


The Use of Aryl-Substituted Homophthalic Anhydrides in the Castagnoli–Cushman Reaction Provides Access to Novel Tetrahydroisoquinolone Carboxylic Acid Bearing an All-Carbon Quaternary Stereogenic Center
 ,
,  and
and
Abstract
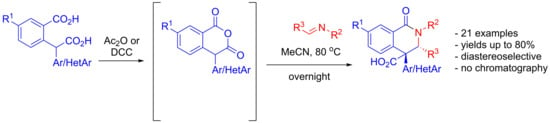
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
3. Conclusions
4. Materials and Methods
4.1. General Information
4.2. Preparation of Arylhomophthalic Acids 10a–10f: General Procedure 1
- Step 1.
- Condensation of arylindanones with diethyl oxalate
- Step 2.
- Oxidation
4.2.1. 2-[Carboxy(4-chlorophenyl)methyl]benzoic Acid (10a)
4.2.2. 2-[Carboxy(phenyl)methyl]benzoic Acid (10b)
4.2.3. 2-[Carboxy(4-methoxyphenyl)methyl]benzoic Acid (10c)
4.2.4. 2-[Carboxy(phenyl)methyl]-5-methylbenzoic Acid (10d)
4.2.5. 2-[Carboxy(4-fluorophenyl)methyl]benzoic Acid (10e)
4.2.6. 2-[Carboxy(4-chlorophenyl)methyl]-5-chlorobenzoic Acid (10f)
4.3. General Procedure for Preparation of Tetrahydroisoquinonolones 9a–9u
- Step 1.
- Anhydride synthesis.
- Step 2.
- The Castagnoli–Cushman reaction
4.3.1. (±)-(3R,4R)-2-Ethyl-1-oxo-4-phenyl-3-(p-tolyl)-1,2,3,4-tetrahydroisoquinoline-4-carboxylic Acid (9a)
4.3.2. (±)-(3R,4R)-3-(4-Nitrophenyl)-1-oxo-4-phenyl-2-propyl-1,2,3,4-tetrahydroisoquinoline-4-carboxylic Acid (9b)
4.3.3. (±)-(3R,4R)-2-Ethyl-7-methyl-1-oxo-4-phenyl-3-(p-tolyl)-1,2,3,4-tetrahydroisoquinoline-4-carboxylic Acid (9c)
4.3.4. (±)-(3R,4R)-2-Ethyl-4-(4-methoxyphenyl)-1-oxo-3-(p-tolyl)-1,2,3,4-tetrahydroisoquinoline-4-carboxylic Acid (9d)
4.3.5. (±)-(3R,4R)-7-Chloro-4-(4-chlorophenyl)-2-ethyl-1-oxo-3-(p-tolyl)-1,2,3,4-tetrahydroisoquinoline-4-carboxylic acid (9e)
4.3.6. (±)-(3R,4R)-3-(4-(Benzyloxy)-3-methoxyphenyl)-4-(4-chlorophenyl)-1-oxo-2-(prop-2-yn-1-yl)-1,2,3,4-tetrahydroisoquinoline-4-carboxylic Acid (9f)
4.3.7. (±)-(3R,4R)-2-Benzyl-4-(4-chlorophenyl)-3-(2-methoxyphenyl)-1-oxo-1,2,3,4-tetrahydroisoquinoline-4-carboxylic Acid (9g)
4.3.8. (±)-(3R,4R)-2-Allyl-4-(4-chlorophenyl)-3-(2,4-dimethoxyphenyl)-1-oxo-1,2,3,4-tetrahydroisoquinoline-4-carboxylic Acid (9h)
4.3.9. (±)-(3R,4R)-4-(4-Chlorophenyl)-1-oxo-3-phenyl-2-(p-tolyl)-1,2,3,4-tetrahydroisoquinoline-4-carboxylic Acid (9i)
4.3.10. (±)-(3R,4R)-4-(4-Chlorophenyl)-3-(4-methoxyphenyl)-1-oxo-2-(4-(trifluoromethyl)phenyl)-1,2,3,4-tetrahydroisoquinoline-4-carboxylic Acid (9j)
4.3.11. (±)-(3S,4R)-4-(4-Chlorophenyl)-1-oxo-3-(thiophen-2-yl)-2-(p-tolyl)-1,2,3,4-tetrahydroisoquinoline-4-carboxylic Acid (9k)
4.3.12. (±)-(3R,4R)-4-(4-Chlorophenyl)-3-(4-fluorophenyl)-2-(4-methoxybenzyl)-1-oxo-1,2,3,4-tetrahydroisoquinoline-4-carboxylic Acid (9l)
4.3.13. (±)-(3R,4R)-4-(4-Chlorophenyl)-1-oxo-2,3-di-p-tolyl-1,2,3,4-tetrahydroisoquinoline-4-carboxylic Acid (9m)
4.3.14. (±)-(3S,4R)-4-(4-Chlorophenyl)-2-(2-(cyclopentylthio)ethyl)-3-(furan-2-yl)-1-oxo-1,2,3,4-tetrahydroisoquinoline-4-carboxylic Acid (9n)
4.3.15. (±)-(3R,4R)-4-(4-Chlorophenyl)-2-ethyl-1-oxo-3-(p-tolyl)-1,2,3,4-tetrahydroisoquinoline-4-carboxylic acid (9o)
4.3.16. (±)-(3S,4R)-4-(4-Chlorophenyl)-1-oxo-2-propyl-3-(pyridin-2-yl)-1,2,3,4-tetrahydroisoquinoline-4-carboxylic Acid (9p)
4.3.17. (±)-(3S,4R)-3-(2-Chlorophenyl)-4-(4-fluorophenyl)-1-oxo-2-propyl-1,2,3,4-tetrahydroisoquinoline-4-carboxylic Acid (9q)
4.3.18. (±)-(3R,4R)-4-(4-Fluorophenyl)-2-isopropyl-3-(4-methoxyphenyl)-1-oxo-1,2,3,4-tetrahydroisoquinoline-4-carboxylic Acid (9r)
4.3.19. (±)-(3R,4R)-2-Butyl-4-(4-fluorophenyl)-3-(4-methoxyphenyl)-1-oxo-1,2,3,4-tetrahydroisoquinoline-4-carboxylic Acid (9s)
4.3.20. (±)-(3R,4R)-4-(4-Fluorophenyl)-3-(2-methoxyphenyl)-1-oxo-2-(p-tolyl)-1,2,3,4-tetrahydroisoquinoline-4-carboxylic Acid (9t)
4.3.21. (±)-(3R,4R)-2-Ethyl-4-(4-fluorophenyl)-1-oxo-3-(p-tolyl)-1,2,3,4-tetrahydroisoquinoline-4-carboxylic acid (9u)
4.4. 2-(Carboxy(4-phenyl-1H-1,2,3-triazol-1-yl)methyl)benzoic Acid (15)
4.5. (±)-(3R,4S)-2-Ethyl-1-oxo-4-(4-phenyl-1H-1,2,3-triazol-1-yl)-3-(p-tolyl)-1,2,3,4-tetrahydroisoquinoline-4-carboxylic Acid (18)
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Howard, S.Y.; Di Maso, M.J.; Shimabukuro, K.; Burlow, N.P.; Tan, D.Q.; Fettinger, J.C.; Malig, T.C.; Hein, J.E.; Shaw, J.T. Mechanistic Investigation of Castagnoli–Cushman Multicomponent Reactions Leading to a Three-Component Synthesis of Dihydroisoquinolones. J. Org. Chem. 2021, 86, 11599–11607. [Google Scholar] [CrossRef] [PubMed]
- Krasavin, M.; Dar’in, D. Current diversity of cyclic anhydrides for the Castagnoli–Cushman-type formal cycloaddition reactions: Prospects and challenges. Tetrahedron Lett. 2016, 57, 1635–1640. [Google Scholar] [CrossRef]
- Lepikhina, A.; Dar’in, D.; Bakulina, O.; Chupakhin, E.; Krasavin, M. Skeletal Diversity in Combinatorial Fashion: A New Format for the Castagnoli–Cushman Reaction. ACS Comb. Sci. 2017, 19, 702–707. [Google Scholar] [CrossRef] [PubMed]
- Chupakhin, E.; Dar’in, D.; Krasavin, M. The Castagnoli–Cushman reaction in a three-component format. Tetrahedron Lett. 2018, 59, 2595–2599. [Google Scholar] [CrossRef]
- Bakulina, O.; Chizhova, M.; Dar’in, D.; Krasavin, M. A General Way to Construct Arene-Fused Seven-Membered Nitrogen Heterocycles. Eur. J. Org. Chem. 2018, 2018, 362–371. [Google Scholar] [CrossRef]
- Adamovskyi, M.I.; Avramenko, M.M.; Volochnyuk, D.M.; Ryabukhin, S.V. Fluoral Hydrate: A Perspective Substrate for the Castagnoli–Cushman Reaction. ACS Omega 2020, 5, 20932–20942. [Google Scholar] [CrossRef]
- Bayles, T.; Guillou, C. Trifluoroethanol Promoted Castagnoli–Cushman Cycloadditions of Imines with Homophthalic Anhydride. Molecules 2022, 27, 844. [Google Scholar] [CrossRef]
- Jarvis, C.L.; Hirschi, J.S.; Vetticatt, M.J.; Seidel, D. Catalytic Enantioselective Synthesis of Lactams through Formal [4 + 2] Cycloaddition of Imines with Homophthalic Anhydride. Angew. Chem. 2017, 56, 2670–2674. [Google Scholar] [CrossRef]
- Ivashchenko, A.V.; Tkachenko, S.Y.; Okun, I.M.R.S.A.; Vladimirovich, K.D.; Khvat, A.V. Pharmaceutical Composition, Method for the Production and the Use Thereof. Patent WO 2007133108, 22 November 2007. [Google Scholar]
- Okun, I.; Balakin, K.V.; Tkachenko, S.E.; Ivachtchenko, A.V. Caspase activity modulators as anticancer agents. Anticancer Agents Med. Chem. 2008, 8, 322–341. [Google Scholar] [CrossRef]
- Pereira, G.A.N.; da Silva, E.B.; Braga, S.F.P.; Leite, P.G.; Martins, L.C.; Vieira, R.P.; Soh, W.T.; Villela, F.S.; Costa, F.M.R.; Ray, D.; et al. Discovery and characterization of trypanocidal cysteine protease inhibitors from the ‘malaria box’. Eur. J. Med. Chem. 2019, 179, 765–778. [Google Scholar] [CrossRef]
- Chen, Y.; Zhu, F.; Hammill, J.; Holbrook, G.; Yang, L.; Freeman, B.; White, K.L.; Shackleford, D.M.; O’Loughlin, K.G.; Charman, S.A.; et al. Selecting an anti-malarial clinical candidate from two potent dihydroisoquinolones. Malar. J. 2021, 20, 107. [Google Scholar] [CrossRef] [PubMed]
- Guranova, N.; Golubev, P.; Bakulina, O.; Dar’in, D.; Kantin, G.; Krasavin, M. Unexpected formal [4 + 2]-cycloaddition of chalcone imines and homophthalic anhydrides: Preparation of dihydropyridin-2(1H)-ones. Org. Biomol. Chem. 2021, 19, 3829–3833. [Google Scholar] [CrossRef] [PubMed]
- Laws, S.W.; Moore, L.C.; Di Maso, M.J.; Nguyen, Q.N.N.; Tantillo, D.J.; Shaw, J.T. Diastereoselective Base-Catalyzed Formal [4 + 2] Cycloadditions of N-Sulfonyl Imines and Cyclic Anhydrides. Org. Lett. 2017, 19, 2466–2469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, P.Y.; Tang, Y.; Knosp, W.M.; Stadler, H.S.; Shaw, J.T. Synthesis of diverse lactam carboxamides leading to the discovery of a new transcription-factor inhibitor. Angew. Chem. 2007, 46, 5352–5355. [Google Scholar] [CrossRef]
- Rendy, R.; Zhang, Y.; McElrea, A.; Gomez, A.; Klumpp, D.A. Superacid-catalyzed reactions of cinnamic acids and the role of superelectrophiles. J. Org. Chem. 2004, 69, 2340–2347. [Google Scholar] [CrossRef]
- Püschl, A.; Rudbeck, H.C.; Faldt, A.; Confante, A.; Kehler, J. Versatile Synthesis of 3-Arylindan-1-ones by Palladium-Catalyzed Intramolecular Reductive Cyclization of Bromochalcones. Synthesis 2005, 2005, 291–295. [Google Scholar] [CrossRef]
- Safrygin, A.; Zhmurov, P.; Dar’in, D.; Silonov, S.; Kasatkina, M.; Zonis, Y.; Gureev, M.; Krasavin, M. Three-component Castagnoli–Cushman reaction with ammonium acetate delivers 2-unsubstituted isoquinol-1-ones as potent inhibitors of poly(ADP-ribose) polymerase (PARP). J. Enzym. Inhib. Med. Chem. 2021, 36, 1916–1921. [Google Scholar] [CrossRef]
- Liu, J.; Wang, Z.; Levin, A.; Emge, T.J.; Rablen, P.R.; Floyd, D.M.; Knapp, S. N-methylimidazole promotes the reaction of homophthalic anhydride with imines. J. Org. Chem. 2014, 79, 7593–7599. [Google Scholar] [CrossRef]
- Scorzelli, F.; Di Mola, A.; De Piano, F.; Tedesco, C.; Palombi, L.; Filosa, R.; Waser, M.; Massa, A. A systematic study on the use of different organocatalytic activation modes for asymmetric conjugated addition reactions of isoindolinones. Tetrahedron 2017, 73, 819–828. [Google Scholar] [CrossRef]
- Yue, G.; Lei, K.; Hirao, H.; Zhou, J.S. Palladium-catalyzed asymmetric reductive heck reaction of aryl halides. Angew. Chem. 2015, 54, 6531–6535. [Google Scholar] [CrossRef]
- Bogeso, K.P. Neuroleptic activity and dopamine-uptake inhibition in 1-piperazino-3-phenylindans. J. Med. Chem. 1983, 26, 935–947. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. A Found Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moshnenko, N.; Kazantsev, A.; Bakulina, O.; Dar’in, D.; Krasavin, M. The Use of Aryl-Substituted Homophthalic Anhydrides in the Castagnoli–Cushman Reaction Provides Access to Novel Tetrahydroisoquinolone Carboxylic Acid Bearing an All-Carbon Quaternary Stereogenic Center. Molecules 2022, 27, 8462. https://doi.org/10.3390/molecules27238462
Moshnenko N, Kazantsev A, Bakulina O, Dar’in D, Krasavin M. The Use of Aryl-Substituted Homophthalic Anhydrides in the Castagnoli–Cushman Reaction Provides Access to Novel Tetrahydroisoquinolone Carboxylic Acid Bearing an All-Carbon Quaternary Stereogenic Center. Molecules. 2022; 27(23):8462. https://doi.org/10.3390/molecules27238462
Chicago/Turabian StyleMoshnenko, Nazar, Alexander Kazantsev, Olga Bakulina, Dmitry Dar’in, and Mikhail Krasavin. 2022. "The Use of Aryl-Substituted Homophthalic Anhydrides in the Castagnoli–Cushman Reaction Provides Access to Novel Tetrahydroisoquinolone Carboxylic Acid Bearing an All-Carbon Quaternary Stereogenic Center" Molecules 27, no. 23: 8462. https://doi.org/10.3390/molecules27238462
APA StyleMoshnenko, N., Kazantsev, A., Bakulina, O., Dar’in, D., & Krasavin, M. (2022). The Use of Aryl-Substituted Homophthalic Anhydrides in the Castagnoli–Cushman Reaction Provides Access to Novel Tetrahydroisoquinolone Carboxylic Acid Bearing an All-Carbon Quaternary Stereogenic Center. Molecules, 27(23), 8462. https://doi.org/10.3390/molecules27238462