




Viruses 2020, 12(11), 1337; https://doi.org/10.3390/v12111337 - 21 Nov 2020
Cited by 26 | Viewed by 6680
Abstract
►
Show Figures
Giant viruses are a group of eukaryotic double-stranded DNA viruses with large virion and genome size that challenged the traditional view of virus. Newly isolated strains and sequenced genomes in the last two decades have substantially advanced our knowledge of their host diversity,
[...] Read more.
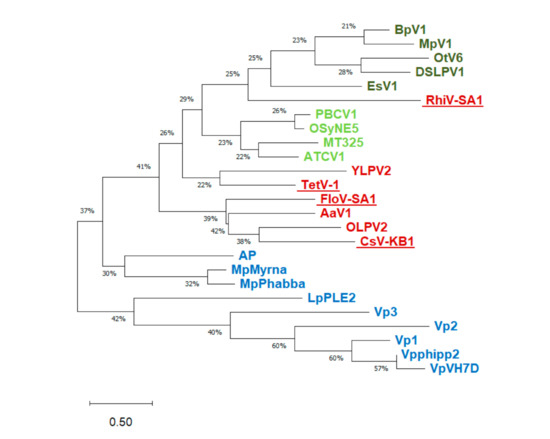
Giant viruses are a group of eukaryotic double-stranded DNA viruses with large virion and genome size that challenged the traditional view of virus. Newly isolated strains and sequenced genomes in the last two decades have substantially advanced our knowledge of their host diversity, gene functions, and evolutionary history. Giant viruses are now known to infect hosts from all major supergroups in the eukaryotic tree of life, which predominantly comprises microbial organisms. The seven well-recognized viral clades (taxonomic families) have drastically different host range. Mimiviridae and Phycodnaviridae, both with notable intrafamilial genome variation and high abundance in environmental samples, have members that infect the most diverse eukaryotic lineages. Laboratory experiments and comparative genomics have shed light on the unprecedented functional potential of giant viruses, encoding proteins for genetic information flow, energy metabolism, synthesis of biomolecules, membrane transport, and sensing that allow for sophisticated control of intracellular conditions and cell-environment interactions. Evolutionary genomics can illuminate how current and past hosts shape viral gene repertoires, although it becomes more obscure with divergent sequences and deep phylogenies. Continued works to characterize giant viruses from marine and other environments will further contribute to our understanding of their host range, coding potential, and virus-host coevolution.
Full article
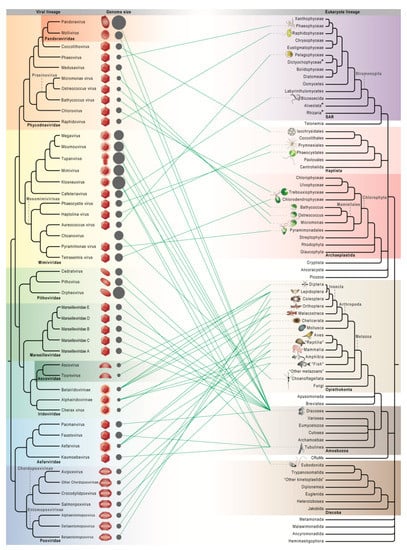
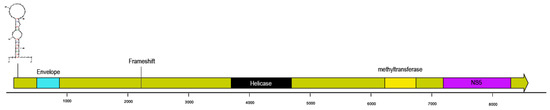
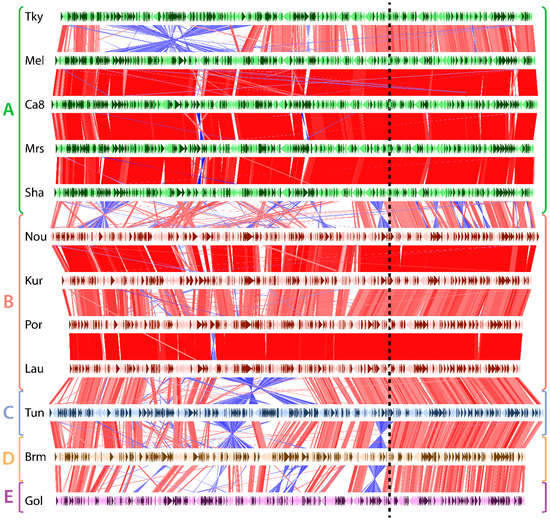
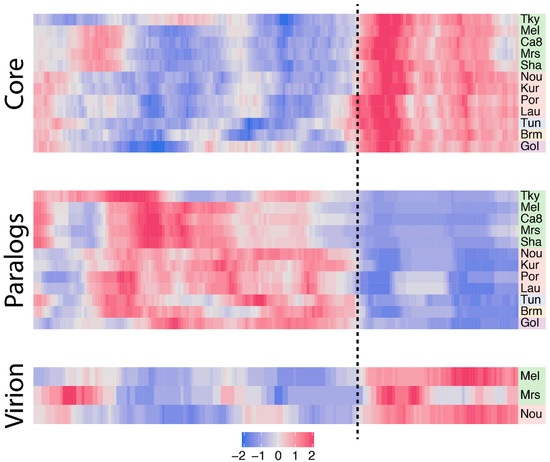
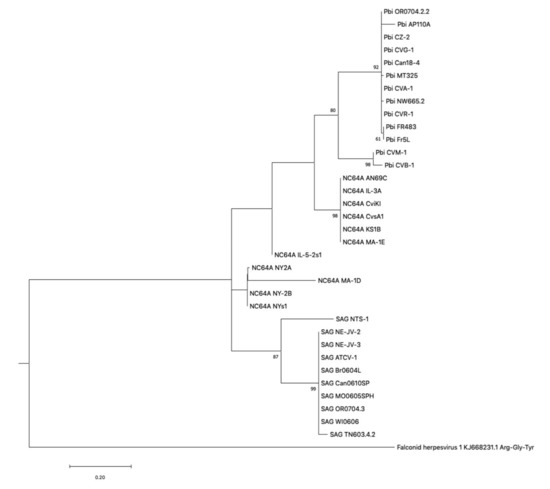
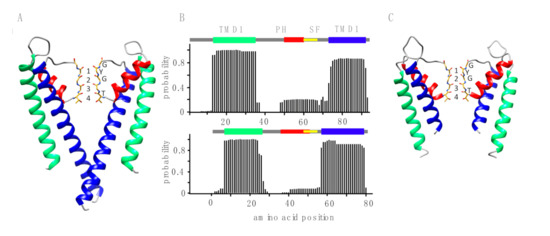
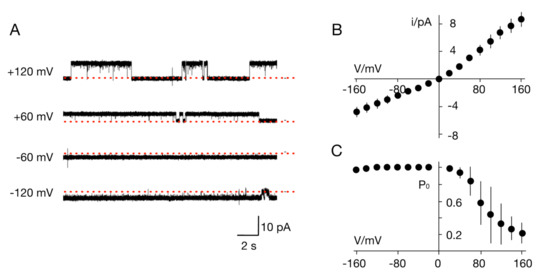
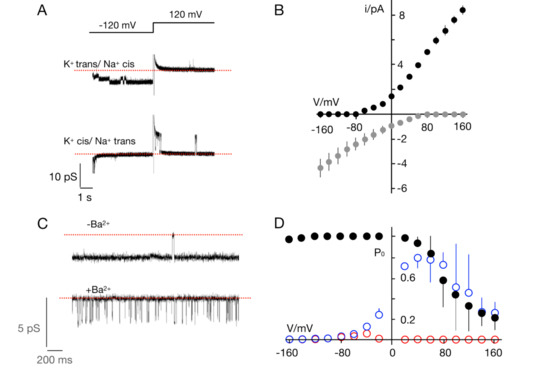
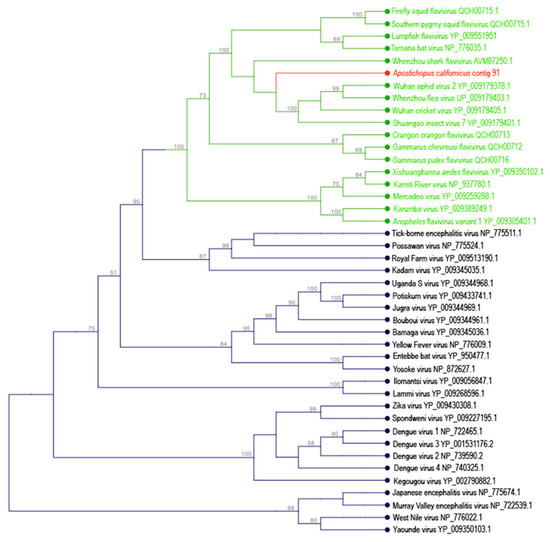
Figure 1



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}