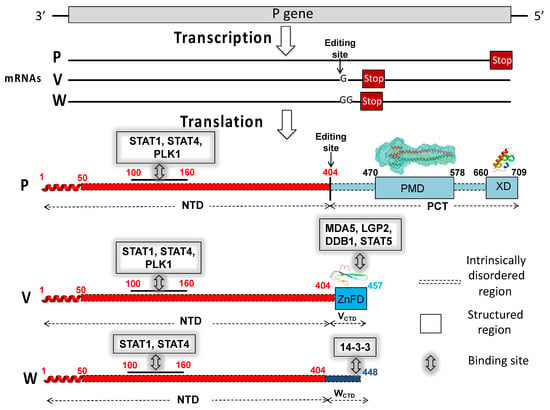
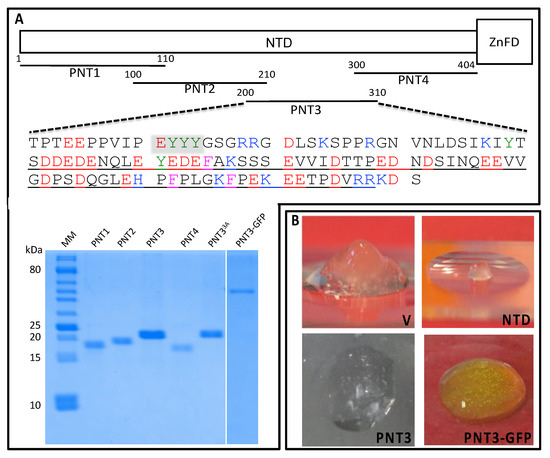
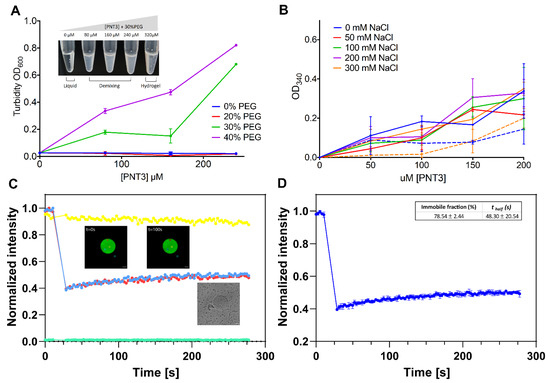
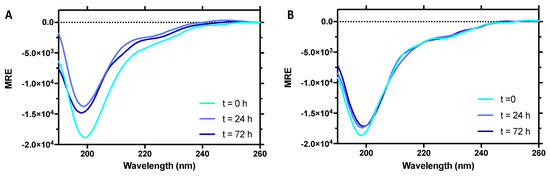
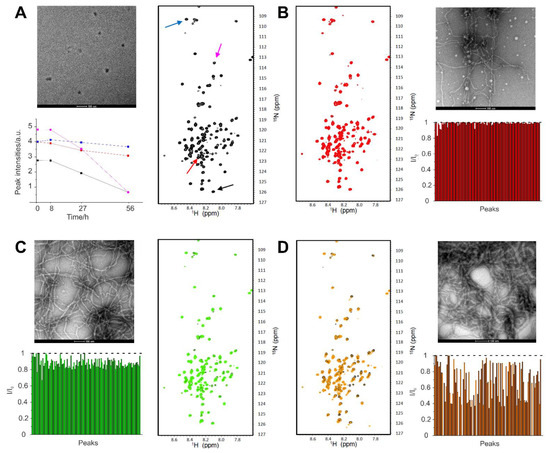
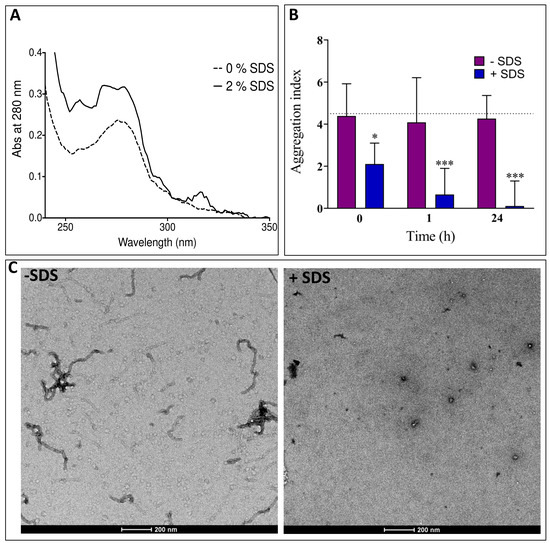
Identification of a Region in the Common Amino-terminal Domain of Hendra Virus P, V, and W Proteins Responsible for Phase Transition and Amyloid Formation
by
Edoardo Salladini, Frank Gondelaud, Juliet F. Nilsson, Giulia Pesce, Christophe Bignon, Maria Grazia Murrali, Roxane Fabre, Roberta Pierattelli, Andrey V. Kajava, Branka Horvat, Denis Gerlier, Cyrille Mathieu and Sonia Longhi
Cited by 13 | Viewed by 4342
Abstract
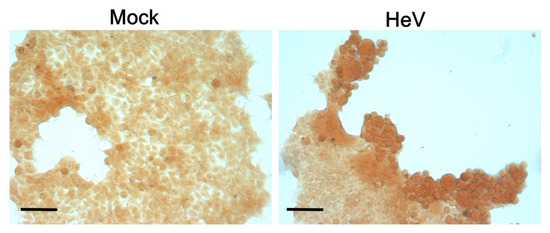
Henipaviruses are BSL-4 zoonotic pathogens responsible in humans for severe encephalitis. Their V protein is a key player in the evasion of the host innate immune response. We previously showed that the
Henipavirus V proteins consist of a long intrinsically disordered N-terminal domain
[...] Read more.
Henipaviruses are BSL-4 zoonotic pathogens responsible in humans for severe encephalitis. Their V protein is a key player in the evasion of the host innate immune response. We previously showed that the
Henipavirus V proteins consist of a long intrinsically disordered N-terminal domain (NTD) and a β-enriched C-terminal domain (CTD). The CTD is critical for V binding to DDB1, which is a cellular protein that is a component of the ubiquitin ligase E3 complex, as well as binding to MDA5 and LGP2, which are two host sensors of viral RNA. Here, we serendipitously discovered that the Hendra virus V protein undergoes a liquid-to-hydrogel phase transition and identified the V region responsible for this phenomenon. This region, referred to as PNT3 and encompassing residues 200–310, was further investigated using a combination of biophysical and structural approaches. Congo red binding assays, together with negative-staining transmisison electron microscopy (TEM) studies, show that PNT3 forms amyloid-like fibrils. Fibrillation abilities are dramatically reduced in a rationally designed PNT3 variant in which a stretch of three contiguous tyrosines, falling within an amyloidogenic motif, were replaced by three alanines. Worthy to note, Congo red staining experiments provided hints that these amyloid-like fibrils form not only in vitro but also in cellula after transfection or infection. The present results set the stage for further investigations aimed at assessing the functional role of phase separation and fibrillation by the
Henipavirus V proteins.
Full article
►▼
Show Figures
Open AccessFeature PaperArticle
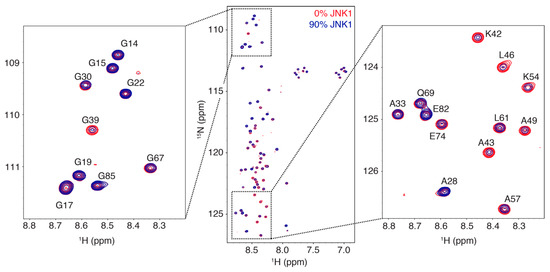
Enthalpy–Entropy Compensation in the Promiscuous Interaction of an Intrinsically Disordered Protein with Homologous Protein Partners
by
Jaka Kragelj, Thibault Orand, Elise Delaforge, Laura Tengo, Martin Blackledge, Andrés Palencia and Malene Ringkjøbing Jensen
Cited by 12 | Viewed by 3812
Abstract
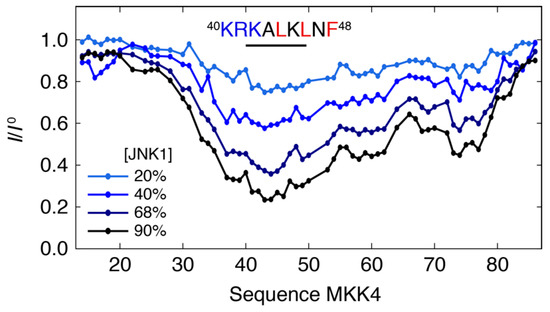
Intrinsically disordered proteins (IDPs) can engage in promiscuous interactions with their protein targets; however, it is not clear how this feature is encoded in the primary sequence of the IDPs and to what extent the surface properties and the shape of the binding
[...] Read more.
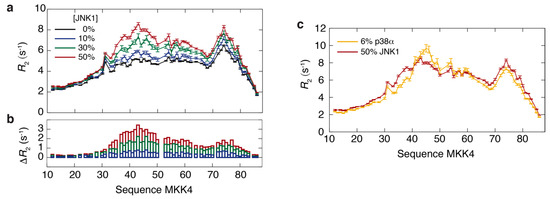
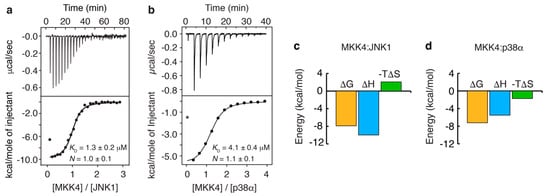
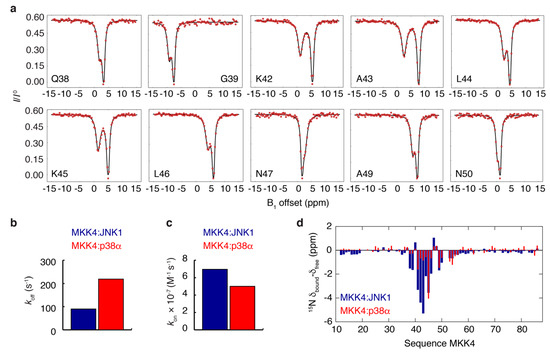
Intrinsically disordered proteins (IDPs) can engage in promiscuous interactions with their protein targets; however, it is not clear how this feature is encoded in the primary sequence of the IDPs and to what extent the surface properties and the shape of the binding cavity dictate the binding mode and the final bound conformation. Here we show, using a combination of nuclear magnetic resonance (NMR) spectroscopy and isothermal titration calorimetry (ITC), that the promiscuous interaction of the intrinsically disordered regulatory domain of the mitogen-activated protein kinase kinase MKK4 with p38α and JNK1 is facilitated by folding-upon-binding into two different conformations, despite the high sequence conservation and structural homology between p38α and JNK1. Our results support a model whereby the specific surface properties of JNK1 and p38α dictate the bound conformation of MKK4 and that enthalpy–entropy compensation plays a major role in maintaining comparable binding affinities for MKK4 towards the two kinases.
Full article
►▼
Show Figures
Open AccessEditor’s ChoiceArticle
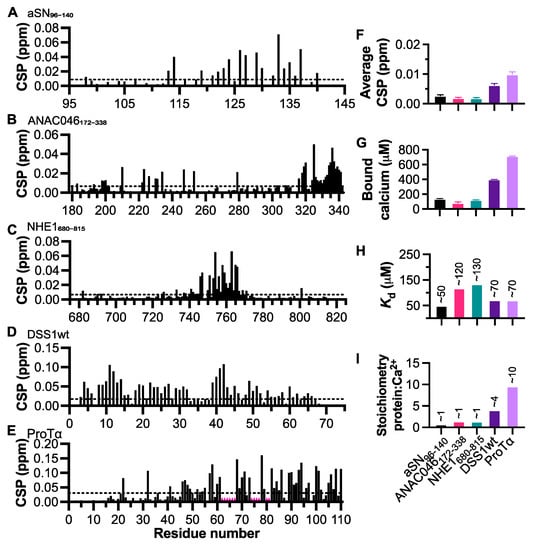
Insight into Calcium-Binding Motifs of Intrinsically Disordered Proteins
by
Estella A. Newcombe, Catarina B. Fernandes, Jeppe E. Lundsgaard, Inna Brakti, Kresten Lindorff-Larsen, Annette E. Langkilde, Karen Skriver and Birthe B. Kragelund
Cited by 13 | Viewed by 4886
Abstract
Motifs within proteins help us categorize their functions. Intrinsically disordered proteins (IDPs) are rich in short linear motifs, conferring them many different roles. IDPs are also frequently highly charged and, therefore, likely to interact with ions. Canonical calcium-binding motifs, such as the EF-hand,
[...] Read more.
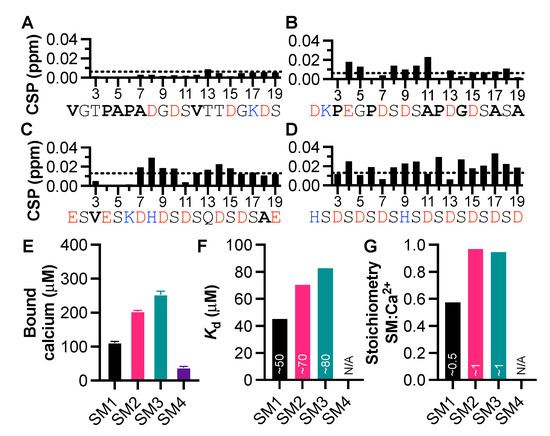
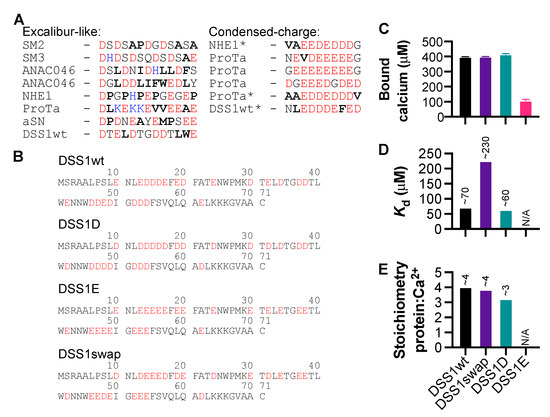
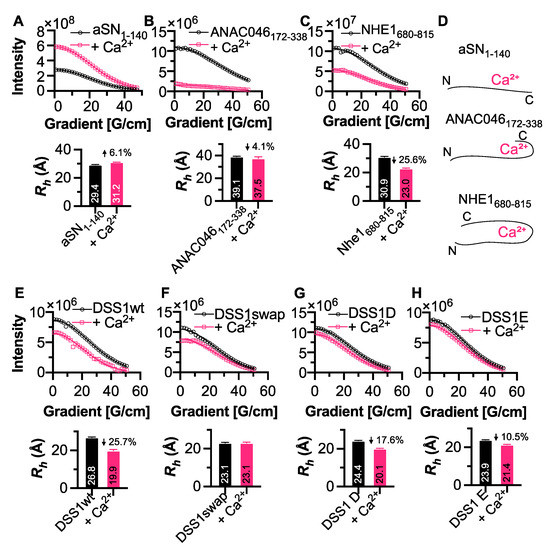
Motifs within proteins help us categorize their functions. Intrinsically disordered proteins (IDPs) are rich in short linear motifs, conferring them many different roles. IDPs are also frequently highly charged and, therefore, likely to interact with ions. Canonical calcium-binding motifs, such as the EF-hand, often rely on the formation of stabilizing flanking helices, which are a key characteristic of folded proteins, but are absent in IDPs. In this study, we probe the existence of a calcium-binding motif relevant to IDPs. Upon screening several carefully selected IDPs using NMR spectroscopy supplemented with affinity quantification by colorimetric assays, we found calcium-binding motifs in IDPs which could be categorized into at least two groups—an Excalibur-like motif, sequentially similar to the EF-hand loop, and a condensed-charge motif carrying repetitive negative charges. The motifs show an affinity for calcium typically in the ~100 μM range relevant to regulatory functions and, while calcium binding to the condensed-charge motif had little effect on the overall compaction of the IDP chain, calcium binding to Excalibur-like motifs resulted in changes in compaction. Thus, calcium binding to IDPs may serve various structural and functional roles that have previously been underreported.
Full article
►▼
Show Figures
Open AccessFeature PaperReview
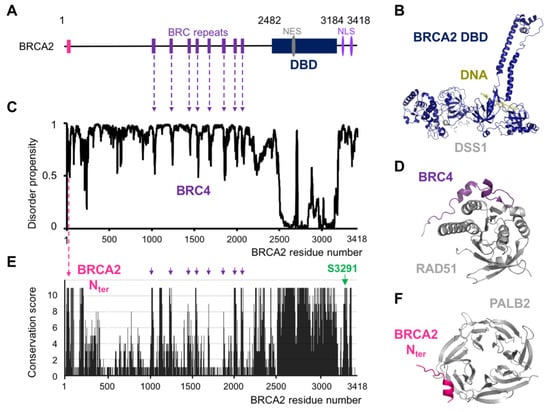
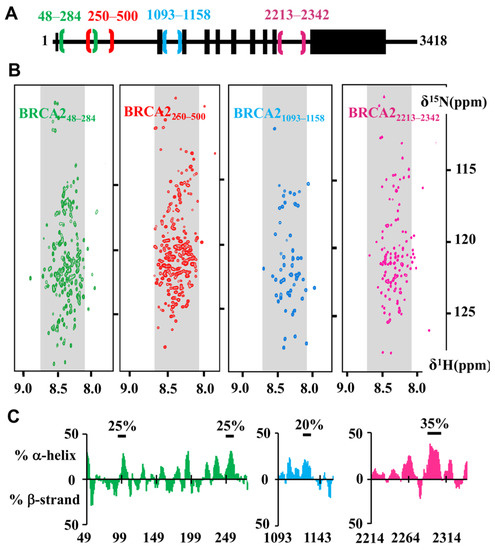
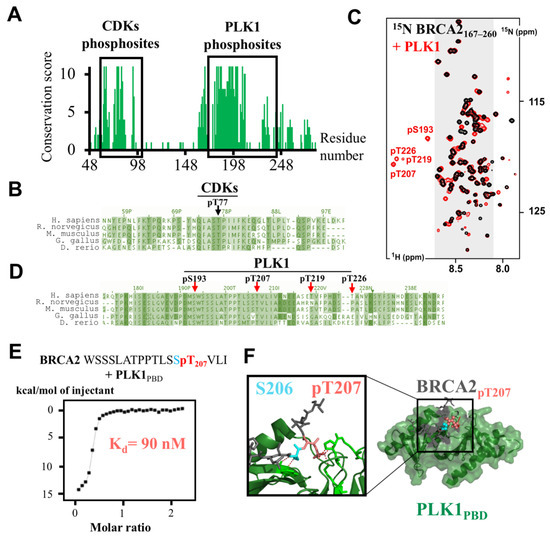
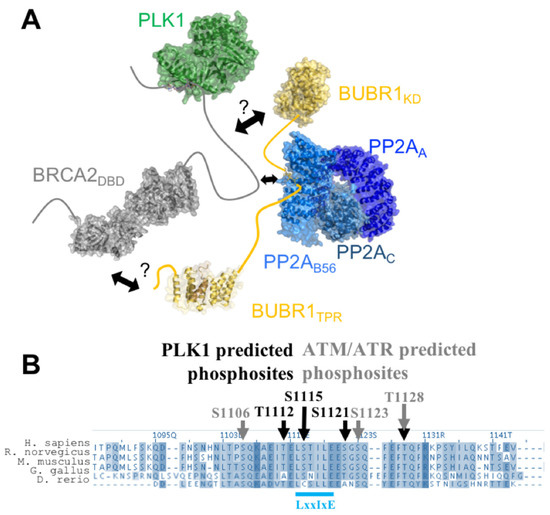
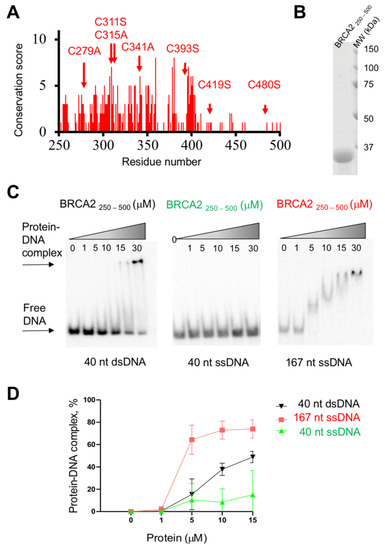
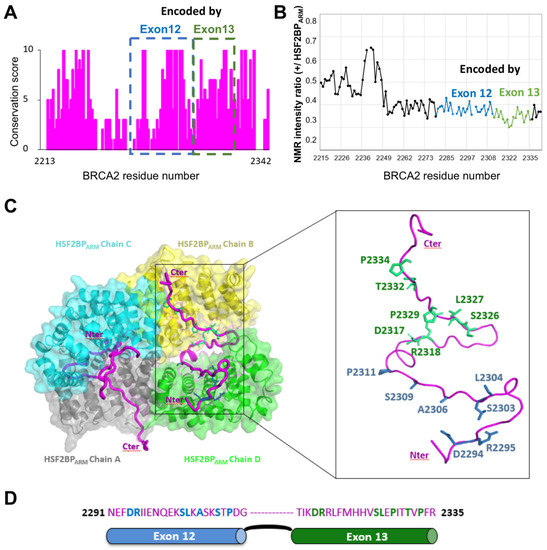
Intrinsic Disorder and Phosphorylation in BRCA2 Facilitate Tight Regulation of Multiple Conserved Binding Events
by
Manon Julien, Rania Ghouil, Ambre Petitalot, Sandrine M. Caputo, Aura Carreira and Sophie Zinn-Justin
Cited by 7 | Viewed by 4298
Abstract
The maintenance of genome integrity in the cell is an essential process for the accurate transmission of the genetic material. BRCA2 participates in this process at several levels, including DNA repair by homologous recombination, protection of stalled replication forks, and cell division. These
[...] Read more.
The maintenance of genome integrity in the cell is an essential process for the accurate transmission of the genetic material. BRCA2 participates in this process at several levels, including DNA repair by homologous recombination, protection of stalled replication forks, and cell division. These activities are regulated and coordinated via cell-cycle dependent modifications. Pathogenic variants in
BRCA2 cause genome instability and are associated with breast and/or ovarian cancers. BRCA2 is a very large protein of 3418 amino acids. Most well-characterized variants causing a strong predisposition to cancer are mutated in the
C-terminal 700 residues DNA binding domain of BRCA2. The rest of the BRCA2 protein is predicted to be disordered. Interactions involving intrinsically disordered regions (IDRs) remain difficult to identify both using bioinformatics tools and performing experimental assays. However, the lack of well-structured binding sites provides unique functional opportunities for BRCA2 to bind to a large set of partners in a tightly regulated manner. We here summarize the predictive and experimental arguments that support the presence of disorder in BRCA2. We describe how BRCA2 IDRs mediate self-assembly and binding to partners during DNA double-strand break repair, mitosis, and meiosis. We highlight how phosphorylation by DNA repair and cell-cycle kinases regulate these interactions. We finally discuss the impact of cancer-associated variants on the function of BRCA2 IDRs and more generally on genome stability and cancer risk.
Full article
►▼
Show Figures
Open AccessArticle
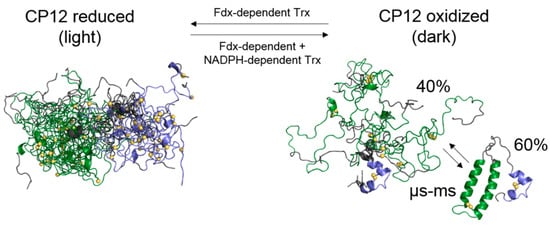
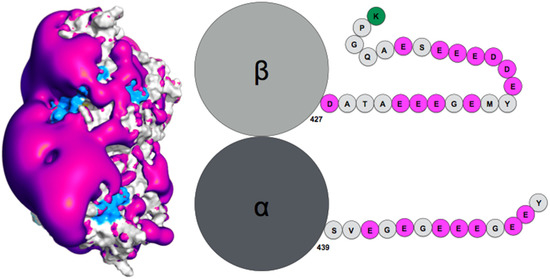
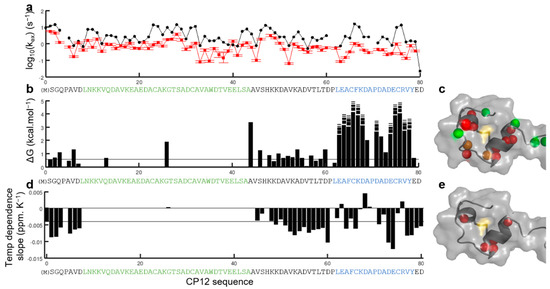
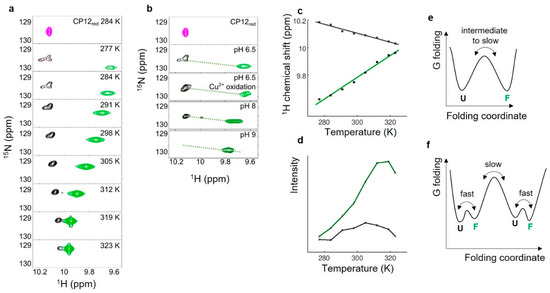
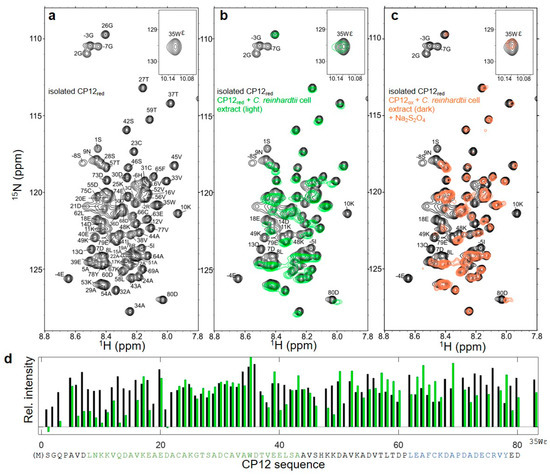
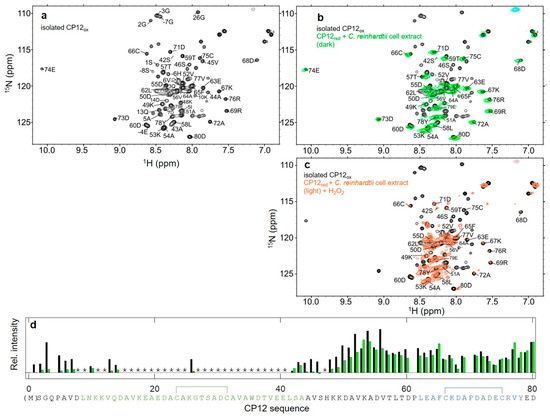
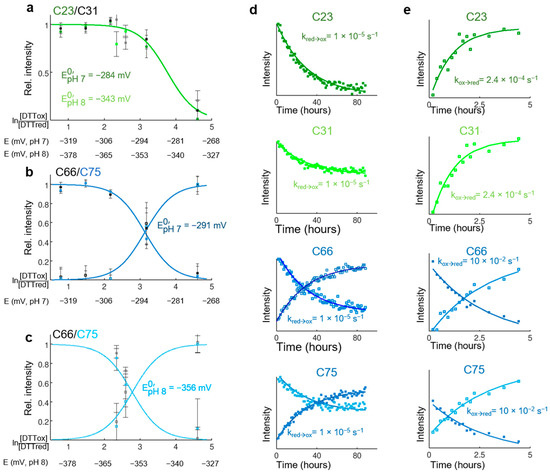
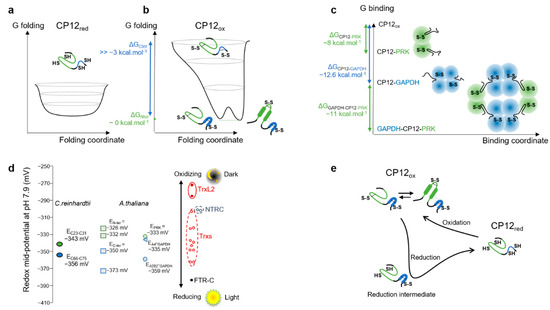
Flexibility of Oxidized and Reduced States of the Chloroplast Regulatory Protein CP12 in Isolation and in Cell Extracts
by
Helene Launay, Hui Shao, Olivier Bornet, Francois-Xavier Cantrelle, Regine Lebrun, Veronique Receveur-Brechot and Brigitte Gontero
Cited by 5 | Viewed by 3086
Abstract
In the chloroplast, Calvin–Benson–Bassham enzymes are active in the reducing environment created in the light by electrons from the photosystems. In the dark, these enzymes are inhibited, mainly caused by oxidation of key regulatory cysteine residues. CP12 is a small protein that plays
[...] Read more.
In the chloroplast, Calvin–Benson–Bassham enzymes are active in the reducing environment created in the light by electrons from the photosystems. In the dark, these enzymes are inhibited, mainly caused by oxidation of key regulatory cysteine residues. CP12 is a small protein that plays a role in this regulation with four cysteine residues that undergo a redox transition. Using amide-proton exchange with solvent, measured by nuclear magnetic resonance (NMR) and mass-spectrometry, we confirmed that reduced CP12 is intrinsically disordered. Using real-time NMR, we showed that the oxidation of the two disulfide bridges is simultaneous. In oxidized CP12, the C
23–C
31 pair is in a region that undergoes a conformational exchange in the NMR-intermediate timescale. The C
66–C
75 pair is in the C-terminus that folds into a stable helical turn. We confirmed that these structural states exist in a physiologically relevant environment: a cell extract from
Chlamydomonas reinhardtii. Consistent with these structural equilibria, the reduction is slower for the C
66–C
75 pair than for the C
23–C
31 pair. The redox mid-potentials for the two cysteine pairs differ and are similar to those found for glyceraldehyde 3-phosphate dehydrogenase and phosphoribulokinase, consistent with the regulatory role of CP12.
Full article
►▼
Show Figures
Open AccessArticle
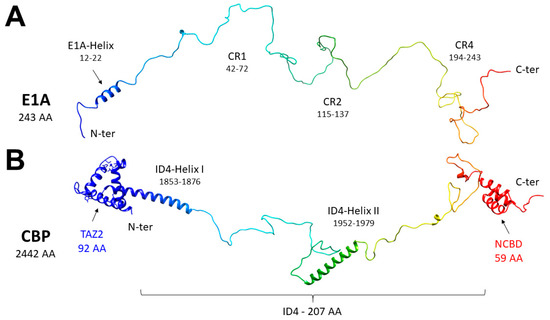
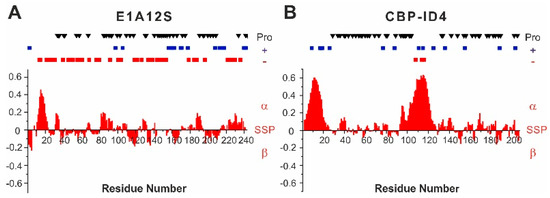
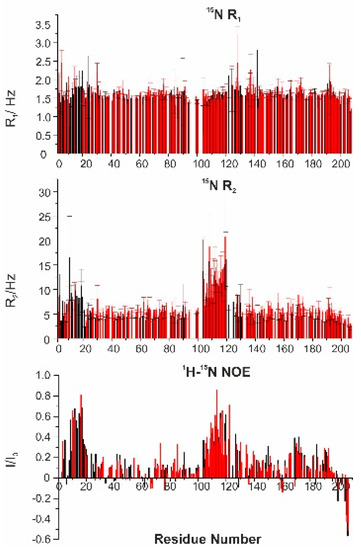
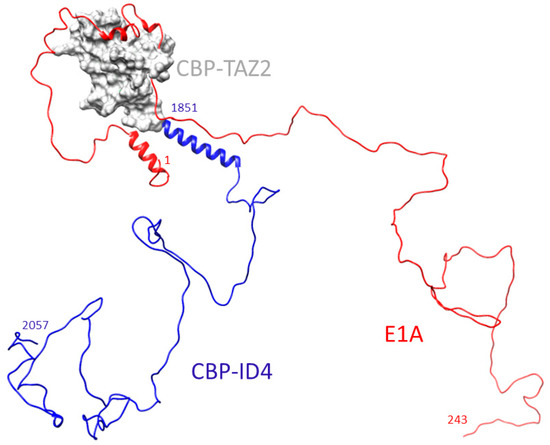
Adenoviral E1A Exploits Flexibility and Disorder to Target Cellular Proteins
by
Maria Grazia Murrali, Isabella C. Felli and Roberta Pierattelli
Cited by 10 | Viewed by 3392
Abstract
Direct interaction between intrinsically disordered proteins (IDPs) is often difficult to characterize hampering the elucidation of their binding mechanism. Particularly challenging is the study of fuzzy complexes, in which the intrinsically disordered proteins or regions retain conformational freedom within the assembly. To date,
[...] Read more.
Direct interaction between intrinsically disordered proteins (IDPs) is often difficult to characterize hampering the elucidation of their binding mechanism. Particularly challenging is the study of fuzzy complexes, in which the intrinsically disordered proteins or regions retain conformational freedom within the assembly. To date, nuclear magnetic resonance spectroscopy has proven to be one of the most powerful techniques to characterize at the atomic level intrinsically disordered proteins and their interactions, including those cases where the formed complexes are highly dynamic. Here, we present the characterization of the interaction between a viral protein, the Early region 1A protein from Adenovirus (E1A), and a disordered region of the human CREB-binding protein, namely the fourth intrinsically disordered linker CBP-ID4. E1A was widely studied as a prototypical viral oncogene. Its interaction with two folded domains of CBP was mapped, providing hints for understanding some functional aspects of the interaction with this transcriptional coactivator. However, the role of the flexible linker connecting these two globular domains of CBP in this interaction was never explored before.
Full article
►▼
Show Figures
Planned Papers
The below list represents only planned manuscripts. Some of these
manuscripts have not been received by the Editorial Office yet. Papers
submitted to MDPI journals are subject to peer-review.
Title: Intrinsic disorder in BRCA2 facilitates tight regulation of multiple conserved binding events
Authors: Julien Manon; Petitalot Ambre; Ghouil Rania; Sandrine Caputo; Carreira Aura; Zinn-Justin Sophie
Affiliation: Institute for Integrative Biology of the Cell (I2BC), CEA, CNRS, Univ Paris-Sud, Université Paris-Saclay, Gif-sur-Yvette Cedex, France Department of Biology, École Normale Supérieure, 94230 Cachan, France Service de génétique, unité de génétique constitutionnelle, Institut Curie, 26 rue d'Ulm, Paris, France Paris Sciences Lettres Research University, Paris, France Institut Curie, PSL Research University, CNRS, UMR3348, F-91405, Orsay, France Paris Sud University, Paris-Saclay University CNRS, UMR3348, F-91405 Orsay, France
Abstract: The maintenance of genome integrity in the cell is an essential process for the accurate transmission of the genetic material. BRCA2 participates in this process at several levels, including DNA repair by homologous recombination, protection of stalled replication forks and cell division. These activities are regulated and coordinated via cell-cycle dependent modifications. Mutations in BRCA2 cause genome instability and are associated with breast and ovarian cancers. BRCA2 is a very large protein of 3418 amino acids. Most well-characterized mutations causing a strong predisposition to cancer are located in the C-terminal 700 amino acids DNA binding domain of BRCA2. The rest of the BRCA2 protein is predicted to be disordered. Interactions involving intrinsically disordered regions (IDRs) remain difficult to identify both using bioinformatics tools and performing experimental assays. Yet, the lack of well-structured binding sites provides unique functional opportunities for BRCA2 to bind to a large set of partners in a tightly regulated manner. We here summarize the predictive and experimental arguments that support the presence of disorder in BRCA2. We describe how BRCA2 IDRs mediate self-assembly and binding to partners during DNA double-strand break repair, mitosis and meiosis. We highlight how phosphorylation by cell-cycle and DNA repair kinases regulates these interactions.
Title: Structural insights into myotilin dimerization and its implication in Z-disc organization
Authors: Kristina Djinovic Carugo; Miha Pavsic; Julius Kostan
Affiliation: Department of Structural and Computational Biology, University of Vienna, Campus-Vienna-Biocenter 5, 1030 Wien, Austria
Abstract: Assembly and integrity of Z-disc—the delimiting unit of sarcomeres—is governed by a network of protein interactions. One of the interaction hubs is the structural protein myotilin, composed of two Ig-like domains and flexible flanking regions. Myotilin forms an antiparallel dimer, however the structural details of dimerization and its implication in myotilin interaction capacity are not yet clear. To elucidate them we generated an integrative three-dimensional model of myotilin dimer by combining known structures of the individual Ig-like domains together with chemical cross-linking, NMR and small angle X-ray scattering data. In this ensemble model, the C-terminal Ig-like domain forms the dimerization core where the predominantly electrostatic interactions are mediated via the ‘BED’-sheet. Region(s) of the domain involved in dimerization lie within the recently characterized myotilin-F-actin interface, however binding of myotilin to F-actin does not have effect on myotilin dimerization. The flexible linker to the preceding Ig-like domain and flexible intrinsically disordered N-terminal region give the dimer a substantial structural plasticity. Thus, the tentacle-like myotilin dimer can span large distances and act as a flexible binding platform for other proteins of the Z-disc.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}