Selective C-N σ Bond Cleavage in Azetidinyl Amides under Transition Metal-Free Conditions


Abstract
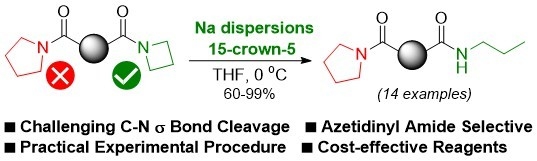
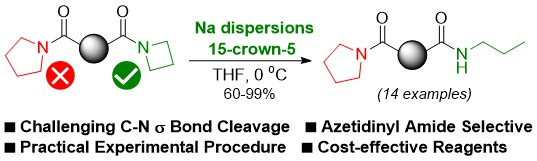
:
1. Introduction
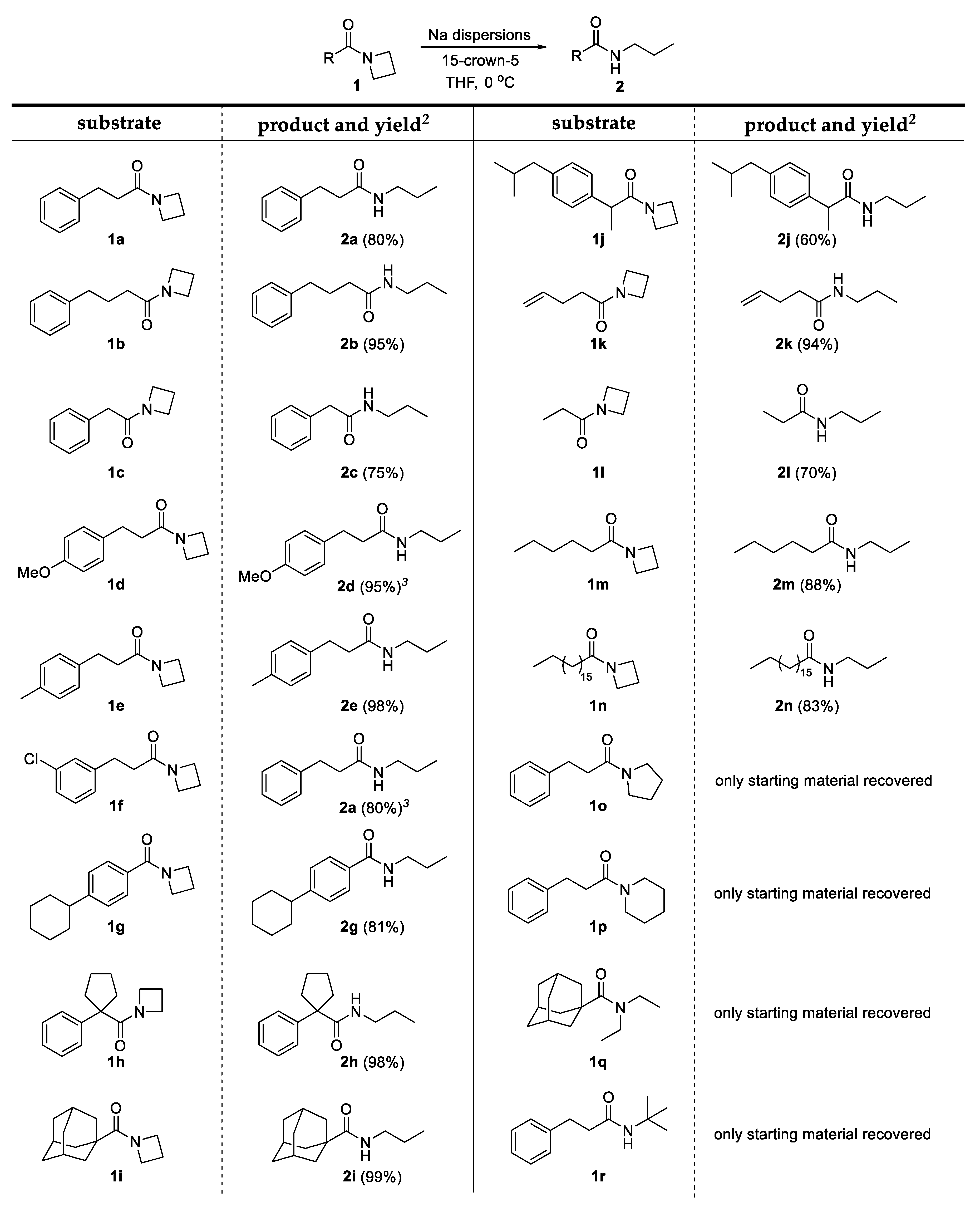
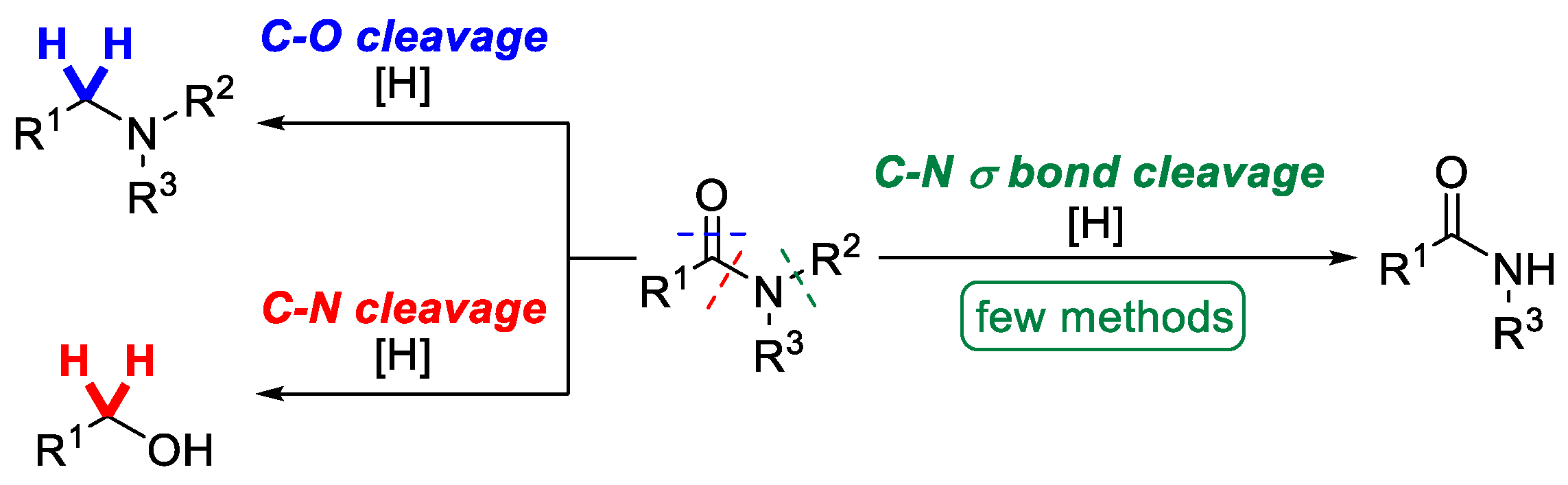
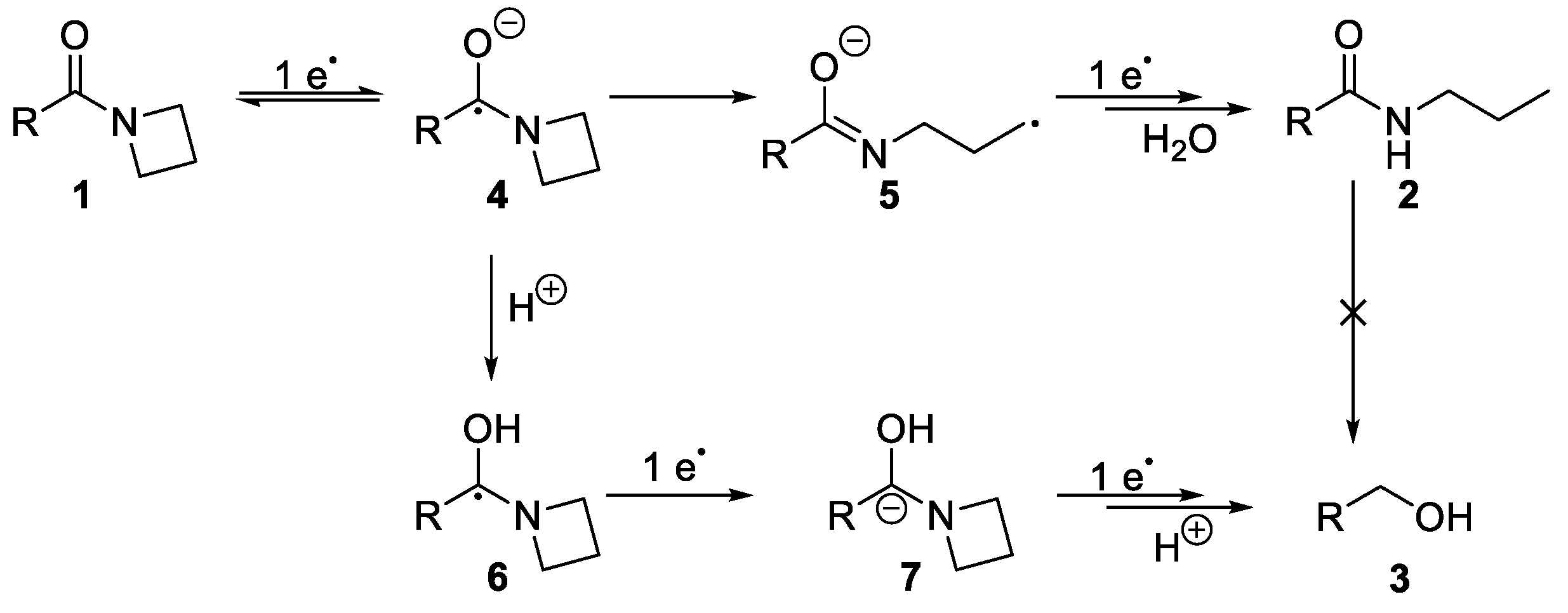
2. Results and Discussion


3. Materials and Methods
3.1. General Information
3.2. Optimization Studies (Table 1)
3.3. General Procedure for the C-N Bond Cleavage in Azetidinyl Amides
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Greenberg, A.; Breneman, C.M.; Liebman, J.F. The Amide Linkage: Structural Significance in Chemistry, Biochemistry and Materials Science; Wiley: Hoboken, NJ, USA, 2000. [Google Scholar]
- Volkov, A.; Tinnis, F.; Slagbrand, T.; Trillo, P.; Adolfsson, H. Chemoselective reduction of carboxamides. Chem. Soc. Rev. 2016, 45, 6685–6697. [Google Scholar] [CrossRef]
- Dub, P.A.; Ikariya, T. Catalytic reductive transformations of carboxylic and carbonic acid derivatives using molecular hydrogen. ACS Catal. 2012, 2, 1718–1741. [Google Scholar] [CrossRef]
- Addis, D.; Das, S.; Junge, K.; Beller, M. Selective reduction of carboxylic acid derivatives by catalytic hydrosilylation. Angew. Chemie Int. Ed. 2011, 50, 6004–6011. [Google Scholar] [CrossRef]
- Zhang, B.; Li, H.; Ding, Y.; Yan, Y.; An, J. Reduction and reductive deuteration of tertiary amides mediated by sodium dispersions with distinct proton donor-dependent chemoselectivity. J. Org. Chem. 2018, 83, 6006–6014. [Google Scholar] [CrossRef] [PubMed]
- Rasu, L.; John, J.M.; Stephenson, E.; Endean, R.; Kalapugama, S.; Clément, R.; Bergens, S.H. Highly enantioselective hydrogenation of amides via dynamic kinetic resolution under low pressure and room temperature. J. Am. Chem. Soc. 2017, 139, 3065–3071. [Google Scholar] [CrossRef] [PubMed]
- Tinnis, F.; Volkov, A.; Slagbrand, T.; Adolfsson, H. Chemoselective reduction of tertiary amides under thermal control: formation of either aldehydes or amines. Angew. Chemie Int. Ed. 2016, 55, 4562–4566. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, D.; Shirase, S.; Mashima, K.; Okuda, J. Chemoselective reduction of tertiary amides to amines catalyzed by triphenylborane. Angew. Chemie Int. Ed. 2016, 55, 13326–13329. [Google Scholar] [CrossRef]
- Cabrero-Antonino, J.R.; Alberico, E.; Drexler, H.-J.; Baumann, W.; Junge, K.; Junge, H.; Beller, M. Efficient base-free hydrogenation of amides to alcohols and amines catalyzed by well-defined pincer imidazolyl–ruthenium complexes. ACS Catal. 2016, 6, 47–54. [Google Scholar] [CrossRef]
- Szostak, M.; Spain, M.; Eberhart, A.J.; Procter, D.J. Highly chemoselective reduction of amides (primary, secondary, tertiary) to alcohols using SmI2 /amine/H2O under mild conditions. J. Am. Chem. Soc. 2014, 136, 2268–2271. [Google Scholar] [CrossRef]
- Das, S.; Wendt, B.; Möller, K.; Junge, K.; Beller, M. Two iron catalysts are better than one: A general and convenient reduction of aromatic and aliphatic primary amides. Angew. Chemie Int. Ed. 2012, 51, 1662–1666. [Google Scholar] [CrossRef]
- Hu, F.; Nareddy, P.; Lalancette, R.; Jordan, F.; Szostak, M. σ N-C Bond Difunctionalization in Bridged Twisted Amides: Sew-and-Cut Activation Approach to Functionalized Isoquinolines. Org. Lett. 2017, 19, 2386–2389. [Google Scholar] [CrossRef] [PubMed]
- Hu, F.; Lalancette, R.; Szostak, M. Structural Characterization of N-Alkylated Twisted Amides: Consequences for Amide Bond Resonance and N-C Cleavage. Angew. Chemie Int. Ed. 2016, 55, 5062–5066. [Google Scholar] [CrossRef]
- Lei, Y.; Wrobleski, A.D.; Golden, J.E.; Powell, D.R.; Aubé, J. Facile C-N cleavage in a series of bridged lactams. J. Am. Chem. Soc. 2005, 127, 4552–4553. [Google Scholar] [CrossRef] [PubMed]
- Szostak, M.; Spain, M.; Procter, D.J. Uncovering the importance of proton donors in TmI2-promoted electron transfer: facile C−N bond cleavage in unactivated amides. Angew. Chemie Int. Ed. 2013, 52, 7237–7241. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.; Szostak, M. Synthesis of nitrogen heterocycles using samarium (II) iodide. Molecules 2017, 22, 2018. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.; Szostak, R.; Szostak, M. Proton-coupled electron transfer in the reduction of carbonyls using SmI2–H2O: implications for the reductive coupling of acyl-type ketyl radicals with SmI2–H2O. Org. Biomol. Chem. 2016, 14, 9151–9157. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.; Szostak, M. Aminoketyl radicals in organic synthesis: Stereoselective cyclization of five- and six-membered cyclic imides to 2-azabicycles using SmI2–H2O. Org. Lett. 2015, 17, 5144–5147. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.C. Electrons in Liquid Ammonia; Oxford University Press: Oxford, UK, 1976. [Google Scholar]
- Thomas, S.J.M.; Edwards, P.P.; Kuznetsov, V.L. Sir Humphry Davy: boundless Chemist, physicist, poet and man of action. Chem. Phys. Chem. 2008, 9, 59–66. [Google Scholar] [CrossRef]
- Rabideau, P.W.; Marcinow, Z. The Birch reduction of aromatic compounds. In Organic Reactions; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 1992; pp. 1–334. [Google Scholar]
- Zimmerman, H.E. A mechanistic analysis of the Birch reduction. Acc. Chem. Res. 2012, 45, 164–170. [Google Scholar] [CrossRef]
- Cossy, J.; Gille, B.; Bellosta, V. Facile synthesis of spirocyclic systems through the intramolecular addition of ketyl radicals via the sodium/ammonia reduction of δ,ε-unsaturated carboxylic esters. J. Org. Chem. 1998, 63, 3141–3146. [Google Scholar] [CrossRef]
- Dye, J.L. Electrons as anions. Science 2003, 301, 607–608. [Google Scholar] [CrossRef] [PubMed]
- Dye, J.L. Electrides: ionic salts with electrons as the anions. Science 1990, 247, 663–668. [Google Scholar] [CrossRef] [PubMed]
- Dye, J.L. Compounds of alkali metal snions. Angew. Chemie Int. Ed. English 1979, 18, 587–598. [Google Scholar] [CrossRef]
- Jedliński, Z. Novel electron-transfer reactions mediated by alkali metals complexed by macrocyclic ligand. Acc. Chem. Res. 1998, 31, 55–61. [Google Scholar] [CrossRef]
- Lei, P.; Ding, Y.; Zhang, X.; Adijiang, A.; Li, H.; Ling, Y.; An, J. A practical and chemoselective ammonia-free Birch reduction. Org. Lett. 2018, 20, 3439–3442. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Luo, S.; Adijiang, A.; Zhao, H.; An, J. Reductive deuteration of nitriles: the synthesis of α,α-dideuterio amines by sodium mediated electron transfer reactions. J. Org. Chem. 2018, 83, 12269–12274. [Google Scholar] [CrossRef] [PubMed]
- Han, M.; Ding, Y.; Yan, Y.; Li, H.; Luo, S.; Adijiang, A.; Ling, Y.; An, J. Transition-metal-free, selective reductive deuteration of terminal alkynes with sodium dispersions and EtOD-d1. Org. Lett. 2018, 20, 3010–3013. [Google Scholar] [CrossRef]
- Li, H.; Zhang, B.; Dong, Y.; Liu, T.; Zhang, Y.; Nie, H.; Yang, R.; Ma, X.; Ling, Y.; An, J. A selective and cost-effective method for the reductive deuteration of activated alkenes. Tetrahedron Lett. 2017, 58, 2757–2760. [Google Scholar] [CrossRef]
- Han, M.; Ma, X.; Yao, S.; Ding, Y.; Yan, Z.; Adijiang, A.; Wu, Y.; Li, H.; Zhang, Y.; Lei, P.; et al. Development of a modified Bouveault–Blanc reduction for the selective synthesis of α,α-dideuterio alcohols. J. Org. Chem. 2017, 82, 1285–1290. [Google Scholar] [CrossRef]
- Liu, C.; Achtenhagen, M.; Szostak, M. Chemoselective ketone synthesis by the addition of organometallics to N-Acylazetidines. Org. Lett. 2016, 18, 2375–2378. [Google Scholar] [CrossRef]
- Štefane, B.; Polanc, S. hydrogenation of BF2 complexes with 1,3-dicarbonyl ligands. Tetrahedron 2009, 65, 2339–2343. [Google Scholar] [CrossRef]
- Ignatenko, V.A.; Deligonul, N.; Viswanathan, R. Branch-Selective Synthesis of Oxindole and Indene Scaffolds: Transition Metal-Controlled Intramolecular Aryl Amidation Leading to C3 Reverse-Prenylated Oxindoles. Org. Lett. 2010, 12, 3594–3597. [Google Scholar] [CrossRef] [PubMed]
- Gajda, T.; Zwierzak, A. Phase-transfer-catalysed N-alkylation of carboxamides and sulfonamides. Synthesis 1981, 1981, 1005–1008. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors |




{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | Sodium Dispersion 1 (Equiv) | 15-crown-5 (Equiv) | Time | Solvent | Yield (%) 2 |
| 1 | 5.0 | 0 | 2.0 h | Et2O | 50 |
| 2 | 5.0 | 0 | 2.0 h | THF | 67 |
| 3 | 5.0 | 5.0 | 2.0 h | Et2O | 79 |
| 4 | 5.0 | 5.0 | 2.0 h | THF | 80 |
| 5 | 4.5 | 4.5 | 2.0 h | Et2O | 65 |
| 6 | 3.0 | 3.0 | 2.0 h | Et2O | 56 |
| 7 | 5.0 | 5.0 | 1.0 h | THF | 71 |
| 8 | 5.0 | 5.0 | 10 min | THF | 47 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, H.; Lai, Z.; Adijiang, A.; Zhao, H.; An, J. Selective C-N σ Bond Cleavage in Azetidinyl Amides under Transition Metal-Free Conditions. Molecules 2019, 24, 459. https://doi.org/10.3390/molecules24030459
Li H, Lai Z, Adijiang A, Zhao H, An J. Selective C-N σ Bond Cleavage in Azetidinyl Amides under Transition Metal-Free Conditions. Molecules. 2019; 24(3):459. https://doi.org/10.3390/molecules24030459
Chicago/Turabian StyleLi, Hengzhao, Zemin Lai, Adila Adijiang, Hongye Zhao, and Jie An. 2019. "Selective C-N σ Bond Cleavage in Azetidinyl Amides under Transition Metal-Free Conditions" Molecules 24, no. 3: 459. https://doi.org/10.3390/molecules24030459
APA StyleLi, H., Lai, Z., Adijiang, A., Zhao, H., & An, J. (2019). Selective C-N σ Bond Cleavage in Azetidinyl Amides under Transition Metal-Free Conditions. Molecules, 24(3), 459. https://doi.org/10.3390/molecules24030459