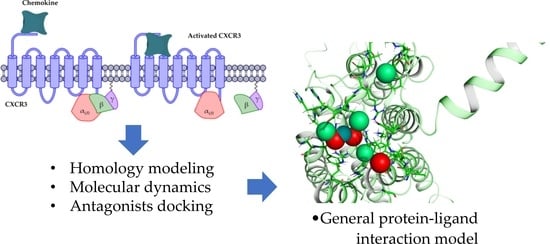
Figure 1.
CXCR3 receptor activation. The first activation step is the arrival of the chemokine and the subsequent orientation towards the transmembrane region. The second step is the release of the βγ complex caused by the conformational change of the receptor.
Figure 1.
CXCR3 receptor activation. The first activation step is the arrival of the chemokine and the subsequent orientation towards the transmembrane region. The second step is the release of the βγ complex caused by the conformational change of the receptor.
Figure 2.
All-atom molecular dynamics (AA-MD) simulation of CXCR3 50 ns. (A) CXCR3 model obtained from I-TASSER (initial confirmations or T0), (B) cluster1 of the simulation, and (C) the alignment of T0 and cluster1.
Figure 2.
All-atom molecular dynamics (AA-MD) simulation of CXCR3 50 ns. (A) CXCR3 model obtained from I-TASSER (initial confirmations or T0), (B) cluster1 of the simulation, and (C) the alignment of T0 and cluster1.
Figure 3.
AA-MD simulation of CXCR3-Gαi/0βγ 100 ns. (A) Structure of the complex at T0, (B) cluster1 of the simulation, and (C) the alignment of T0 and cluster1.
Figure 3.
AA-MD simulation of CXCR3-Gαi/0βγ 100 ns. (A) Structure of the complex at T0, (B) cluster1 of the simulation, and (C) the alignment of T0 and cluster1.
Figure 4.
Interactions between CXCL9 and CXCR3. (A) CXCR3-CXCL9_T0. (B) Cluster1_50-ns AA-MD. The R chain corresponds to CXCR3 and the Q chain to CXCL9. An increase of hydrogen bonds in cluster1 of the 50-ns simulation can be observed, as well as an interaction between the Q99 residue of CXCL9 and the glycosylation present in the N32 residue of CXCR3. The residues in red and purple represent hydrophobic interactions between proteins.
Figure 4.
Interactions between CXCL9 and CXCR3. (A) CXCR3-CXCL9_T0. (B) Cluster1_50-ns AA-MD. The R chain corresponds to CXCR3 and the Q chain to CXCL9. An increase of hydrogen bonds in cluster1 of the 50-ns simulation can be observed, as well as an interaction between the Q99 residue of CXCL9 and the glycosylation present in the N32 residue of CXCR3. The residues in red and purple represent hydrophobic interactions between proteins.
Figure 5.
Interactions between CXCR3 and CXCL10. (A) CXCR3-CXCL10_T0. (B) Cluster1_50 ns. The R chain corresponds to CXCR3 and the Q chain to CXCL10. An increase of hydrogen bonds can be observed, along with a decrease of the hydrophobic environment involved and an increase in CXCL10 residues that interact with CXCR3 in cluster1 of the 50-ns simulation.
Figure 5.
Interactions between CXCR3 and CXCL10. (A) CXCR3-CXCL10_T0. (B) Cluster1_50 ns. The R chain corresponds to CXCR3 and the Q chain to CXCL10. An increase of hydrogen bonds can be observed, along with a decrease of the hydrophobic environment involved and an increase in CXCL10 residues that interact with CXCR3 in cluster1 of the 50-ns simulation.
Figure 6.
Interactions of CXCR3 and CXCL11. (A) CXCR3-CXCL11_T0. (B) Cluster1_50 ns. The R chain corresponds to CXCR3 and the Q chain to CXCL11. A decrease of interactions between CXCR3 and CXCL11 is observed. Glycosylation in the N22 participates in the hydrophobic environment for the interaction of CXCL11 T0 and cluster1.
Figure 6.
Interactions of CXCR3 and CXCL11. (A) CXCR3-CXCL11_T0. (B) Cluster1_50 ns. The R chain corresponds to CXCR3 and the Q chain to CXCL11. A decrease of interactions between CXCR3 and CXCL11 is observed. Glycosylation in the N22 participates in the hydrophobic environment for the interaction of CXCL11 T0 and cluster1.
Figure 7.
AA-MD simulation of CXCR3-CXCL9. (A) Alignment of CXCR3-CXCL9_T0 (blue) and CXCR3-CXCL9_50 ns (red). (B) Alignment of CXCL9_T0 (blue) and CXCL9_50 ns (magenta). There is a root mean standard deviation (RMSD) of 2.841 Å between T0 and cluster1 of the 50-ns simulation, and changes in the Gαi/0βγ are observed; in addition, there is a conformational change of CXCL9 with an RMSD of 2.972 Å, which indicates that the system evolves into a more favorable state for interactions between CXCL9 and CXCR3.
Figure 7.
AA-MD simulation of CXCR3-CXCL9. (A) Alignment of CXCR3-CXCL9_T0 (blue) and CXCR3-CXCL9_50 ns (red). (B) Alignment of CXCL9_T0 (blue) and CXCL9_50 ns (magenta). There is a root mean standard deviation (RMSD) of 2.841 Å between T0 and cluster1 of the 50-ns simulation, and changes in the Gαi/0βγ are observed; in addition, there is a conformational change of CXCL9 with an RMSD of 2.972 Å, which indicates that the system evolves into a more favorable state for interactions between CXCL9 and CXCR3.
Figure 8.
AA-MD simulation of CXCR3_CXCL10. (A) Alignment of CXCR3-CXCL10_T0 (blue) and CXCR3-CXCL10_50 ns (green). (B) Alignment of CXCL10_T0 (blue) and CXCL10_50 ns (cyan). There is an RMSD of 2.646 Å between T0 and cluster1 of the 50-ns simulation; changes in the Gαi/0βγ are observed; additionally, a conformational change of CXCL10 exists with an RMSD of 4.745 Å, which shows that the system evolves into a more favorable state for interactions between CXCL10 and CXCR3.
Figure 8.
AA-MD simulation of CXCR3_CXCL10. (A) Alignment of CXCR3-CXCL10_T0 (blue) and CXCR3-CXCL10_50 ns (green). (B) Alignment of CXCL10_T0 (blue) and CXCL10_50 ns (cyan). There is an RMSD of 2.646 Å between T0 and cluster1 of the 50-ns simulation; changes in the Gαi/0βγ are observed; additionally, a conformational change of CXCL10 exists with an RMSD of 4.745 Å, which shows that the system evolves into a more favorable state for interactions between CXCL10 and CXCR3.
Figure 9.
AA-MD simulation of CXCR3_CXCL11. (A) Alignment of CXCR3-CXCL11_T0 (blue) and CXCR3-CXCL11_50 ns (yellow). (B) Alignment of CXCL11_T0 (blue) and CXCL11_50 ns (orange). There is an RMSD of 2.538 Å between T0 and cluster1 of the 50-ns simulation; changes in the Gαi/0βγ are observed in addition to a conformational change of CXCL10 with an RMSD of 6.339 Å; however, unlike CXCL9 and 10, the hydrogen bonds and hydrophobic-type interactions decreased.
Figure 9.
AA-MD simulation of CXCR3_CXCL11. (A) Alignment of CXCR3-CXCL11_T0 (blue) and CXCR3-CXCL11_50 ns (yellow). (B) Alignment of CXCL11_T0 (blue) and CXCL11_50 ns (orange). There is an RMSD of 2.538 Å between T0 and cluster1 of the 50-ns simulation; changes in the Gαi/0βγ are observed in addition to a conformational change of CXCL10 with an RMSD of 6.339 Å; however, unlike CXCL9 and 10, the hydrogen bonds and hydrophobic-type interactions decreased.
Figure 10.
Coarse-grained molecular dynamics (CG-MD) simulation of CXCR3-CXCL9. (A) Alignment between CXCR3-CXCL9_T0 (blue) and cluster1_CG-MD (red), with an RMSD of 9.409 Å. (B) Distances among the CXCR3-CXCL9 complex chains. The distance decreases during the simulation by approximately 2 to 3 Å.
Figure 10.
Coarse-grained molecular dynamics (CG-MD) simulation of CXCR3-CXCL9. (A) Alignment between CXCR3-CXCL9_T0 (blue) and cluster1_CG-MD (red), with an RMSD of 9.409 Å. (B) Distances among the CXCR3-CXCL9 complex chains. The distance decreases during the simulation by approximately 2 to 3 Å.
Figure 11.
CG-MD simulation of CXCR3-CXCL10. (A) Alignment of CXCR3-CXCL10_T0 (blue) and cluster1_CG-MD (green), with an RMSD of 9.409 Å. (B) Distances among the CXCR3-CXCL10 complex chains. The distance decreases during the simulation by approximately 2 to 3 Å.
Figure 11.
CG-MD simulation of CXCR3-CXCL10. (A) Alignment of CXCR3-CXCL10_T0 (blue) and cluster1_CG-MD (green), with an RMSD of 9.409 Å. (B) Distances among the CXCR3-CXCL10 complex chains. The distance decreases during the simulation by approximately 2 to 3 Å.
Figure 12.
CG-MD simulation of CXCR3-CXCL11. (A) Alignment of CXCR3-CXCL11_T0 (blue) and cluster1_CG-MD (yellow), with an RMSD of 9.853 Å. (B) Distances among the CXCR3-CXCL11 complex chains. The distance decreases during the simulation by approximately 1 Å less than CXCL9 and CXCL10.
Figure 12.
CG-MD simulation of CXCR3-CXCL11. (A) Alignment of CXCR3-CXCL11_T0 (blue) and cluster1_CG-MD (yellow), with an RMSD of 9.853 Å. (B) Distances among the CXCR3-CXCL11 complex chains. The distance decreases during the simulation by approximately 1 Å less than CXCL9 and CXCL10.
Figure 13.
Interactions between CXCR3-CXCL9 from the CG-MD simulation. There is an increase in hydrogen bonds and hydrophobic interactions of CXCR3 with CXCL9 after the CG-MD simulation.
Figure 13.
Interactions between CXCR3-CXCL9 from the CG-MD simulation. There is an increase in hydrogen bonds and hydrophobic interactions of CXCR3 with CXCL9 after the CG-MD simulation.
Figure 14.
Interactions between CXCR3 and CXCL10 from the CG-MD simulation. An increase in hydrogen bonds and hydrophobic interactions is observed, although, in comparison with CXCL9, the number of polar interactions is lower. The interactions are maintained with the first residues of CXCR3.
Figure 14.
Interactions between CXCR3 and CXCL10 from the CG-MD simulation. An increase in hydrogen bonds and hydrophobic interactions is observed, although, in comparison with CXCL9, the number of polar interactions is lower. The interactions are maintained with the first residues of CXCR3.
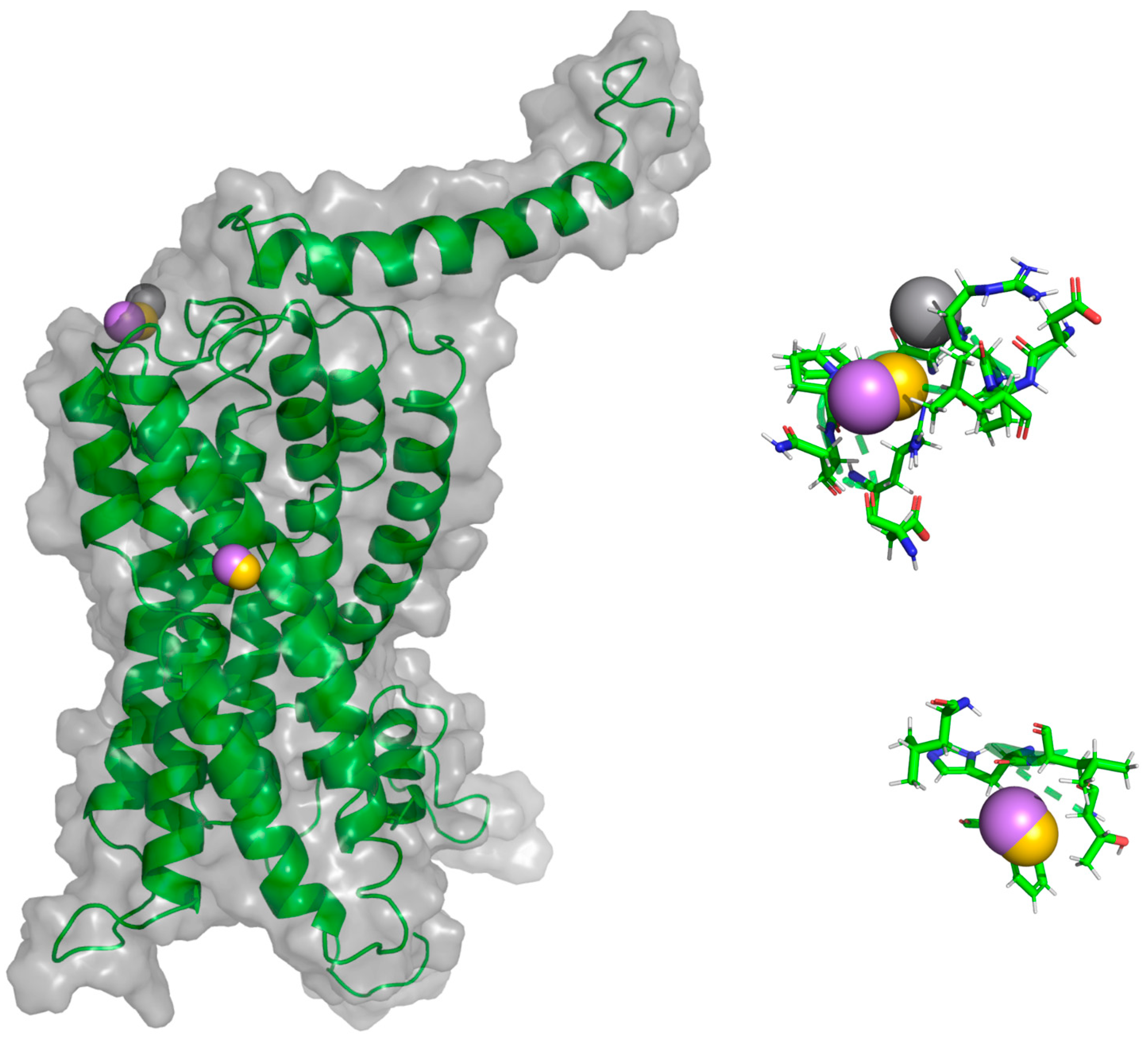
Figure 15.
Interactions between CXCR3-CXCL11 from the CG-MD simulation. An increase of hydrogen bonds and hydrophobic interactions is observed. The diagram shows the interaction with the R197 residue of CXCR3, a residue reported to be important for the binding of CXCL11, and interactions with E293, which is reported to be necessary for the stabilization of the CXCL11-CXCR3 complex.
Figure 15.
Interactions between CXCR3-CXCL11 from the CG-MD simulation. An increase of hydrogen bonds and hydrophobic interactions is observed. The diagram shows the interaction with the R197 residue of CXCR3, a residue reported to be important for the binding of CXCL11, and interactions with E293, which is reported to be necessary for the stabilization of the CXCL11-CXCR3 complex.
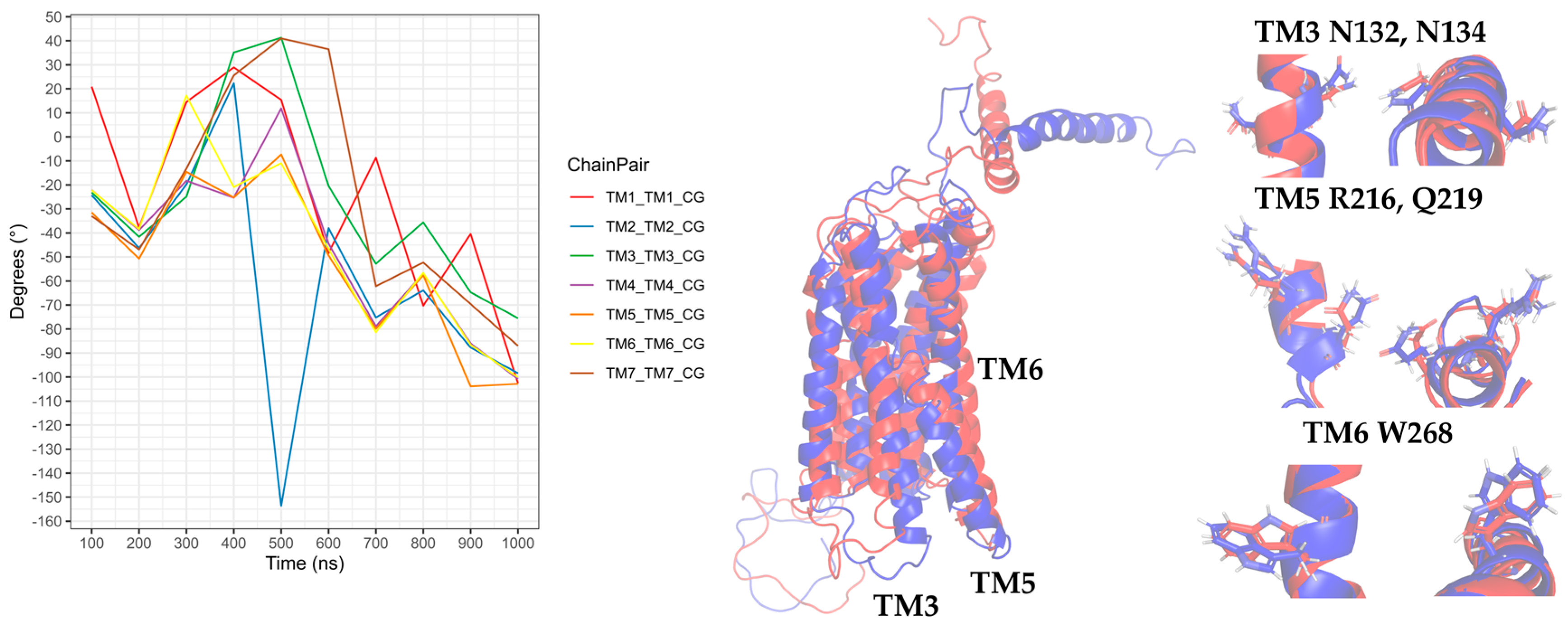
Figure 16.
Rotation of the transmembrane segments (TMs) from the CG-MD simulation of CXCR3_CXCL9. The rotations of the TMs throughout the simulation are seen, with intense rotations of TM1, TM5, and TM6. In blue is shown the initial conformation; in red, the conformation at 800 ns.
Figure 16.
Rotation of the transmembrane segments (TMs) from the CG-MD simulation of CXCR3_CXCL9. The rotations of the TMs throughout the simulation are seen, with intense rotations of TM1, TM5, and TM6. In blue is shown the initial conformation; in red, the conformation at 800 ns.
Figure 17.
Rotation of the TMs from the CG-MD simulation of CXCR3-CXCL10. The rotations of the TMs throughout the simulation are seen, with intense rotations of TM1, TM2, TM5, and TM6. In blue, the initial conformation is shown; in red is the conformation at 700 ns.
Figure 17.
Rotation of the TMs from the CG-MD simulation of CXCR3-CXCL10. The rotations of the TMs throughout the simulation are seen, with intense rotations of TM1, TM2, TM5, and TM6. In blue, the initial conformation is shown; in red is the conformation at 700 ns.
Figure 18.
Rotation of the TMs from the CG-MD simulation of CXCR3_CXCL11. Rotations of the TMs throughout the simulation are present, with relevant rotations of TM1, TM2, TM5, and TM6, starting at 200 ns. In blue, the initial conformation is shown; in red is the conformation at 600 ns.
Figure 18.
Rotation of the TMs from the CG-MD simulation of CXCR3_CXCL11. Rotations of the TMs throughout the simulation are present, with relevant rotations of TM1, TM2, TM5, and TM6, starting at 200 ns. In blue, the initial conformation is shown; in red is the conformation at 600 ns.
Figure 19.
Rotation of TMs from the 250-ns AA-MD simulation of CXCR3-CXCL10-Gi/0. Rotations of the TMs throughout the simulation are presented, with the relevant rotations of TM1, TM2, TM3, TM5, and TM6. TM7 has an intense rotation at 50 ns. In blue, the initial conformation; in red is the conformation at 600 ns.
Figure 19.
Rotation of TMs from the 250-ns AA-MD simulation of CXCR3-CXCL10-Gi/0. Rotations of the TMs throughout the simulation are presented, with the relevant rotations of TM1, TM2, TM3, TM5, and TM6. TM7 has an intense rotation at 50 ns. In blue, the initial conformation; in red is the conformation at 600 ns.
Figure 20.
The distance of complex proteins in the AA-MD 250-ns simulation of CXCR3-CXCL10. The separation of the βγ complex to the α subunit occurs at 175 ns.
Figure 20.
The distance of complex proteins in the AA-MD 250-ns simulation of CXCR3-CXCL10. The separation of the βγ complex to the α subunit occurs at 175 ns.
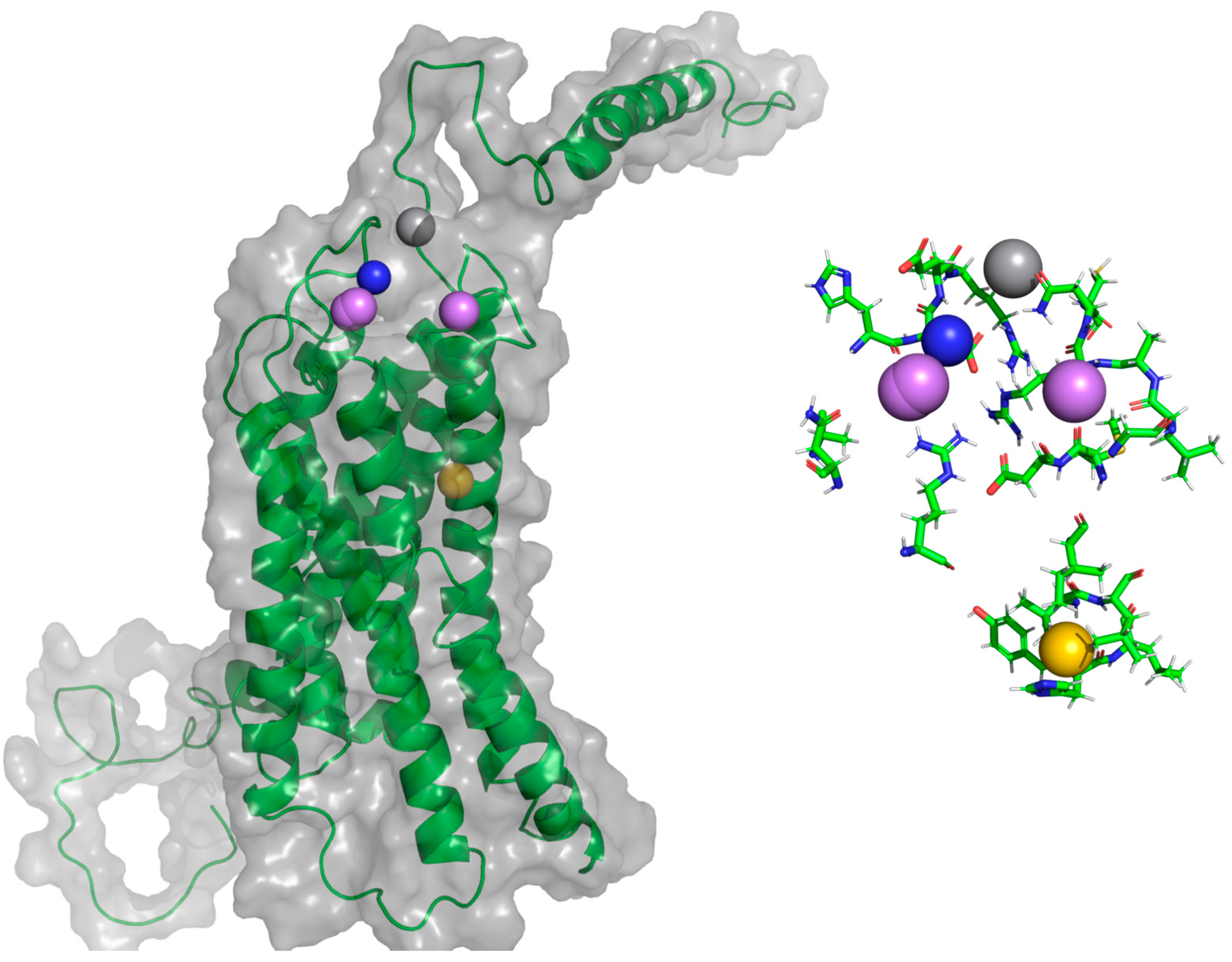
Figure 21.
General protein–ligand interaction model of F0 CXCR3-CXCL10_CG-MD. In magenta: hydrogen bond acceptors (HAc), gray: hydrogen bond donors (HDn), yellow: hydrophobic groups (Hph), and blue: positive-charged groups (Pin).
Figure 21.
General protein–ligand interaction model of F0 CXCR3-CXCL10_CG-MD. In magenta: hydrogen bond acceptors (HAc), gray: hydrogen bond donors (HDn), yellow: hydrophobic groups (Hph), and blue: positive-charged groups (Pin).
Figure 22.
General protein–ligand interaction model of C1 CXCR3-CXCL10_CG-MD. In magenta: HAc, gray: HDn, yellow: Hph, and blue: PIn.
Figure 22.
General protein–ligand interaction model of C1 CXCR3-CXCL10_CG-MD. In magenta: HAc, gray: HDn, yellow: Hph, and blue: PIn.
Figure 23.
General protein–ligand interaction model of F7 CXCR3-CXCL10_CG-MD. In magenta: HAc, gray: HDn, yellow: Hph, green: aromatic rings (Arm), and red: negative-charged groups (Nin).
Figure 23.
General protein–ligand interaction model of F7 CXCR3-CXCL10_CG-MD. In magenta: HAc, gray: HDn, yellow: Hph, green: aromatic rings (Arm), and red: negative-charged groups (Nin).
Table 1.
Timestep and clustering of the AA-MD simulations.
Table 1.
Timestep and clustering of the AA-MD simulations.
| | Most Representative Conformation Timestep (ns) | | |
|---|
| Complex | 5 ns | 50 ns | 100 ns | Cutoff (nm) | Clusters |
|---|
| CXCR3 | | 32.1 | 86.9 | 0.4 */0.2 ** | 3 */10 ** |
| CXCL9 | 3.8 | | | 0.2 | 14 |
| CXCR3-CXCL9 | | 31.1 | | 0.4 | 9 |
| CXCR3-CXCL10 | | 33.3 | | 0.51 | 3 |
| CXCR3-CXCL11 | | 26.6 | | 0.4 | 10 |
Table 2.
Timestep and clustering of the coarse-grained molecular dynamics (CG-MD) simulations.
Table 2.
Timestep and clustering of the coarse-grained molecular dynamics (CG-MD) simulations.
| | Most Representative Conformation Timestep (ns) | | |
|---|
| Complex | Cluster1 | Cluster2 | Cluster3 | Cutoff (nm) | Clusters |
|---|
| CXCR3-CXCL9 | 607.0 | 131.0 | 39.0 | 0.6 | 10 |
| CXCR3-CXCL10 | 787.9 | 159.6 | 51.2 | 0.6 | 10 |
| CXCR3-CXCL11 | 532.7 | 171.3 | 979.3 | 0.6 | 10 |
Table 3.
Presence of the so-called arginine cage in the CG-MD simulation.
Table 3.
Presence of the so-called arginine cage in the CG-MD simulation.
| Time Frame (ns) | CXCR3_CXCL9 | CXCR3_CXCL10 | CXCR3_CXCL11 |
|---|
| 0 | X | X | X |
| 100 | X | | X |
| 200 | X | | X |
| 300 | X | | X |
| 400 | X | | |
| 500 | X | | |
| 600 | | | |
| 700 | X | X | |
| 800 | | | X |
| 900 | | | |
| 1000 | X | | |
Table 4.
Transmembrane segments in CXCR3.
Table 4.
Transmembrane segments in CXCR3.
| TM1 | TM2 | TM3 * | TM4 | TM5 * | TM6 | TM7 |
|---|
| 54–80 | 90–110 | 126–147 | 170–189 | 213–233 | 256–277 | 299–321 |
Table 5.
Pharmacophoric elements of F0 CXCR3-CXCL10_CG-MD.
Table 5.
Pharmacophoric elements of F0 CXCR3-CXCL10_CG-MD.
| Pharmacophoric Element | Residues (5 Å) |
|---|
| HAc | A192, H194, D195, R212, M281, D282, G284, L286, A287, R288 |
| HDn | D195, E196, R197, N289, C290 |
| Hph | Y271, H272, L273, V275, L276, I279 |
| PIn | H194, D195, E196, R197, R288 |
Table 6.
Pharmacophoric elements of C1 CXCR3-CXCL10_CG-MD.
Table 6.
Pharmacophoric elements of C1 CXCR3-CXCL10_CG-MD.
| Pharmacophoric Element | Residues (5 Å) |
|---|
| HAc | R197, L198, P208, Q209, F224, T269, H272, L273, L276, D282 |
| HDn | D195, E196, R197, N206, F207, R212 |
| Hph | E196, R197, L198, P208, Q209, R212, F224, T269, H272, L273 |
Table 7.
Pharmacophoric elements of F7 CXCR3-CXCL10_AA-MD.
Table 7.
Pharmacophoric elements of F7 CXCR3-CXCL10_AA-MD.
| Pharmacophoric Element | Residues (5 Å) |
|---|
| Arm | A129, I133, L180, A183, L184, F187 |
| HAc | V126, A129, L130, I133, A183, F187, L198, N199, L286, A287, R288 |
| HDn | S36, L198, N199, L286, R288, N289 |
| Hph | V136, A129, L130, I133, A183, F187 |
| NIn | E31, L286, A287, R288 |
Table 8.
Relevant amino acids for the interactions between CXCR3 and its chemokines.
Table 8.
Relevant amino acids for the interactions between CXCR3 and its chemokines.
| Residues | Observation |
|---|
| Residues 1–16 | Relevant for interactions between the CXCR3 receptor and its chemokines [28]. |
| R216 | Fundamental for chemotaxis and the mobilization of Ca+2 but not in the union of chemokines [59]. |
| Y27, Y28 | Sulfation in these residues is necessary for the interactions of the chemokines [28,31,59]. |
| N197, N212 | Essential for the union of CXCL10 and CXCL11 [59]. |
| D112, D278 | Relevant in the interactions of the three chemokines, but only CXL10 and CXCL11 have an influence on the activity [59]. |
| D282, E293 | Influence on the activity of CXCL9 and CXCL10 and the interaction of CXCL11 [59]. |



























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}