
Design and Synthesis of Pyrrolidinyl Ferrocene-Containing Ligands and Their Application in Highly Enantioselective Rhodium-Catalyzed Olefin Hydrogenation

Abstract
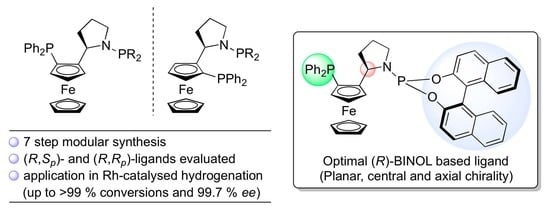
:
1. Introduction
2. Results and Discussion
2.1. Synthesis and Characterization of N-Phosphorus-Substituted Pyrrolidine Based Ligands
2.1.1. Rhodium-Catalyzed Asymmetric Hydrogenation of Dehydroamino Acid Esters
Reaction Condition Optimization
Substrate Scope
2.1.2. Rhodium-Catalyzed Asymmetric Hydrogenation of α-Aryl Enamides
Reaction Condition Optimization
Substrate Scope
3. Materials and Methods—Chemistry
3.1. 4-Chloro-Ferrocenylbutanone (10)
3.2. (R)-4-Chloro-1-Ferrocenylbutanol ((R)-11)
3.3. (R)-4-Chloro-2-Acetoxy-1-Ferrocenylbutane ((R)-12)
3.4. (R)-N-Allyl-Pyrrolidin-2′-ylferrocene ((R)-13)
3.5. 2-[(2R)-N-Allyl-Pyrrolidin-2′-yl]-(1S)-Diphenylphosphineferrocene (14) and 2-[(2R)-N-Allyl-Pyrrolidin-2′-yl]-(1R)-Diphenylphosphineferrocene (14)
3.5.1. Spectroscopic Analysis of (R,Sp)-14
3.5.2. Spectroscopic Analysis of (R,Rp)-14
3.6. 2-[(2R)-N-H-Pyrrolidin-2′-yl]-(1S)-Diphenylphosphineferrocene ((R,Sp)-15)
Spectroscopic Analysis of (R,Sp)-15)
3.7. 2-[(2R)-N-H-Pyrrolidin-2′-yl]-(1R)-Diphenylphosphineferrocene)-(R,Rp)-15)
3.7.1. Spectroscopic Analysis of (R,Rp)-15
3.7.2. Typical Procedure A: Phosphine-Coupling
3.8. 2-[(2R)-N-Diphenylphosphine-Pyrrolidin-2′-yl]-(1S)-Diphenylphosphineferrocene (L1)
Spectroscopic Analysis of L1
3.9. 2-[(2R)-N-Bis(2-Methylphenyl)phosphine-Pyrrolidin-2′-yl]-(1S)-Diphenylphosphineferrocene (L2)
Spectroscopic Analysis of L2
3.10. 2-[(2R)-N-Bis(4-fluorophenyl)phosphine-Pyrrolidin-2′-yl]-(1S)-Diphenylphosphineferrocene (L3)
Spectroscopic Analysis of L3
3.11. 2-[(2R)-N-Bis(3,5-Di-Trifluoromethylphenyl)phosphine-Pyrrolidin-2′-yl]-(1S)-Diphenylphosphineferrocene (L4)
Spectroscopic Analysis of L4
3.12. 2-[(2R)-N-(R)-1,1′-Binaphthyl-2,2′-Diylphosphoro-Pyrrolidin-2′-yl]-(1S)-Diphenylphosphineferrocene (L5)
Spectroscopic Analysis of L5
3.13. 2-[(2R)-N-(S)-1,1′-Binaphthyl-2,2′-Diylphosphoro-Pyrrolidin-2′-yl]-(1S)-Diphenylphosphineferrocene (L6)
Spectroscopic Analysis of L6
3.14. 2-[(2R)-N-(R)-1,1′-Binaphthyl-2,2′-Diylphosphoro-Pyrrolidin-2′-yl]-(1R)-Diphenylphosphineferrocene (L7)
Spectroscopic Analysis of L7
3.15. 2-[(2R)-N-(S)-1,1′-Binaphthyl-2,2′-Diylphosphoro-Pyrrolidin-2′-yl]-(1R)-Diphenylphosphineferrocene (L8)
3.15.1. Spectroscopic Analysis of L8
3.15.2. Rhodium-Catalyzed Asymmetric Hydrogenation
Preparation of Substrates/Characterization Data for Substrates and Products
3.15.3. Rhodium-Catalyzed Asymmetric Hydrogenation of Dehydroamino Acid Esters
Typical Procedure B: Optimization and Substrate Scope
3.16. (S)-Methyl 2-Acetamido-3-Phenylpropanoate (17a)
3.17. (S)-Methyl 2-Acetamido-3-(4-Methoxyphenyl)propanoate (17b)
3.18. (S)-Methyl 2-Acetamido-3-(p-Tolyl)propanoate (17c)
3.19. (S)-Methyl 2-Acetamido-3-(4-Chlorophenyl)propanoate (17d)
3.20. (S)-Methyl 2-Acetamido-3-(4-Fluorophenyl)propanoate (17e)
3.21. (S)-Methyl 2-Acetamido-3-(4-Nitrophenyl)propanoate (17f)
3.22. (S)-Methyl 2-Acetamido-3-(3-Chlorophenyl)propanoate (17g)
3.23. (S)-Methyl 2-Acetamido-3-(3-Bromophenyl)propanoate (17h)
3.24. (S)-Methyl 2-Acetamido-3-(2-Chlorophenyl)propanoate (17i)
3.25. (S)-Methyl 2-Acetamido-3-(Naphthalen-1-yl)propanoate (17j)
3.26. (S)-Methyl 2-Acetamidopropanoate (17k)
3.26.1. Rhodium-Catalyzed Asymmetric Hydrogenation of α-Aryl Enamides
Typical Procedure C: Optimization and Substrate Scope
3.27. (S)-N-(1-Phenylethyl)acetamide (19a)
3.28. (S)-N-(1-(Naphthalen-2-yl)ethyl)acetamide (19b)
3.29. (S)-N-(1-(4-Chlorophenyl)ethyl)acetamide (19c)
3.30. (S)-N-(1-(4-Methoxyphenyl)ethyl)acetamide (19d)
3.31. (S)-N-(1,2,3,4-Tetrahydronaphthalen-1-yl)acetamide (19e)
3.32. (S)-Ethyl 3-Acetamido-3-Phenylpropanoate (19f)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Jacobsen, E.N.; Pfaltz, A.; Yamamoto, H. (Eds.) Comprehensive Asymmetric Catalyses; Springer: Berlin, Heilderberg, Germany, 1999. [Google Scholar]
- Ojima, I. (Ed.) Catalytic Asymmetric Synthesis, 3rd ed.; Wiley & Sons: New York, NY, USA, 2010. [Google Scholar]
- Lin, G.-Q.; Li, Y.-M.; Chan, A.S.C. Principles and Applications of Asymmetric Synthesis; Wiley & Sons: New York, NY, USA, 2001. [Google Scholar]
- Yoon, T.P.; Jacobsen, E.N. Privileged Chiral Catalysts. Science 2003, 299, 1691–1693. [Google Scholar] [CrossRef] [PubMed]
- Togni, A.; Breutel, C.; Schnyder, A.; Spindler, F.; Landert, H.; Tijani, A. A Novel Easily Accessible Chiral Ferrocenyldiphosphine for Highly Enantioselective Hydrogenation, Allylic Alkylation, and Hydroboration Reactions. J. Am. Chem. Soc. 1994, 116, 4062–4066. [Google Scholar] [CrossRef]
- Tang, W.; Zhang, X. New Chiral Phosphorus Ligands for Enantioselective Hydrogenation. Chem. Rev. 2003, 103, 3029–3070. [Google Scholar] [CrossRef] [PubMed]
- Blaser, H. The Chiral Switch of (S)-Metolachlor: A Personal Account of an Industrial Odyssey in Asymmetric Catalysis. Adv. Synth. Catal. 2002, 344, 17–31. [Google Scholar] [CrossRef]
- Knochel, P.; Polborn, K.; Lotz, M. New Ferrocenyl Ligands with Broad Applications in Asymmetric Catalysis. Angew. Chem. Int. Ed. 2002, 41, 4708–4711. [Google Scholar] [CrossRef]
- Cunningham, L.; Benson, A.; Guiry, P.J. Recent developments in the synthesis and applications of chiral ferrocene ligands and organocatalysts in asymmetric catalysis. Org. Biomol. Chem. 2020, 18, 9329–9370. [Google Scholar] [CrossRef]
- Farrell, A.; Goddard, R.; Guiry, P.J. The preparation of ferrocene-containing phosphinamine ligands possessing central and planar chirality and their application in palladium-catalysed asymmetric allylic alkylation. J. Org. Chem. 2002, 67, 4209–4217. [Google Scholar] [CrossRef]
- Cahill, J.P.; Guiry, P.J. The Application of Pd-Complexes of trans-2,5-Dialkylpyrrolidinylbenzyldiphenyl phosphines to Enantioselective Allylic Alkylation. Tetrahedron Asymmetry 1998, 9, 4301–4306. [Google Scholar] [CrossRef]
- Cahill, J.P.; Bohnen, F.; Goddard, R.; Krüger, C.; Guiry, P.J. The Preparation of trans-2,5-Dialkylpyrrolidinylbenzyldiphenylphosphines: New Phosphinamine Ligands for Asymmetric Catalysis. Tetrahedron Asymmetry 1998, 9, 3831–3839. [Google Scholar] [CrossRef]
- Cahill, J.P.; Cunneen, D.; Guiry, P.J. trans-2,5-Dialkylpyrrolidinyl-containing Phosphinamines; Synthetic and Mechanistic Studies in Pd-Catalysed Asymmetric Allylic Alkylation. Tetrahedron Asymmetry 1999, 10, 4157–4173. [Google Scholar] [CrossRef]
- Ahern, T.; Müller-Bunz, H.; Guiry, P.J. The Synthesis of N,O-Ferrocenyl Pyrrolidine-Containing Ligands And Their Application in Diethyl- and Diphenylzinc Addition to Aromatic Aldehydes. J. Org. Chem. 2006, 71, 7596–7602. [Google Scholar] [CrossRef] [PubMed]
- Meaney, K.; Goddard, R.; Bronger, R.P.J.; Guiry, P.J. The preparation of ferrocene-containing phosphinamine ligands possessing central and planar chirality and their application in palladium-catalysed allylic substitution. Tetrahedron 2021, 90, 132088. [Google Scholar] [CrossRef]
- Boaz, N.W.; Debenham, S.D.; MacKenzie, E.B.; Large, S.E. Phosphinoferrocenylaminophosphines as Novel and Practical Ligands for Asymmetric Catalysis. Org. Lett. 2002, 4, 2421–2424. [Google Scholar] [CrossRef] [PubMed]
- Boaz, N.W.; MacKenzie, E.B.; Debenham, S.D.; Large, S.E.; Ponasik, J.A. Synthesis and Application of Phosphinoferrocenylaminophosphine Ligands for Asymmetric Catalysis. J. Org. Chem. 2005, 70, 1872–1880. [Google Scholar] [CrossRef]
- Krabbe, S.W.; Hatcher, M.A.; Bowman, R.K.; Mitchell, M.B.; McClure, M.S.; Johnson, J.S. Copper-Catalyzed Asymmetric Hydrogenation of Aryl and Heteroaryl Ketones. Org. Lett. 2013, 15, 4560–4563. [Google Scholar] [CrossRef]
- Deng, J.; Hu, X.-P.; Huang, J.-D.; Yu, S.-B.; Wang, D.-Y.; Duan, Z.-C.; Zheng, Z. Enantioselective Synthesis of β2-Amino Acids via Rh-Catalyzed Asymmetric Hydrogenation with BoPhoz-Type Ligands: Important Influence of an N−H Proton in the Ligand on the Enantioselectivity. J. Org. Chem. 2008, 73, 2015–2017. [Google Scholar] [CrossRef]
- Duan, Z.-C.; Hu, X.-P.; Zhang, C.; Zheng, Z. Enantioselective Rh-Catalyzed Hydrogenation of 3-Aryl-4-phosphonobutenoates with a P-Stereogenic BoPhoz-Type Ligand. J. Org. Chem. 2010, 75, 8319–8321. [Google Scholar] [CrossRef]
- Pan, C.; Gu, Z.; Zhang, M.; Zhu, Z. Palladium-Catalyzed Enantioselective Synthesis of 2-Aryl Cyclohex-2-enone Atropisomers: Platform Molecules for the Divergent Synthesis of Axially Chiral Biaryl Compounds. Angew. Chem. Int. Ed. 2017, 56, 4777–4781. [Google Scholar] [CrossRef]
- Hu, X.-P.; Zheng, Z. Unsymmetrical Hybrid Ferrocene-Based Phosphine-Phosphoramidites: A New Class of Practical Ligands for Rh-Catalyzed Asymmetric Hydrogenation. Org. Lett. 2004, 6, 3585–3588. [Google Scholar] [CrossRef]
- Jia, X.; Li, X.; Lam, W.S.; Kok, S.H.L.; Xu, L.; Lu, G.; Yeung, C.-H.; Chan, A.S.C. The synthesis of new chiral phosphine–phosphinites, phosphine–phosphoramidite, and phosphine–phosphite ligands and their applications in asymmetric hydrogenation. Tetrahedron Asymmetry 2004, 15, 2273–2278. [Google Scholar] [CrossRef]
- Hu, X.-P.; Zheng, Z. Practical Rh(I)-Catalyzed Asymmetric Hydrogenation of β-(Acylamino)acrylates Using a New Unsymmetrical Hybrid Ferrocenylphosphine−Phosphoramidite Ligand: Crucial Influence of an N−H Proton in the Ligand. Org. Lett. 2005, 7, 419–422. [Google Scholar] [CrossRef] [PubMed]
- Garro-Helion, F.; Merzouk, A.; Guibe, F. Mild and Selective Palladium(0)-Catalyzed Deallylation of Allylic Amines. Allylamine and Diallylamine as Very Convenient Ammonia Equivalents for the Synthesis of Primary Amines. J. Org. Chem. 1993, 58, 6109–6113. [Google Scholar] [CrossRef]
- Reetz, M.T.; Gosberg, A.; Goddard, R.; Kyung, S.-H. Diphosphonites as highly efficient ligands for enantioselective rhodium-catalyzed hydrogenation. Chem. Commun. 1998, 2077–2078. [Google Scholar] [CrossRef]
- Ouyang, G.; Xiang, J.; Li, Y.; He, Y.; Fan, Q. Cation-Triggered Switchable Asymmetric Catalysis with Chiral Aza-CrownPho. Angew. Chem. Int. Ed. 2015, 54, 4334–4337. [Google Scholar] [CrossRef] [PubMed]
- Meetsma, A.; de Vries, J.G.; Giacomina, F.; de Vries, A.H.M.; Lefort, L.; Panella, L. High Enantioselectivity Is Induced by a Single Monodentate Phosphoramidite Ligand in Iridium-Catalyzed Asymmetric Hydrogenation. Angew. Chem. Int. Ed. 2007, 46, 1497–1500. [Google Scholar] [CrossRef]
- Doskocz, M.; Malinowska, B.; Młynarz, P.; Lejczak, B.; Kafarski, P. Long range phosphorus–phosphorus coupling constants in bis(phosphorylhydroxymethyl)benzene derivatives. Tetrahedron Lett. 2010, 51, 3406–3411. [Google Scholar] [CrossRef]
- Hierso, J.-C.; Fihri, A.; Ivanov, V.V.; Hanquet, B.; Pirio, N.; Donnadieu, B.; Rebière, B.; Amardeil, R.; Meunier, P. “Through-Space” Nuclear Spin−Spin JPP Coupling in Tetraphosphine Ferrocenyl Derivatives: A 31P NMR and X-ray Structure Correlation Study for Coordination Complexes. J. Am. Chem. Soc. 2004, 126, 11077–11087. [Google Scholar] [CrossRef]
- Dai, L.-X.; Tu, T.; You, S.-L.; Deng, W.-P.; Hou, X.-L. Asymmetric Catalysis with Chiral Ferrocene Ligands. Acc. Chem. Res. 2003, 36, 659–667. [Google Scholar] [CrossRef]
- Vineyard, B.D.; Knowles, W.S.; Sabacky, M.J. Catalytic Asymmetric Hydrogenation. J. Chem. Soc. Chem. Commun. 1972, 10–11. [Google Scholar] [CrossRef]
- Vineyard, B.D.; Knowles, W.S.; Sabacky, M.J.; Bachman, G.L.; Weinkauff, D.J. Asymmetric Hydrogenation. Rhodium Chiral Bisphosphine Catalyst. J. Am. Chem. Soc. 1977, 99, 5946. [Google Scholar] [CrossRef]
- Kagan, H.; Dang, T.-P. Asymmetric Catalytic Reduction with Transition Metal Complexes. I. A Catalytic System of Rh (I) with (−)-2,3-O−Isopropylidene-2,3-dihydroxy-1,4-bis(diphenylphosphino)butane, a New Chiral Diphosphine. J. Am. Chem. Soc. 1972, 94, 6429. [Google Scholar] [CrossRef]
- Burk, M.J. C2-Symmetric Bis(phospholanes) and Their Use in Highly Enantioselective Hydrogenation Reactions. J. Am. Chem. Soc. 1991, 113, 8518. [Google Scholar] [CrossRef]
- Burk, M.J.; Wang, Y.M.; Lee, J.R. A Convenient Asymmetric Synthesis of α-1-Arylalkylamines Through the Enantioselective Hydrogenation of Enamides. J. Am. Chem. Soc. 1996, 118, 5142. [Google Scholar] [CrossRef]
- Mohara, B.; Stephan, M. Practical Enantioselective Hydrogenation of α-Aryl- and α-Carboxyamidoethylenes by Rhodium(I)-{1,2-Bis[(o-tert-butoxyphenyl)(phenyl)phosphino]ethane}. Adv. Synth. Catal. 2013, 355, 594–600. [Google Scholar] [CrossRef]
- Li, X.; You, C.; Li, S.; Lv, H.; Zhang, Z. Nickel-Catalyzed Enantioselective Hydrogenation of β-(Acylamino) acrylates: Synthesis of Chiral β-Amino Acid Derivatives. J. Org. Chem. 2004, 69, 8157–8160. [Google Scholar] [CrossRef]
- Tang, W.; Capacci, A.G.; White, A.; Ma, S.; Rodriguez, S.; Qu, B.; Savoie, J.; Patel, N.D.; Wei, X.; Haddad, N.; et al. Novel and Efficient Chiral Bisphosphorus Ligands for Rhodium-Catalyzed Asymmetric Hydrogenation. Org. Lett. 2010, 12, 1104–1107. [Google Scholar] [CrossRef]
- Kohara, T.; Hashimoto, Y.; Saigo, K. Design, synthesis, and optical resolution of a novel non-natural chiral auxiliary, 1-(2,5-dimethoxyphenyl)ethylamine. Application to diastereoselective alkylation of aldimines. Tetrahedron 1999, 55, 6453–6464. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand | tsJPP (Hz) |
|---|---|
| L1 | 16.4 |
| L2 | 26.0 |
| L3 | 19.5 |
| L4 | 26.2 |
| L5 | 56.2 |
| L6 | 29.3 |
| L7 | - |
| L8 | - |

| Entry | Ligand | Rh Source | Solvent | H2 Pressure (bar) | Time (h) | Conv. (%) [a] | Ee (%) [b] |
|---|---|---|---|---|---|---|---|
| 1 | (±)-BINAP | Rh(COD)2OTf | THF | 2.3 | 15 | >99 | - |
| 2 | L1 | Rh(COD)2OTf | THF | 1 | 12 | >99 | 89.0 |
| 3 | L1 | Rh(COD)2BF4 | THF | 1 | 12 | 78.7 | 90.0 |
| 4 | L1 | Rh(COD)2OTf | THF | 20 | 2 | >99 | 76.3 |
| 5 | L1 | Rh(COD)2OTf | THF | 40 | 1 | >99 | 71.2 |
| 6 | L1 | Rh(COD)2OTf | DCM | 1 | 12 | >99 | 85.3 |
| 7 | L1 | Rh(COD)2OTf | MeOH | 1 | 12 | >99 | 85.7 |
| 8 | L1 | Rh(COD)2OTf | DMF | 1 | 12 | 42.5 | 80.2 |
| 9 | L1 | Rh(COD)2OTf | 1,4-Dioxane | 1 | 12 | 66 | 82.8 |
| 10 | L1 | Rh(COD)2OTf | Toluene | 1 | 12 | 50 | 80.3 |
| 11 | L1 | Rh(COD)2BF4 | Toluene | 1 | 12 | 38.3 | 81.8 |
| 12 | L2 | Rh(COD)2OTf | THF | 1 | 12 | 70.6 | 27.3 |
| 13 | L2 | Rh(COD)2OTf | DCM | 1 | 10 | 64.3 | 17.6 |
| 14 | L3 | Rh(COD)2OTf | THF | 1 | 12 | >99 | 88.9 |
| 15 | L4 | Rh(COD)2OTf | THF | 1 | 2 | >99 | 92.2 |
| 16 [c] | L4 | Rh(COD)2OTf | THF | 1 | 3 | >99 | 95.5 |
| 17 [c] | L4 | Rh(COD)2BF4 | THF | 1 | 4 | 97 | 95.4 |
| 18 | L5 | Rh(COD)2OTf | THF | 1 | 12 | >99 | 97.9 |
| 19 | L5 | Rh(COD)2BF4 | THF | 1 | 12 | >99 | 97.7 |
| 20 | L5 | Rh(COD)2OTf | THF | 10 | 2 | >99 | 95.3 |
| 21 | L5 | Rh(COD)2OTf | DCM | 1 | 12 | >99 | 97.7 |
| 22 | L5 | Rh(COD)2OTf | MeOH | 1 | 12 | >99 | 92.3 |
| 23 | L6 | Rh(COD)2OTf | THF | 1 | 12 | >99 | 86.7 |
| 24 | L7 | Rh(COD)2OTf | THF | 1 | 1 | >99 | 99.5 |
| 25 [d] | L7 | Rh(COD)2OTf | THF | 20 | 4 | >99 | 97.7 |
| 26 | L8 | Rh(COD)2OTf | THF | 1 | 12 | >99 | 46.7 (R) |
| 27 [5] | (R,S)-Josiphos | Rh(COD)2OTf | MeOH | 1 | 0.33 | >99 | 96 |
| 28 [35] | DuPhos | Rh(COD)2OTf | MeOH | 2 (atm) | 1 | >99 | 85 |
| 29 [16] | BoPhoz | Rh(COD)2OTf | THF | 1 | 1 | 96 | 99.5 |

| Entry | Ligand | Solvent | H2 Pressure (bar) | Time (h) | Conv. (%) [a] | Ee (%) [b] |
|---|---|---|---|---|---|---|
| 1 | (±)-BINAP | THF | 40 | 18 | >99 | - |
| 2 | L1 | THF | 40 | 2 | >99 | 33.6 |
| 3 | L1 | THF | 20 | 2 | >99 | 33.4 |
| 4 | L1 | CH2Cl2 | 20 | 2 | >99 | 29.5 |
| 5 | L1 | MeOH | 20 | 1 | >99 | 46.0 |
| 6 | L2 | MeOH | 20 | 1 | >99 | 12.5 |
| 7 | L3 | MeOH | 20 | 1 | >99 | 45.6 |
| 8 | L4 | MeOH | 20 | 1 | >99 | 91.3 |
| 9 | L5 | THF | 10 | 2 | >99 | 92.0 |
| 10 | L5 | THF | 1 | 3 | 42 | 89.2 |
| 11 | L7 | THF | 1 | 2.5 | >99 | 96.4 |
| 12 | L7 | THF | 10 | 1 | >99 | 96.0 |
| 13 [c] | L7 | THF | 10 | 1 | >99 | 91.0 |
| 14 | L8 | THF | 1 | 2 | 59 | 31.7 (R) |
| 15 [36] | Me-BPE | MeOH | 4 (atm) | 12 | >99 | 95.2 |
| 16 [22] | BoPhoz | CH2Cl2 | 10 | 1 | 99.5 | 61.8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, X.; Brennan, T.B.; Kingston, C.; Ortin, Y.; Guiry, P.J. Design and Synthesis of Pyrrolidinyl Ferrocene-Containing Ligands and Their Application in Highly Enantioselective Rhodium-Catalyzed Olefin Hydrogenation. Molecules 2022, 27, 6078. https://doi.org/10.3390/molecules27186078
Li X, Brennan TB, Kingston C, Ortin Y, Guiry PJ. Design and Synthesis of Pyrrolidinyl Ferrocene-Containing Ligands and Their Application in Highly Enantioselective Rhodium-Catalyzed Olefin Hydrogenation. Molecules. 2022; 27(18):6078. https://doi.org/10.3390/molecules27186078
Chicago/Turabian StyleLi, Xin, Therese B. Brennan, Cian Kingston, Yannick Ortin, and Patrick J. Guiry. 2022. "Design and Synthesis of Pyrrolidinyl Ferrocene-Containing Ligands and Their Application in Highly Enantioselective Rhodium-Catalyzed Olefin Hydrogenation" Molecules 27, no. 18: 6078. https://doi.org/10.3390/molecules27186078
APA StyleLi, X., Brennan, T. B., Kingston, C., Ortin, Y., & Guiry, P. J. (2022). Design and Synthesis of Pyrrolidinyl Ferrocene-Containing Ligands and Their Application in Highly Enantioselective Rhodium-Catalyzed Olefin Hydrogenation. Molecules, 27(18), 6078. https://doi.org/10.3390/molecules27186078