

Novel 1,2,3-Triazole-Based Benzothiazole Derivatives: Efficient Synthesis, DFT, Molecular Docking, and ADMET Studies
 ,
, 
Abstract
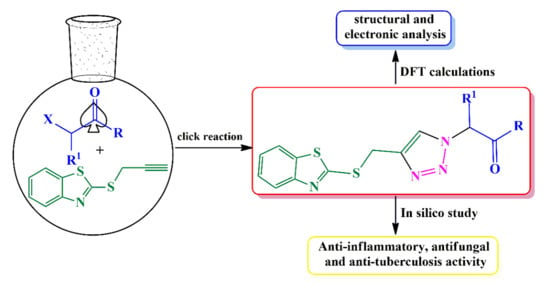
:
1. Introduction
2. Results and Discussion
2.1. Synthesis and Spectroscopic Characterization of 1,2,3-Triazoles 5a–f
2.2. Theoretical Studies on the 1,2,3-Triazole 5e
2.2.1. Benchmarking of the Computational Method

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Atom a | δB3LYP | δexp | Atom a | δB3LYP | δexp | Atom a | δB3LYP | δexp |
|---|---|---|---|---|---|---|---|---|
| H35−37 | 1.27 | 1.01 | H19 | 8.15 | 8.01 | C4 | 119.53 | 121.82 |
| H27−29 | 1.83 | 1.72 | H7 | 8.17 | 8.24 | C6 | 120.86 | 123.85 |
| H32 | 4.10 | 4.10 | C34 | 16.54 | 13.80 | C18 | 123.18 | 124.59 |
| H16 | 4.55 | 4.71 | C26 | 20.67 | 17.05 | C5 | 123.45 | 126.41 |
| H25 | 5.55 | 5.62 | C15 | 35.50 | 27.44 | C2 | 139.12 | 134.76 |
| H10 | 7.62 | 7.35 | C24 | 64.14 | 57.53 | C20 | 140.05 | 142.29 |
| H9 | 7.77 | 7.49 | C31 | 66.31 | 61.66 | |||
| H8 | 8.09 | 7.90 | C1 | 118.96 | 120.31 |

2.2.2. Structural Characterization of 5a and 5e
2.2.3. Electronic Characterization of 5e
2.3. Molecular Docking and ADMET Studies
3. Experimental Section
3.1. General Information
3.2. Computational Details
3.3. Molecular Docking and ADMET Details
3.4. General Synthesis Procedure for 2-Mercaptobenzothiazole 2
3.5. General Synthesis Procedure for S-Propargylated Mercaptobenzothiazole 3
3.6. General Synthesis Procedure for 1,2,3-Triazole Derivatives 5a–f
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Jampilek, J. Heterocycles in medicinal chemistry. Molecules 2019, 24, 3839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arora, P.; Arora, V.; Lamba, H.S.; Wadhwa, D. Importance of heterocyclic chemistry: A review. Int. J. Pharm. Sci. 2012, 3, 2947–2954. [Google Scholar]
- Breugst, M.; Reissig, H.U. The Huisgen Reaction: Milestones of the 1, 3-Dipolar Cycloaddition. Angew. Chem. Int. Ed. 2020, 59, 12293–12307. [Google Scholar] [CrossRef] [Green Version]
- Matin, M.M.; Matin, P.; Rahman, M.; Ben Hadda, T.; Almalki, F.A.; Mahmud, S.; Ghoneim, M.M.; Alruwaily, M.; Alshehri, S. Triazoles and Their Derivatives: Chemistry, Synthesis, and Therapeutic Applications. Front. Mol. Biosci. 2022, 9, 303–310. [Google Scholar] [CrossRef]
- Liang, L.; Astruc, D. The copper (I)-catalyzed alkyne-azide cycloaddition (CuAAC)“click” reaction and its applications. An overview. Coord. Chem. Rev. 2011, 255, 2933–2945. [Google Scholar] [CrossRef]
- Bedi, A.; Manor Armon, A.; Diskin-Posner, Y.; Bogosalvsky, B.; Gidron, O. Controlling the helicity of π-conjugated oligomers by tuning the aromatic backbone twist. Nat. Commun. 2022, 13, 451. [Google Scholar] [CrossRef]
- Tornøe, C.W.; Christensen, C.; Meldal, M. Peptidotriazoles on solid phase: [1, 2, 3]-triazoles by regiospecific copper (I)-catalyzed 1, 3-dipolar cycloadditions of terminal alkynes to azides. J. Org. Chem. 2002, 67, 3057–3064. [Google Scholar] [CrossRef]
- Rostovtsev, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A stepwise huisgen cycloaddition process: Copper (I)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angew. Chem. 2002, 114, 2708–2711. [Google Scholar] [CrossRef]
- Dheer, D.; Singh, V.; Shankar, R. Medicinal attributes of 1, 2, 3-triazoles: Current developments. Bioorg. Chem. 2017, 71, 30–54. [Google Scholar] [CrossRef]
- Dixit, D.; Verma, P.K.; Marwaha, R.K. A review on ‘triazoles’: Their chemistry, synthesis and pharmacological potentials. J. Iran. Chem. Soc. 2021, 18, 2535–2565. [Google Scholar] [CrossRef]
- Tratrat, C.; Haroun, M.; Paparisva, A.; Geronikaki, A.; Kamoutsis, C.; Ćirić, A.; Glamočlija, J.; Soković, M.; Fotakis, C.; Zoumpoulakis, P.; et al. Design, synthesis and biological evaluation of new substituted 5-benzylideno-2-adamantylthiazol [3, 2-b][1, 2, 4] triazol-6 (5H) ones. Pharmacophore models for antifungal activity. Arab. J. Chem. 2018, 11, 573–590. [Google Scholar] [CrossRef]
- Sharma, M.K.; Parashar, S.; Chahal, M.; Lal, K.; Pandya, N.U.; Om, H. Antimicrobial and in-silico evaluation of novel chalcone and amide-linked 1, 4-disubstituted 1, 2, 3 triazoles. J. Mol. Struct. 2022, 1257, 132632. [Google Scholar] [CrossRef]
- Chu, X.M.; Wang, C.; Wang, W.L.; Liang, L.L.; Liu, W.; Gong, K.K.; Sun, K.L. riazole derivatives and their antiplasmodial and antimalarial activities. Eur. J. Med. Chem. 2019, 166, 206–223. [Google Scholar] [CrossRef]
- Carvalho da Silva, F.D.; Cardoso, M.F.D.C.; Ferreira, P.G.; Ferreira, V.F. Biological Properties of 1H-1, 2, 3-and 2H-1, 2, 3-Triazoles. In Chemistry of 1, 2, 3-Triazoles; Springer-Verlag Berlin Heidelberg: Berlin, Germany, 2014; pp. 117–165. [Google Scholar]
- Fallah, Z.; Tajbakhsh, M.; Alikhani, M.; Larijani, B.; Faramarzi, M.A.; Hamedifar, H.; Mohammadi-Khanaposhtani, M.; Mahdavi, M. A review on synthesis, mechanism of action, and structure-activity relationships of 1, 2, 3-triazole-based α-glucosidase inhibitors as promising anti-diabetic agents. J. Mol. Struct. 2022, 1255, 132469. [Google Scholar] [CrossRef]
- Petrou, A.; Fesatidou, M.; Geronikaki, A. Thiazole ring—A biologically active scaffold. Molecules 2021, 26, 3166. [Google Scholar] [CrossRef]
- Bedi, A.; Senanayak, S.P.; Das, S.; Narayan, K.S.; Zade, S.S. Cyclopenta [c] thiophene oligomers based solution processable D–A copolymers and their application as FET materials. Polym. Chem. 2012, 3, 1453–1460. [Google Scholar] [CrossRef]
- Rana, A.; Siddiqui, N.; Khan, S.A. Benzothiazoles: A new profile of biological activities. Indian J. Pharm. Sci. 2007, 69, 10–17. [Google Scholar]
- Noolvi, M.N.; Patel, H.M.; Kaur, M. Benzothiazoles: Search for anticancer agents. Eur. J. Med. Chem. 2012, 54, 447–462. [Google Scholar] [CrossRef]
- Ali, R.; Siddiqui, N. Biological aspects of emerging benzothiazoles: A short review. J. Chem. 2013, 2013, 345198. [Google Scholar] [CrossRef]
- Gill, R.K.; Rawal, R.K.; Bariwal, J. Recent advances in the chemistry and biology of benzothiazoles. Arch. Pharm. 2015, 348, 155–178. [Google Scholar] [CrossRef]
- Kamal, A.; Syed, M.A.H.; Mohammed, S.M. Therapeutic potential of benzothiazoles: A patent review (2010–2014). Expert Opin. Ther. Pat. 2015, 25, 335–349. [Google Scholar] [CrossRef] [PubMed]
- Kabatc, J.; Jurek, K. Synthesis, properties, and application of new benzothiazole-based sensitisers in polymer chemistry. Color. Technol. 2015, 131, 183–191. [Google Scholar] [CrossRef]
- Henary, M.; Paranjpe, S.; Owens, E.A. Synthesis and applications of benzothiazole containing cyanine dyes. Heterocycl. Comm. 2013, 19, 1–11. [Google Scholar] [CrossRef]
- Debnath, S.; Bedi, A.; Zade, S.S. Thienopentathiepine: A sulfur containing fused heterocycle for conjugated systems and their electrochemical polymerization. Polym. Chem. 2015, 6, 7658–7665. [Google Scholar] [CrossRef] [Green Version]
- Fel’dshteĭn, M.S.; Eĭtingon, I.I.; Pevzner, D.M.; Strel’nikova, N.P.; Dogadkin, B.A. Derivatives of 2-Mercaptobenzothiazole and Dimethyldithiocarbamic Acid as Vulcanization Accelerators. Rubber Chem. Technol. 1959, 32, 983–991. [Google Scholar] [CrossRef]
- Nehra, N.; Tittal, R.K.; Ghule Vikas, D.; Naveen; Lal, K. Synthesis, Antifungal Studies, Molecular Docking, ADME and DNA Interaction Studies of 4-Hydroxyphenyl Benzothiazole Linked 1,2,3-Triazoles. J. Mol. Struct. 2021, 1245, 131013. [Google Scholar] [CrossRef]
- Fahim, A.M.; Tolan, H.E.; El-Sayed, W.A. Synthesis of novel 1, 2, 3-triazole based acridine and benzothiazole scaffold N-glycosides with anti-proliferative activity, docking studies, and comparative computational studies. J. Mol. Struct. 2022, 1251, 131941. [Google Scholar] [CrossRef]
- Mirjafary, Z.; Ahmadi, L.; Moradi, M.; Saeidian, H. A copper (II)–thioamide combination as a robust heterogeneous catalytic system for green synthesis of 1, 4-disubstituted-1, 2, 3-triazoles under click conditions. RSC Adv. 2015, 5, 78038–78046. [Google Scholar] [CrossRef]
- Saeidian, H.; Sadighian, H.; Abdoli, M.; Sahandi, M. Versatile and green synthesis, spectroscopic characterizations, crystal structure and DFT calculations of 1, 2, 3-triazole-based sulfonamides. J. Mol. Struct. 2017, 1131, 73–78. [Google Scholar] [CrossRef]
- Saeidian, H.; Sadighian, H.; Arabgari, M.; Mirjafary, Z.; Ayati, S.E.; Najafi, E.; Moghaddam, F.M. Organocopper-based magnetically recoverable and reusable nanocatalyst for efficient synthesis of novel 1, 2, 3-triazole-based sulfonamides in green medium. Res. Chem. Intermed. 2018, 44, 601–612. [Google Scholar] [CrossRef]
- Paghandeh, H.; Saeidian, H. Expedient and click synthesis, spectroscopic characterizations and DFT calculations of novel 1, 5-bis (N-substituted 1, 2, 3-triazole) benzodiazepinedione scaffolds. J. Mol. Struct. 2018, 1157, 560–566. [Google Scholar] [CrossRef]
- Karbasi, M.M.; Mirjafary, Z.; Saeidian, H.; Mokhtari, J. Efficient synthesis and DFT analysis of novel 1, 2, 3-triazole-based dithiocarbamates. J. Mol. Struct. 2021, 1227, 129535. [Google Scholar] [CrossRef]
- Esmaeeli, Z.; Khodabakhshi, M.R.; Mirjafary, Z.; Saeidian, H. Efficient synthesis of novel 1, 2, 3-triazole-based diazepam derivatives by click CuAAC reaction: Spectroscopic characterizations and DFT studies. J. Mol. Struct. 2021, 1246, 131206. [Google Scholar] [CrossRef]
- Liu, B.; Qu, L.; Yan, S. Cyclooxygenase-2 Promotes Tumor Growth and Suppresses Tumor Immunity. Cancer Cell Int. 2015, 15, 106. [Google Scholar] [CrossRef] [Green Version]
- Pang, L.Y.; Hurst, E.A.; Argyle, D.J. Cyclooxygenase-2: A Role in Cancer Stem Cell Survival and Repopulation of Cancer Cells during Therapy. Stem Cells Int. 2016, 2016, 2048731. [Google Scholar] [CrossRef] [Green Version]
- Chow, L.W.C.; Loo, W.T.Y.; Toi, M. Current Directions for COX-2 Inhibition in Breast Cancer. Biomed. Pharmacother. 2005, 59, 281–284. [Google Scholar] [CrossRef]
- Li, S.M.; Tsai, S.E.; Chiang, C.Y.; Chung, C.Y.; Chuang, T.J.; Tseng, C.C.; Jiang, W.P.; Huang, G.J.; Lin, C.Y.; Yang, Y.-C.; et al. New Methyl -5-(Halomethyl)-1-Aryl-1H-1,2,4-Triazole-3-Carboxylates as Selective COX-2 Inhibitors and Anti-Inflammatory Agents: Design, Synthesis, Biological Evaluation, and Docking Study. Bioorg. Chem. 2020, 104, 104333–104346. [Google Scholar] [CrossRef]
- Mohassab, A.M.; Hassan, H.A.; Abdelhamid, D.; Gouda, A.M.; Gomaa, H.A.M.; Youssif, B.G.M.; Radwan, M.O.; Fujita, M.; Otsuka, M.; Abdel-Aziz, M. New Quinoline/1,2,4-Triazole Hybrids as Dual Inhibitors of COX-2/5-LOX and Inflammatory Cytokines: Design, Synthesis, and Docking Study. J. Mol. Struct. 2021, 1244, 130948. [Google Scholar] [CrossRef]
- Venkateswarlu, K.; Kelly, D.E.; Kelly, S.L. Characterization of Saccharomyces Cerevisiae CYP51 and a CYP51 Fusion Protein with NADPH Cytochrome P-450 Oxidoreductase Expressed in Escherichia Coli. Antimicrob. Agents Chemother. 1997, 41, 776–780. [Google Scholar] [CrossRef] [Green Version]
- Baptista, R.; Bhowmick, S.; Shen, J.; Mur, L.A.J. Molecular Docking Suggests the Targets of Anti-Mycobacterial Natural Products. Molecules 2021, 26, 475. [Google Scholar] [CrossRef]
- Enugopala, K.N.; Chandrashekharappa, S.; Deb, P.K.; Tratrat, C.; Pillay, M.; Chopra, D.; Al-Shar’i, N.A.; Hourani, W.; Dahabiyeh, L.A.; Borah, P.; et al. Anti-Tubercular Activity and Molecular Docking Studies of Indolizine Derivatives Targeting Mycobacterial InhA Enzyme. J. Enzyme Inhib. Med. Chem. 2021, 36, 1471–1486. [Google Scholar] [CrossRef] [PubMed]
- Ghattas, M.A.V.; Mansour, R.A.; Atatreh, N.; Bryce, R.A. Analysis of Enoyl-Acyl Carrier Protein Reductase Structure and Interactions Yields an Efficient Virtual Screening Approach and Suggests a Potential Allosteric Site. Chem. Biol. Drug. Des. 2015, 87, 131–142. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Alian, A.; Ortiz de Montellano, P.R. Inhibition of the Mycobacterium Tuberculosis Enoyl Acyl Carrier Protein Reductase InhA by Arylamides. Bioorg. Med. Chem. 2007, 15, 6649–6658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabbagh, G.; Berakdar, N. Docking Studies of Flavonoid Compounds as Inhibitors of β-Ketoacyl Acyl Carrier Protein Synthase I (Kas I) of Escherichia Coli. J. Mol. Graph. Model. 2015, 61, 214–223. [Google Scholar] [CrossRef] [PubMed]
- Martel, C.M.; Parker, J.E.; Warrilow, A.G.S.; Rolley, N.J.; Kelly, S.L.; Kelly, D.E. Complementation of Saccharomyces Cerevisiae ERG11/CYP51 (Sterol 14α-Demethylase) Doxycycline-Regulated Mutant and Screening of the Azole Sensitivity of Aspergillus Fumigatus Isoenzymes CYP51A and CYP51B. Antimicrob. Agents Chemother. 2010, 54, 4920–4923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Li, L.; Lv, Q.; Yan, L.; Wang, Y.; Jiang, Y. The Fungal CYP51s: Their Functions, Structures, Related Drug Resistance, and Inhibitors. Front. Microbiol. 2019, 10, 691–699. [Google Scholar] [CrossRef] [Green Version]
- Price, C.L.; Warrilow, A.G.S.; Parker, J.E.; Mullins, J.G.L.; Nes, W.D.; Kelly, D.E.; Kelly, S.L. Novel Substrate Specificity and Temperature-Sensitive Activity of Mycosphaerella Graminicola CYP51 Supported by the Native NADPH Cytochrome P450 Reductase. Appl. Environ. Microbiol. 2015, 81, 3379–3386. [Google Scholar] [CrossRef] [Green Version]
- Yadav, D.K.; Ahmad, I.; Shukla, A.; Khan, F.; Negi, A.S.; Gupta, A. QSAR and Docking Studies on Chalcone Derivatives for Antitubercular Activity AgainstM. TuberculosisH37Rv. J. Chemom. 2014, 28, 499–507. [Google Scholar] [CrossRef]
- Mir, F.; Shafi, S.; Zaman, M.S.; Kalia, N.P.; Rajput, V.S.; Mulakayala, C.; Mulakayala, N.; Khan, I.A.; Alam, M.S. Sulfur Rich 2-Mercaptobenzothiazole and 1,2,3-Triazole Conjugates as Novel Antitubercular Agents. Eur. J. Med. Chem. 2014, 76, 274–283. [Google Scholar] [CrossRef]
- Shafi, S.; Mahboob Alam, M.; Mulakayala, N.; Mulakayala, C.; Vanaja, G.; Kalle, A.M.; Pallu, R.; Alam, M.S. Synthesis of Novel 2-Mercapto Benzothiazole and 1,2,3-Triazole Based Bis-Heterocycles: Their Anti-Inflammatory and Anti-Nociceptive Activities. Eur. J. Med. Chem. 2012, 49, 324–333. [Google Scholar] [CrossRef]
- Ballabeni, M.; Ballini, R.; Bigi, F.; Maggi, R.; Parrini, M.; Predieri, G.; Sartori, G. Synthesis of Symmetrical N, N’-Disubstituted Thioureas and Heterocyclic Thiones from Amines and CS (2) over a ZnO/Al2O3 Composite as Heterogeneous and Reusable Catalyst. J. Org. Chem. Res. 1999, 64, 1029–1032. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.B. Conceptual density functional theory and some recent developments. Acta. Phys. Sin. 2009, 25, 590–600. [Google Scholar]
- Saeidian, H.; Sahandi, M. Comprehensive DFT study on molecular structures of Lewisites in support of the Chemical Weapons Convention. J. Mol. Struct. 2015, 1100, 486–495. [Google Scholar] [CrossRef]
- Ghiasi, M.; Oskouie, A.A.; Saeidian, H. Dynamic stereochemistry of Topiramate (anticonvulsant drug) in solution: Theoretical approaches and experimental validation. Carbohydr. Res. 2012, 348, 47–54. [Google Scholar] [CrossRef] [Green Version]
- Shams, B.; Saeidian, H. Design of the novel neutral organic superacids by comprehensive DFT study on organic fluorosulfuric acids. Comput. Theor. Chem. 2018, 1135, 48–55. [Google Scholar] [CrossRef]
- Saeidian, H.; Mirjafary, Z. Engineering non-ionic carbon super-and hyperbases by a computational DFT approach: Substituted allenes have unprecedented cation affinities. New J. Chem. 2020, 44, 12967–12977. [Google Scholar] [CrossRef]
- Dennington, R.I.I.; Keith, T.; Millam, J. GaussView, Version 4.1.2; Semichem Inc.: Shawnee Mission, KS, USA, 2007.
- Weinhold, F.; Landis, C.R.; Glendening, E.D. What is NBO analysis and how is it useful? Int. Rev. Phys. Chem. 2016, 35, 399–440. [Google Scholar] [CrossRef]
- Jupp, A.R.; Johnstone, T.C.; Stephan, D.W. The global electrophilicity index as a metric for Lewis acidity. Dalton Trans. 2018, 47, 7029–7035. [Google Scholar] [CrossRef]
- Parthasarathi, R.; Subramanian, V.; Roy, D.R.; Chattaraj, P.K. Electrophilicity index as a possible descriptor of biological activity. Bioorg. Med. Chem. 2004, 12, 5533–5543. [Google Scholar] [CrossRef]
- Parr, R.G.; Szentpály, L.V.; Liu, S. Electrophilicity index. J. Am. Chem. Soc. 1999, 121, 1922–1924. [Google Scholar] [CrossRef]
- Pérez, P.; Domingo, L.R.; Aizman, A.; Contreras, R. The electrophilicity index in organic chemistry. Comput. Theor. Chem. 2007, 19, 139–201. [Google Scholar]
- Frisch, J.M.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Zakrzewski, V.G.; Montgomery, J.A.; Stratmann, R.E., Jr.; Burant, J.C.; et al. Gaussian 09; Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Wolinski, K.; Hinton, J.F.; Pulay, P. Efficient implementation of the gauge-independent atomic orbital method for NMR chemical shift calculations. J. Am. Chem. Soc. 1990, 112, 8251–8260. [Google Scholar] [CrossRef]
- Takano, Y.; Houk, K.N. Benchmarking the conductor-like polarizable continuum model (CPCM) for aqueous solvation free energies of neutral and ionic organic molecules. J. Chem. Theory Comput. 2005, 1, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Dallakyan, S.; Olson, A.J. Small-Molecule Library Screening by Docking with PyRx. Methods Mol. Biol. 2014, 1263, 243–250. [Google Scholar]
- Home Page of Cheminfo Website. Available online: https://www.cheminfo.org/ (accessed on 5 July 2022).
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera-A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An Open Chemical Toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [Green Version]
- Xiong, G.; Wu, Z.; Yi, J.; Fu, L.; Yang, Z.; Hsieh, C.; Yin, M.; Zeng, X.; Wu, C.; Lu, A.; et al. ADMETlab 2.0: An Integrated Online Platform for Accurate and Comprehensive Predictions of ADMET Properties. Nucleic Acids Res. 2021, 49, 5–14. [Google Scholar] [CrossRef]








 | ||||
|---|---|---|---|---|
| Entry | Catalyst | Solvent | T (°C) | Yield |
| 1 | CuCl (50 mol%) | H2O | 60 | trace |
| 2 | CuI | H2O/EtOH | 60 | 14 |
| 3 | CuSO4·5H2O (50 mol%)/sodium ascorbate (50 mol%) | H2O | 60 | trace |
| 4 | CuSO4·5H2O (50 mol%)/sodium ascorbate (50 mol%) | H2O/t-BuOH | r.t | 75 |
| 5 | CuSO4·5H2O (50 mol%)/sodium ascorbate (50 mol%) | H2O/t-BuOH | 60 | 30 |
| 6 | CuCl (50 mol%) | H2O/t-BuOH | r.t | 22 |
| Geometrical Parameters a | Theoretical Value |
|---|---|
| Bond lengths (Å) | |
| C1—C2 | 1.397 |
| C2—S12 | 1.757 |
| N13—C11 | 1.298 |
| C11—S14 | 1.761 |
| S14—C15 | 1.856 |
| N21—N22 | 1.306 |
| C30—O39 | 1.215 |
| C38—C31 | 1.463 |
| C20—C18 | 1.382 |
| Bond angles (°) | |
| C1—C2—C3 | 121.7 |
| H10—C6—C5 | 119.7 |
| C11—S14—C15 | 102.8 |
| N21—N22—N23 | 107.6 |
| N23—C24—H25 | 105.2 |
| H16—C15—H17 | 109.8 |
| Dihedral angles (°) | |
| C11—S14—C15—H17 | 58.5 |
| H37—C34—C31—H32 | 61.1 |
| O39—C30—C24—H29 | 85.5 |
| Parameter | Value |
|---|---|
| Energy (Hartree) | −1747.11 |
| μD (Deby) | 8.26 |
| EHOMO (eV) | −6.32 |
| ELUMO (eV) | −1.34 |
| Eg (eV) | 4.98 |
| Chemical potential (μ) (eV) | −3.83 |
| Chemical hardness (η) (eV) | 2.49 |
| Electrophilicity (ω) (eV) | 2.94 |
| Compound ID | Protein PDB ID | |||||
|---|---|---|---|---|---|---|
| 1G3U | 1QSG | 4P8N | 5F19 | 5V3X | 4ZE2 | |
| 5a | −9.6 | −8.5 | −9.1 | −9.3 | −9.4 | −9.4 (1) a |
| 5b | −8.2 | −9.0 | −9 | −9.2 | −9.2 | −9.0 |
| 5c | −9.3 | −8.8 | −9.1 | −9.6 | −8.5 | −8.9 |
| 5d | −9.7 (2) | −9.1 | −9.5 (1) | −9.7 (1) | −9.7 (3) | −9.4 |
| 5e-R | −7.3 | −7.8 | −7.7 | −7.6 | −7.8 | −7.6 |
| 5e-S | −9.3 | −7.7 | −9.2 | −8.3 | −7.8 | −8.5 |
| 5f | −6.3 | −8 | −7.7 | −8 | −6.8 | −7.7 |
| Isoniazid b | −6.5 | −5.8 | - | - | - | - |
| BTZ043 b | - | - | −10.6 | - | - | - |
| Ibuprofen b | - | - | - | −7.4 | - | - |
| I28 b | - | - | - | - | −10.1 | |
| Posaconazole b | - | - | - | - | - | −10.1 |
| Compound | Log S a | Log P b | Pfizer Rule | Lipinski Rule | GSK Rule |
|---|---|---|---|---|---|
| 5a | −4.305 | 3.455 | Rejected | Accepted | Accepted |
| 5b | −4.463 | 3.518 | Accepted | Accepted | Rejected |
| 5c | −4.892 | 4.127 | Rejected | Accepted | Rejected |
| 5d | −4.607 | 3.912 | Rejected | Accepted | Accepted |
| 5e | −3.944 | 3.127 | Rejected | Accepted | Accepted |
| 5f | −3.362 | 2.257 | Accepted | Accepted | Accepted |
| Isoniazid | −0.495 | −0.813 | Accepted | Accepted | Rejected |
| BTZ043 | −5.876 | 3.713 | Accepted | Rejected | Accepted |
| Ibuprofen | −3.701 | 3.687 | Rejected | Accepted | Accepted |
| I28 | −4.885 | 5.264 | Rejected | Rejected | Accepted |
| Posaconazole | −3.014 | 1.451 | Accepted | Rejected | Rejected |
| Protein Name | PDB ID | Control Molecule |
|---|---|---|
| DPRE1 | 4P8N | BTZ043 |
| Saccharomyces cerevisiae CYP51 | 4ZE2 | Posaconazole |
| COX-2 | 5F19 | Ibuprofen |
| Pks13 | 5V3X | I28 |
| Anti-tuberculosis target protein ENR | 1QSG | Isoniazid |
| Thymidylate kinase of Mycobacterium tuberculosis | 1G3U | Isoniazid |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mirjafary, Z.; Mohammad Karbasi, M.; Hesamzadeh, P.; Shaker, H.R.; Amiri, A.; Saeidian, H. Novel 1,2,3-Triazole-Based Benzothiazole Derivatives: Efficient Synthesis, DFT, Molecular Docking, and ADMET Studies. Molecules 2022, 27, 8555. https://doi.org/10.3390/molecules27238555
Mirjafary Z, Mohammad Karbasi M, Hesamzadeh P, Shaker HR, Amiri A, Saeidian H. Novel 1,2,3-Triazole-Based Benzothiazole Derivatives: Efficient Synthesis, DFT, Molecular Docking, and ADMET Studies. Molecules. 2022; 27(23):8555. https://doi.org/10.3390/molecules27238555
Chicago/Turabian StyleMirjafary, Zohreh, Mahdieh Mohammad Karbasi, Parsa Hesamzadeh, Hamid Reza Shaker, Asghar Amiri, and Hamid Saeidian. 2022. "Novel 1,2,3-Triazole-Based Benzothiazole Derivatives: Efficient Synthesis, DFT, Molecular Docking, and ADMET Studies" Molecules 27, no. 23: 8555. https://doi.org/10.3390/molecules27238555
APA StyleMirjafary, Z., Mohammad Karbasi, M., Hesamzadeh, P., Shaker, H. R., Amiri, A., & Saeidian, H. (2022). Novel 1,2,3-Triazole-Based Benzothiazole Derivatives: Efficient Synthesis, DFT, Molecular Docking, and ADMET Studies. Molecules, 27(23), 8555. https://doi.org/10.3390/molecules27238555