
Virtual Screening of Natural Compounds as Potential PI3K-AKT1 Signaling Pathway Inhibitors and Experimental Validation




Abstract
:1. Introduction
2. Results
2.1. Computational Analysis
2.2. Experimental Validation
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Culture and Treatment
4.3. Immunoblotting
4.4. Computational Procedure
- (a)
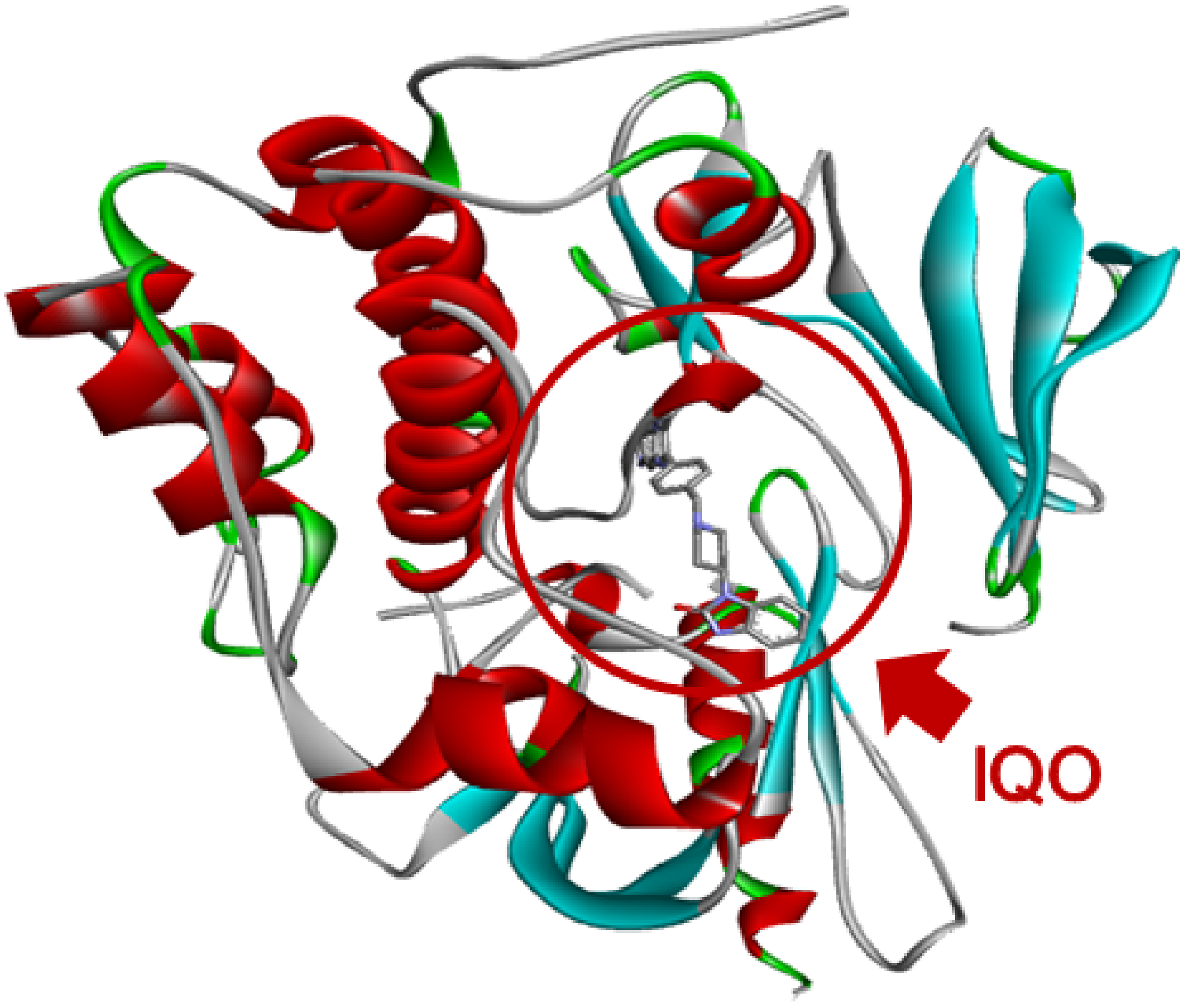
- Selection of the structural model of AKT1. Search for a suitable structural model of AKT1 has been performed in PDB Protein Data Bank [34], looking for structures of AKT1 complexed with an inhibitor [35]. Among the available structures, by excluding those with mutations and limited portions of the protein, the most suitable resulted the PDB file with code 3O96, as the model crystal structure characterized by AKT1 protein linked to the interface to its selective allosteric inhibitor known as IQO (Inhibitor VIII of AKT1/2) [29]. Visual analysis of the 3D structure has been performed by means of Discovery Studio 4.5 (Biovia, San Diego, CA, USA). Analysis of the amino acids interacting with IQO has been performed by DiscoveryStudio and Ligplus [36].
- (b)–(c)
- Pharmacophore modeling and virtual computational screening; the pharmacophore models have been obtained by integrating PHARMIT (http://pharmit.csb.pitt.edu) [37] and Discovery Studio 4.5 tools. ZINC12 database [38] has been searched for compounds of natural and bioactive origin and using Pharmer [39]. The molecular weight has been restricted to the range from 200 to 800 g/mol, RMSD to the range from 0.300 to 0.900 Å, RBnds (rotational angle) not exceeding 15.
- Functional groups on which to implement pharmacophoric models (bonds that act as hydrogen acceptors/donors, bonds for positive/negative ions, hydrophobic bonds and presence of aromatic rings). A maximum of 10 hypotheses (or pharmacophore models) are generated for each run. The HypoGen algorithm used develops models with different pharmacophore features. The hypotheses generated are analyzed in terms of their correlation coefficients and the cost function values.
- Application of automatic parameter minimization based on Best/Fast rigid conformation characteristics. So, the FAST conformation generation method searches conformations only in the torsion space and takes less time. While, the BEST method provides a complete and improved coverage of conformational space by performing a rigorous energy minimization and optimizing the conformations in both torsional and Cartesian space using the Poling algorithm to assess the quality of pharmacophore hypotheses.
- Chemical–physical characteristics of the compounds were selected to perform the subsequent steps of work.
- Validation of the pharmacophore model: the pharmacophore models selected based on the acceptable correlation coefficient (R) and cost analysis should be validated in three subsequent steps: Fischer’s randomization test, test set prediction and the Güner–Henry (GH) scoring method. The method involves evaluation of the following: the percent yield of actives in a database (%Y, recall), the percent ratio of actives in the hit list (%A, precision), the enrichment factor E and the GH score. The GH score ranges from 0 to 1, where a value of 1 signifies the ideal model [40].
- (d)
- After selecting a number of good candidates, blind and focused-rigid direct molecular docking has been performed by using AutoDock tools 4.2 [41], evaluating all the molecular interactions that may exist between the ligand and our AKT1 protein, taking into account the lower binding energy values to identify the most stable complex, and the estimated inhibition constant (Ki), to estimate which compounds are able to inhibit more AKT1 at lower experimental concentrations. Docking procedure has been applied according to protocols in use in our laboratory and described in previous articles [42,43,44]. Redocking procedure has been applied to evaluate the binding energy value of the IQO inhibitor as described in previous studies of our group [44].
- The number of pharmacophore features must be included into the range between 4–6, taking into account that no pharmacophore has more than six functional groups present at the same time [45]). A pharmacophore model consisting of too many chemical features (e.g., more than six or seven) is not appropriate for practical applications. Therefore, it is always important to pick a restricted number of chemical features (usually four to seven) to create a reliable pharmacophore hypothesis. One more significant drawback is that the obtained pharmacophore hypothesis cannot replicate the quantitative structure–activity relationship (QSAR) because the model is generated based just on a single macromolecule–ligand complex or a single macromolecule.
- The selectivity score is a selectivity parameter of a ligand for a specific target to evaluate the quality of the pharmacophore models. There is no maximum limit (the higher the better).
- The method of generating the conformation of pharmacophores (FAST/BEST because they give us in a short time what are considered to be the best pharmacophores for the applied method) is used to then perform other internal screening and select only good candidates. The choice of these parameters is essential, to designate the subsequent steps of biological-molecular evaluation, taking only the best ones from the pharmacophores.
- (e)
- The selected best candidates are then analyzed on a chemical–physical level using: Chemical vendors in Pubchem Compound [46] to identify the specifications of each single compound; SciFinder (scifinder.cas.org) to evaluate the presence or absence of preliminary tests already conducted on these compounds and what is already known in the literature on such compounds; FooDB/HMDB [47] to investigate food properties if there are ones and the origins of such chemical compounds and Chemicalize (chemaxon.com) to calculate the most important chemical parameters related to the stability of the compound. Afterwards, all the information collected were used to trace the origin of the compounds analyzed, underlining the most important features such as:
- Solubility in organic solvents such as DMSO, ethanol, methanol and in inorganic solvents such as water;
- LogD, pKa and the chemical stability of compounds.
- (f)
- Molecular–biological evaluation of the selected compounds with the realization of the pharmacokinetic/pharmacodynamics (PK/PD) models. To select the “hit” compounds by virtual screening, it has been necessary to understand the features of pocket involved in ligand–protein interactions and to underline the amino acids involved in molecular interactions, trying to identify the compounds that can be considered good lead compounds. This step plays a critical role for the choice of the best candidates to calculate the physical–chemical properties and create PK/PD models, for characterizing the good lead compounds for next experimental assays.
- (g)
- All this information are essential to realize the ADMET profile through ADMET/Toxicity predictor server (implemented in Discovery Studio), allowing the application of the Lipinski-Veber five rule, on which the bioavailability and the specific ADMET profile of each individual compound is calculated, and applying the TOPKAT software (implemented in Discovery Studio), useful for identifying and evaluating the toxicity profile of each compound in different conditions and systems.
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Staal, S.P. Molecular cloning of the akt oncogene and its human homologues AKT1 and AKT2: Amplification of AKT1 in a primary human gastric adenocarcinoma. Proc. Natl. Acad. Sci. USA 1987, 84, 5034–5037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellacosa, A.; Testa, J.R.; Staal, S.P.; Tsichlis, P.N. A retroviral oncogene, akt, encoding a serine-threonine kinase containing an SH2-like region. Science 1991, 254, 274–277. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.F.; Jakubowicz, T.; Pitossi, F.J.; Maurer, F.; Hemmings, B.A. Molecular cloning and identification of a serine/threonine protein kinase of the second-messenger subfamily. Proc. Natl. Acad. Sci. USA 1991, 88, 4171–4175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, J.; Weiskirchen, R. What Does the “AKT” Stand for in the Name “AKT Kinase”? Some Historical Comments. Front. Oncol. 2020, 10, 1329. [Google Scholar] [CrossRef] [PubMed]
- Hers, I.; Vincent, E.E.; Tavare, J.M. Akt signalling in health and disease. Cell. Signal. 2011, 23, 1515–1527. [Google Scholar] [CrossRef]
- Hoxhaj, G.; Manning, B.D. The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nat. Rev. Cancer 2020, 20, 74–88. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Bode, A.M.; Dong, Z.; Lee, M.H. AKT as a Therapeutic Target for Cancer. Cancer Res. 2019, 79, 1019–1031. [Google Scholar] [CrossRef] [Green Version]
- Song, M.; Liu, X.; Liu, K.; Zhao, R.; Huang, H.; Shi, Y.; Zhang, M.; Zhou, S.; Xie, H.; Chen, H.; et al. Targeting AKT with Oridonin Inhibits Growth of Esophageal Squamous Cell Carcinoma In Vitro and Patient-Derived Xenografts In Vivo. Mol. Cancer Ther. 2018, 17, 1540–1553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Liu, H.; Liu, J. Akt activation: A potential strategy to ameliorate insulin resistance. Diabetes Res. Clin. Pract. 2019, 156, 107092. [Google Scholar] [CrossRef]
- Toker, A.; Marmiroli, S. Signaling specificity in the Akt pathway in biology and disease. Adv. Biol. Regul. 2014, 55, 28–38. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Chen, X.; Hay, N. Akt as a target for cancer therapy: More is not always better (lessons from studies in mice). Br. J. Cancer 2017, 117, 159–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellacosa, A.; Chan, T.O.; Ahmed, N.N.; Datta, K.; Malstrom, S.; Stokoe, D.; McCormick, F.; Feng, J.; Tsichlis, P. Akt activation by growth factors is a multiple-step process: The role of the PH domain. Oncogene 1998, 17, 313–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manning, B.D.; Cantley, L.C. AKT/PKB signaling: Navigating downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef] [Green Version]
- Link, W. Introduction to FOXO Biology. Methods Mol. Biol. 2019, 1890, 1–9. [Google Scholar]
- Herman, S.E.; Gordon, A.L.; Wagner, A.J.; Heerema, N.A.; Zhao, W.; Flynn, J.M.; Jones, J.; Andritsos, L.; Puri, K.D.; Lannutti, B.J.; et al. Phosphatidylinositol 3-kinase-delta inhibitor CAL-101 shows promising preclinical activity in chronic lymphocytic leukemia by antagonizing intrinsic and extrinsic cellular survival signals. Blood 2010, 116, 2078–2088. [Google Scholar] [CrossRef] [Green Version]
- Zirlik, K.; Veelken, H. Idelalisib. Recent Results Cancer Res. 2018, 212, 243–264. [Google Scholar]
- Tewari, D.; Patni, P.; Bishayee, A.; Sah, A.N.; Bishayee, A. Natural products targeting the PI3K-Akt-mTOR signaling pathway in cancer: A novel therapeutic strategy. Semin. Cancer Biol. 2019, in press. [Google Scholar] [CrossRef]
- Russo, M.; Spagnuolo, C.; Volpe, S.; Tedesco, I.; Bilotto, S.; Russo, G.L. ABT-737 resistance in B-cells isolated from chronic lymphocytic leukemia patients and leukemia cell lines is overcome by the pleiotropic kinase inhibitor quercetin through Mcl-1 down-regulation. Biochem. Pharmacol. 2013, 85, 927–936. [Google Scholar] [CrossRef]
- Russo, M.; Milito, A.; Spagnuolo, C.; Carbone, V.; Rosen, A.; Minasi, P.; Lauria, F.; Russo, G.L. CK2 and PI3K are direct molecular targets of quercetin in chronic lymphocytic leukaemia. Oncotarget 2017, 8, 42571–42587. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.O.; Lee, M.H.; Oi, N.; Kim, S.H.; Bae, K.B.; Huang, Z.; Kim, D.J.; Reddy, K.; Lee, S.Y.; Park, S.J.; et al. (6)-shogaol inhibits growth and induces apoptosis of non-small cell lung cancer cells by directly regulating Akt1/2. Carcinogenesis 2014, 35, 683–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.J.; Lee, M.H.; Liu, K.; Lim, D.Y.; Roh, E.; Chen, H.; Kim, S.H.; Shim, J.H.; Kim, M.O.; Li, W.; et al. Herbacetin suppresses cutaneous squamous cell carcinoma and melanoma cell growth by targeting AKT and ODC. Carcinogenesis 2017, 38, 1136–1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seidel, T.; Schuetz, D.A.; Garon, A.; Langer, T. The Pharmacophore Concept and Its Applications in Computer-Aided Drug Design. Prog. Chem. Org. Nat. Prod. 2019, 110, 99–141. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, L.G.; Dos Santos, R.N.; Oliva, G.; Andricopulo, A.D. Molecular docking and structure-based drug design strategies. Molecules 2015, 20, 13384–13421. [Google Scholar] [CrossRef] [PubMed]
- Alzahrani, A.S. PI3K/Akt/mTOR inhibitors in cancer: At the bench and bedside. Semin. Cancer Biol. 2019, 59, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.S.; Banerji, U. Maximising the potential of AKT inhibitors as anti-cancer treatments. Pharmacol. Ther. 2017, 172, 101–115. [Google Scholar] [CrossRef]
- Longo, P.G.; Laurenti, L.; Gobessi, S.; Sica, S.; Leone, G.; Efremov, D.G. The Akt/Mcl-1 pathway plays a prominent role in mediating antiapoptotic signals downstream of the B-cell receptor in chronic lymphocytic leukemia B cells. Blood 2008, 111, 846–855. [Google Scholar] [CrossRef]
- Lindsley, C.W.; Zhao, Z.; Leister, W.H.; Robinson, R.G.; Barnett, S.F.; Defeo-Jones, D.; Jones, R.E.; Hartman, G.D.; Huff, J.R.; Huber, H.E.; et al. Allosteric Akt (PKB) inhibitors: Discovery and SAR of isozyme selective inhibitors. Bioorganic Med. Chem. Lett. 2005, 15, 761–764. [Google Scholar] [CrossRef]
- Wu, W.I.; Voegtli, W.C.; Sturgis, H.L.; Dizon, F.P.; Vigers, G.P.; Brandhuber, B.J. Crystal structure of human AKT1 with an allosteric inhibitor reveals a new mode of kinase inhibition. PLoS ONE 2010, 5, e12913. [Google Scholar] [CrossRef]
- Rosen, A.; Bergh, A.C.; Gogok, P.; Evaldsson, C.; Myhrinder, A.L.; Hellqvist, E.; Rasul, A.; Bjorkholm, M.; Jansson, M.; Mansouri, L.; et al. Lymphoblastoid cell line with B1 cell characteristics established from a chronic lymphocytic leukemia clone by in vitro EBV infection. Oncoimmunology 2012, 1, 18–27. [Google Scholar] [CrossRef] [Green Version]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Dubey, A.; Facchiano, A.; Ramteke, P.W.; Marabotti, A. In silico approach to find chymase inhibitors among biogenic compounds. Future Med. Chem. 2016, 8, 841–851. [Google Scholar] [CrossRef] [PubMed]
- Dubey, A.; Dotolo, S.; Ramteke, P.W.; Facchiano, A.; Marabotti, A. Searching for Chymase Inhibitors among Chamomile Compounds Using a Computational-Based Approach. Biomolecules 2018, 9, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fei, J.; Zhou, L.; Liu, T.; Tang, X.Y. Pharmacophore modeling, virtual screening, and molecular docking studies for discovery of novel Akt2 inhibitors. Int. J. Med. Sci. 2013, 10, 265–275. [Google Scholar] [CrossRef] [Green Version]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Modeling 2011, 51, 2778–2786. [Google Scholar] [CrossRef]
- Sunseri, J.; Koes, D.R. Pharmit: Interactive exploration of chemical space. Nucleic Acids Res. 2016, 44, W442–W448. [Google Scholar] [CrossRef] [Green Version]
- Irwin, J.J.; Shoichet, B.K. ZINC—A free database of commercially available compounds for virtual screening. J. Chem. Inf. Modeling 2005, 45, 177–182. [Google Scholar] [CrossRef] [Green Version]
- Koes, D.R.; Camacho, C.J. Pharmer: Efficient and exact pharmacophore search. J. Chem. Inf. Modeling 2011, 51, 1307–1314. [Google Scholar] [CrossRef] [Green Version]
- Gupta, N.; Sitwala, N.; Patel, K. Pharmacophore modelling, validation, 3D virtual screening, docking, design and in silico ADMET simulation study of histone deacetylase class-1 inhibitors. Med. Chem. Res. 2014, 23, 4853–4864. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Olson, A.J. Using AutoDock for ligand-receptor docking. Curr. Protoc. Bioinform. 2008, 24. [Google Scholar] [CrossRef] [PubMed]
- Scafuri, B.; Marabotti, A.; Carbone, V.; Minasi, P.; Dotolo, S.; Facchiano, A. A theoretical study on predicted protein targets of apple polyphenols and possible mechanisms of chemoprevention in colorectal cancer. Sci. Rep. 2016, 6, 32516. [Google Scholar] [CrossRef] [PubMed]
- Scafuri, B.; Varriale, A.; Facchiano, A.; D’Auria, S.; Raggi, M.E.; Marabotti, A. Binding of mycotoxins to proteins involved in neuronal plasticity: A combined in silico/wet investigation. Sci. Rep. 2017, 7, 15156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scafuri, B.; Bontempo, P.; Altucci, L.; De Masi, L.; Facchiano, A. Molecular Docking Simulations on Histone Deacetylases (HDAC)-1 and -2 to Investigate the Flavone Binding. Biomedicines 2020, 8, 568. [Google Scholar] [CrossRef]
- Qing, X.; Lee, X.Y.; De Raeymaecker, J.; Tame, J.; Zhang, K.; De Maeyer, M.; Voet, A. Pharmacophore modeling: Advances, limitations, and current utility in drug discovery. J. Recept. Ligand Channel Res. 2014, 7, 81–92. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A.; et al. PubChem Substance and Compound databases. Nucleic Acids Res. 2016, 44, D1202–D1213. [Google Scholar] [CrossRef]
- Wishart, D.S.; Tzur, D.; Knox, C.; Eisner, R.; Guo, A.C.; Young, N.; Cheng, D.; Jewell, K.; Arndt, D.; Sawhney, S.; et al. HMDB: The Human Metabolome Database. Nucleic Acids Res. 2007, 35, D521–D526. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Binding Energy (Kcal/mol) | Ki |
|---|---|---|
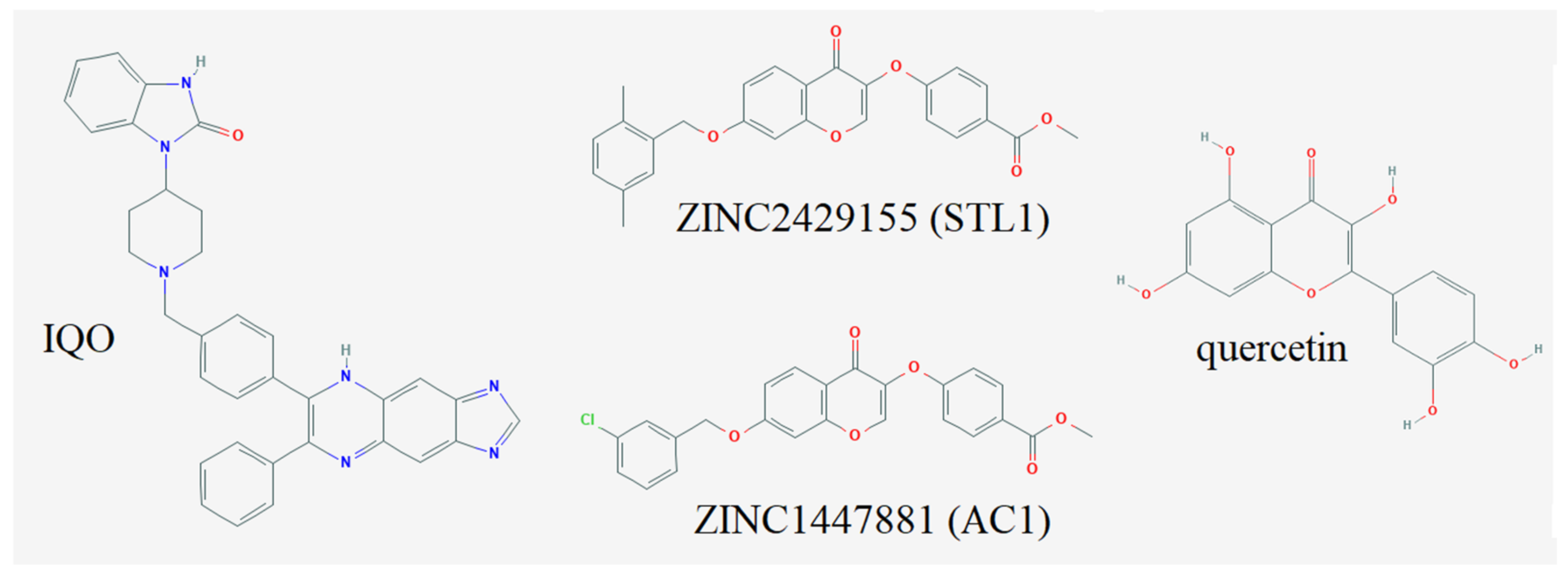
| IQO (iAKT-S) | −12.56 | 635.40 pM |
| ZINC4259855 | −11.00 | 737.43 µM |
| ZINC2161363 | −10.11 | 1.41 µM |
| ZINC2429155 (STL1) | −10.00 | 2.21 µM |
| ZINC1237912 | −10.00 | 1.12 µM |
| ZINC1447881 (AC1) | −9.36 | 1.36 µM |
| ZINC13691379 | −9.30 | 1.13 µM |
| ZINC02154548 | −9.16 | 1.04 µM |
| ZINC03851635 | −9.00 | 99.14 µM |
| ZINC02666313 | −8.91 | 1.60 µM |
| ZINC14611917 | −8.73 | 2.16 µM |
| ZINC54307082 | −8.72 | 2.20 µM |
| Quercetin | −6.55 | 15.85 µM |
| Biodegradability | Test Ames: mutagen | Toxicity | FDA Mouse Female: Carginogen | FDA Mouse Male: Carginogen | RAT Female FDA: Carcinogen | RAT Male FDA: Carcinogen | Skin Irritancy | Skin Sensitizer | Predicted LD50 (mg/Kg) | Prediction Accuracy (%) | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Compound | |||||||||||
| ZINC4259855 | no | no | no | no | yes | no | yes | mild | yes | 100 | 67.4 |
| ZINC2161363 | yes | no | no | no | no | no | no | m/s a | yes | 100 | 67.4 |
| ZINC2429155 | no | no | yes | no | no | no | no | mild | yes | 500 | 68.1 |
| ZINC1237912 | yes | no | no | no | no | no | no | m/s a | yes | 4400 | 54.3 |
| ZINC1447881 | no | no | no | no | no | no | no | mild | yes | 500 | 68.1 |
| ZINC13691379 | no | no | yes | no | yes | yes | no | mild | yes | 1000 | 67.4 |
| ZINC02154548 | yes | no | no | no | yes | yes | no | m/s a | yes | 600 | 54.3 |
| ZINC3869685 | no | yes | no | no | no | no | no | mild | yes | 159 | 100.0 |
| ZINC02666313 | no | no | yes | no | no | no | no | mild | yes | 100 | 67.4 |
| ZINC14611917 | yes | no | no | no | yes | yes | yes | m/s a | yes | 500 | 68.1 |
| ZINC54307082 | no | no | yes | no | no | no | no | m/s a | yes | 2875 | 68.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dotolo, S.; Cervellera, C.; Russo, M.; Russo, G.L.; Facchiano, A. Virtual Screening of Natural Compounds as Potential PI3K-AKT1 Signaling Pathway Inhibitors and Experimental Validation. Molecules 2021, 26, 492. https://doi.org/10.3390/molecules26020492
Dotolo S, Cervellera C, Russo M, Russo GL, Facchiano A. Virtual Screening of Natural Compounds as Potential PI3K-AKT1 Signaling Pathway Inhibitors and Experimental Validation. Molecules. 2021; 26(2):492. https://doi.org/10.3390/molecules26020492
Chicago/Turabian StyleDotolo, Serena, Carmen Cervellera, Maria Russo, Gian Luigi Russo, and Angelo Facchiano. 2021. "Virtual Screening of Natural Compounds as Potential PI3K-AKT1 Signaling Pathway Inhibitors and Experimental Validation" Molecules 26, no. 2: 492. https://doi.org/10.3390/molecules26020492
APA StyleDotolo, S., Cervellera, C., Russo, M., Russo, G. L., & Facchiano, A. (2021). Virtual Screening of Natural Compounds as Potential PI3K-AKT1 Signaling Pathway Inhibitors and Experimental Validation. Molecules, 26(2), 492. https://doi.org/10.3390/molecules26020492