
Mechanisms in the Catalytic Reduction of N2O by CO over the M13@Cu42 Clusters of Aromatic-like Inorganic and Metal Compounds


Abstract
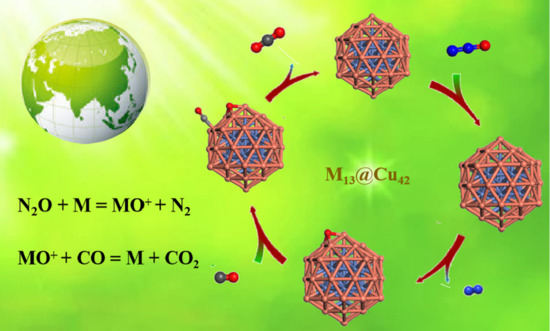
:
1. Introduction
2. Computational Details
3. Results and Discussion
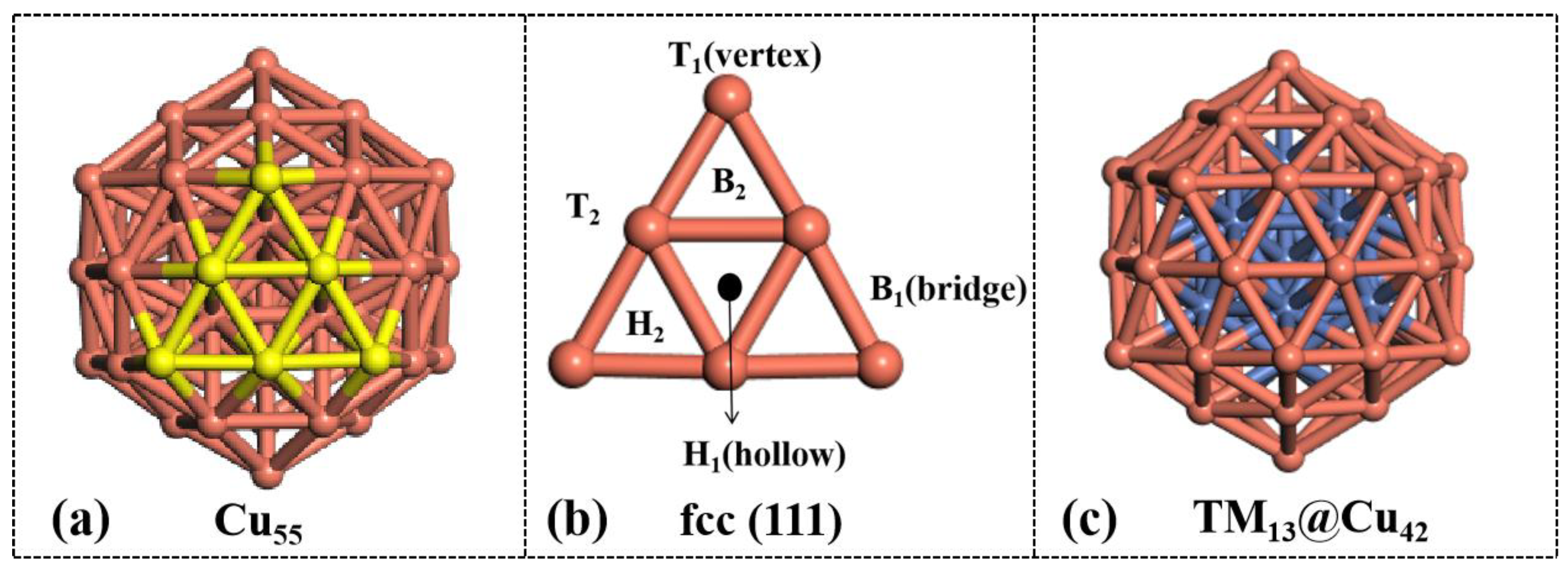
3.1. Structure
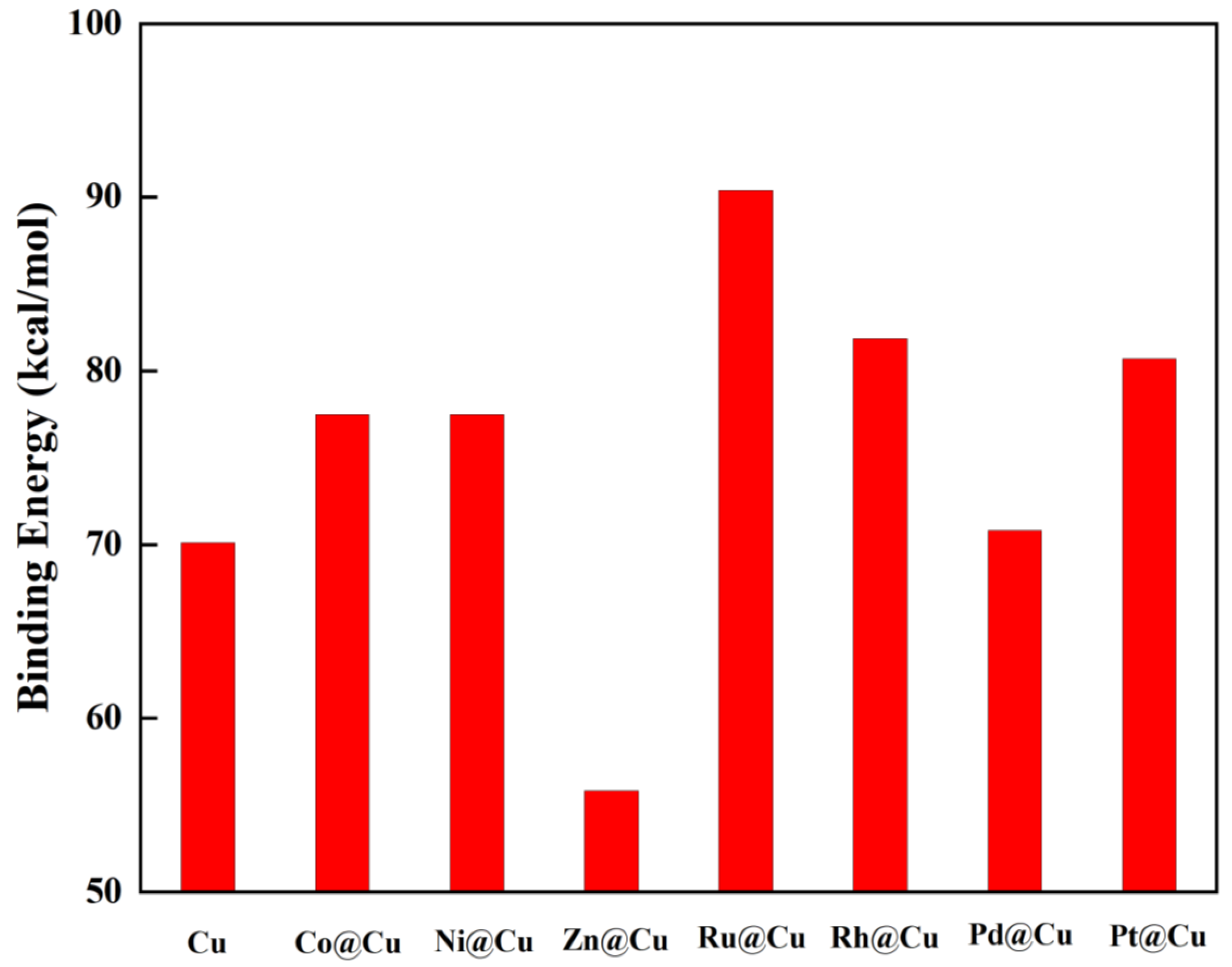
3.2. Catalytic Properties of M13@Cu42 Cluster
3.3. Electronic Structure Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Trogler, W.C. Physical properties and mechanisms of formation of nitrous oxide. Coord. Chem. Rev. 1999, 187, 303–327. [Google Scholar] [CrossRef]
- Ravishankara, A.; Daniel, J.S.; Portmann, R.W. Nitrous oxide (N2O): The dominant ozone-depleting substance emitted in the 21st century. Science 2009, 326, 123–125. [Google Scholar] [CrossRef] [PubMed]
- Dameris, M. Depletion of the ozone layer in the 21st century. Angew. Chem. Int. Ed. 2010, 49, 489–491. [Google Scholar] [CrossRef] [PubMed]
- Esrafili, M.D.; Janebi, H.; Mousavian, P. Single Al atom anchored on defective MoS2: An efficient catalytic site for reduction of greenhouse N2O gas by CO or C2H4 molecules. Appl. Surf. Sci. 2021, 569, 151001. [Google Scholar] [CrossRef]
- Shao, L.; Chen, J.; Wang, K.; Mei, J.; Tan, T.; Wang, G.; Liu, K.; Gao, X. Highly precise measurement of atmospheric N2O and CO using improved White cell and RF current perturbation. Sens. Actuators B Chem. 2022, 352, 130995. [Google Scholar] [CrossRef]
- Esrafili, M.D.; Nejadebrahimi, B. N2O reduction over a porous Si-decorated carbon nitride fullerene: A DFT study. Chem. Phys. Lett. 2019, 716, 11–16. [Google Scholar] [CrossRef]
- Sittiwong, J.; Jaturajamrenchai, T.; Wongkampuan, P.; Somwatcharajit, N.; Impeng, S.; Maihom, T.; Probst, M.; Limtrakul, J. Modulating the catalytic activity of metal-organic frameworks for CO oxidation with N2O through an oriented external electric field. Mol. Catal. 2021, 516, 111970. [Google Scholar] [CrossRef]
- Amaya-Roncancio, S.; Reinaudi, L.; Gimenez, M.C. Adsorption and dissociation of CO on metal clusters. Mater. Today Commun. 2020, 24, 101158. [Google Scholar] [CrossRef]
- Chen, P.; Gu, M.; Chen, G.; Liu, F.; Lin, Y. DFT study on the reaction mechanism of N2O reduction with CO catalyzed by char. Fuel 2019, 254, 115666. [Google Scholar] [CrossRef]
- Khan, A.A.; Ahmad, R.; Ahmad, I. Removal of nitrous and carbon mono oxide from flue gases by Si-coordinated nitrogen doped C60-fullerene: A DFT approach. Mol. Catal. 2021, 509, 111674. [Google Scholar] [CrossRef]
- You, Y.; Chen, S.; Li, J.; Zeng, J.; Chang, H.; Ma, L.; Li, J. Low-temperature selective catalytic reduction of N2O by CO over Fe-ZSM-5 catalysts in the presence of O2. J. Hazard. Mater. 2020, 383, 121117. [Google Scholar] [CrossRef] [PubMed]
- Boehme, D.K.; Schwarz, H. Gas-phase catalysis by atomic and cluster metal ions: The ultimate single-site catalysts. Angew. Chem. Int. Ed. 2005, 44, 2336–2354. [Google Scholar] [CrossRef]
- Delabie, A.; Vinckier, C.; Flock, M.; Pierloot, K. Evaluating the activation barriers for transition metal N2O reactions. J. Phys. Chem. A 2001, 105, 5479–5485. [Google Scholar] [CrossRef]
- Campa, M.C.; Doyle, A.M.; Fierro, G.; Pietrogiacomi, D. Simultaneous abatement of NO and N2O with CH4 over modified Al2O3 supported Pt, Pd, Rh. Catal. Today 2022, 384, 76–87. [Google Scholar] [CrossRef]
- Okemoto, A.; Harada, M.R.; Ishizaka, T.; Hiyoshi, N.; Sato, K. Catalytic performance of MoO3/FAU zeolite catalysts modified by Cu for reverse water gas shift reaction. Appl. Catal. A Gen. 2020, 592, 117415. [Google Scholar] [CrossRef]
- Kim, K.; Baek, S.; Kim, J.J.; Han, J.W. Catalytic decomposition of N2O on PdxCuy alloy catalysts: A density functional theory study. Appl. Surf. Sci. 2020, 510, 145349. [Google Scholar] [CrossRef]
- Richards, N.; Carter, J.H.; Parker, L.A.; Pattisson, S.; Hewes, D.G.; Morgan, D.J.; Davies, T.E.; Dummer, N.F.; Golunski, S.; Hutchings, G.J. Lowering the operating temperature of perovskite catalysts for N2O decomposition through control of preparation methods. ACS Catal. 2020, 10, 5430–5442. [Google Scholar] [CrossRef]
- Li, X.; Kuznetsov, A.E.; Zhang, H.-F.; Boldyrev, A.I.; Wang, L.-S. Observation of all-metal aromatic molecules. Science 2001, 291, 859–861. [Google Scholar] [CrossRef]
- Zhu, C.; Li, S.; Luo, M.; Zhou, X.; Niu, Y.; Lin, M.; Zhu, J.; Cao, Z.; Lu, X.; Wen, T. Stabilization of anti-aromatic and strained five-membered rings with a transition metal. Nat. Chem. 2013, 5, 698–703. [Google Scholar] [CrossRef]
- Niu, T.; Liu, G.; Chen, Y.; Yang, J.; Wu, J.; Cao, Y.; Liu, Y. Hydrothermal synthesis of graphene-LaFeO3 composite supported with Cu-Co nanocatalyst for higher alcohol synthesis from syngas. Appl. Surf. Sci. 2016, 364, 388–399. [Google Scholar] [CrossRef]
- Wang, X.; Qiu, S.; Feng, J.; Tong, Y.; Zhou, F.; Li, Q.; Song, L.; Chen, S.; Wu, K.H.; Su, P. Confined Fe–Cu clusters as sub-nanometer reactors for efficiently regulating the electrochemical nitrogen reduction reaction. Adv. Mater. 2020, 32, 2004382. [Google Scholar] [CrossRef]
- Nicholas, K.M.; Lander, C.; Shao, Y. Computational Evaluation of Potential Molecular Catalysts for Nitrous Oxide Decomposition. Inorg. Chem. 2022, 61, 14591–14605. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Huang, L.; Zhu, X.; Fang, Y.; Dong, S. Coupling Cu with Au for enhanced electrocatalytic activity of nitrogen reduction reaction. Nanoscale 2020, 12, 1811–1816. [Google Scholar] [CrossRef]
- Kim, C.; Kim, J. Comparative evaluation of artificial neural networks for the performance prediction of Pt-based catalysts in water gas shift reaction. Int. J. Energy Res. 2022, 46, 9602–9620. [Google Scholar] [CrossRef]
- Ko, B.H.; Hasa, B.; Shin, H.; Jeng, E.; Overa, S.; Chen, W.; Jiao, F. The impact of nitrogen oxides on electrochemical carbon dioxide reduction. Nat. Commun. 2020, 11, 5856. [Google Scholar] [CrossRef] [PubMed]
- Pan, K.L.; Young, C.W.; Pan, G.T.; Chang, M.B. Catalytic reduction of NO by CO with Cu-based and Mn-based catalysts. Catal. Today 2020, 348, 15–25. [Google Scholar] [CrossRef]
- Andana, T.; Rappé, K.G.; Nelson, N.C.; Gao, F.; Wang, Y. Selective catalytic reduction of NOx with NH3 over Ce-Mn oxide and Cu-SSZ-13 composite catalysts–Low temperature enhancement. Appl. Catal. B Environ. 2022, 316, 121522. [Google Scholar] [CrossRef]
- Barabás, J.L.; Höltzl, T. Reaction of N2O and CO catalyzed with small copper clusters: Mechanism and design. J. Phys. Chem. A 2016, 120, 8862–8870. [Google Scholar] [CrossRef]
- Lian, X.; Guo, W.; He, B.; Yu, B.; Chen, S.; Qin, D.; Chen, F. Insights of the mechanisms for CO oxidation by N2O over M@Cu12 (M = Cu, Pt, Ru, Pd, Rh) core-shell clusters. Mol. Catal. 2020, 494, 111126. [Google Scholar] [CrossRef]
- Jiang, T.; Mowbray, D.; Dobrin, S.; Falsig, H.; Hvolbæk, B.; Bligaard, T.; Nørskov, J.K. Trends in CO oxidation rates for metal nanoparticles and close-packed, stepped, and kinked surfaces. J. Phys. Chem. C 2009, 113, 10548–10553. [Google Scholar] [CrossRef]
- Mao, X.; Wang, L.; Xu, Y.; Wang, P.; Li, Y.; Zhao, J. Computational high-throughput screening of alloy nanoclusters for electrocatalytic hydrogen evolution. npj Comput. Mater. 2021, 7, 46. [Google Scholar] [CrossRef]
- Cao, X.; Ji, Y.; Luo, Y. Dehydrogenation of propane to propylene by a Pd/Cu single-atom catalyst: Insight from first-principles calculations. J. Phys. Chem. C 2015, 119, 1016–1023. [Google Scholar] [CrossRef]
- Liu, Q.; Wang, X.; Li, L.; Song, K.; Qian, P.; Feng, Y.P. Design of platinum single-atom doped metal nanoclusters as efficient oxygen reduction electrocatalysts by coupling electronic descriptor. Nano Res. 2022, 15, 7016–7025. [Google Scholar] [CrossRef]
- Liu, C.; Dong, H.; Ji, Y.; Rujisamphan, N.; Li, Y. High-performance hydrogen evolution reaction catalysis achieved by small core-shell copper nanoparticles. J. Colloid Interface Sci. 2019, 551, 130–137. [Google Scholar] [CrossRef]
- Zhang, R.; Wang, Y.; Wang, B.; Ling, L. Probing into the effects of cluster size and Pd ensemble as active center on the activity of H2 dissociation over the noble metal Pd-doped Cu bimetallic clusters. Mol. Catal. 2019, 475, 110457. [Google Scholar] [CrossRef]
- Zhao, B.; Zhang, R.; Huang, Z.; Wang, B. Effect of the size of Cu clusters on selectivity and activity of acetylene selective hydrogenation. Appl. Catal. A Gen. 2017, 546, 111–121. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef]
- Henkelman, G.; Uberuaga, B.P.; Jónsson, H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 2000, 113, 9901–9904. [Google Scholar] [CrossRef]
- Tang, D.; Chen, Z.; Hu, J.; Sun, G.; Lu, S.; Hu, C. CO oxidation catalyzed by silver nanoclusters: Mechanism and effects of charge. Phys. Chem. Chem. Phys. 2012, 14, 12829–12837. [Google Scholar] [CrossRef]
- Austin, N.; Butina, B.; Mpourmpakis, G. CO2 activation on bimetallic CuNi nanoparticles. Prog. Nat. Sci. Mater. Int. 2016, 26, 487–492. [Google Scholar] [CrossRef]
- McEuen, P.; Kittel, C. Introduction to Solid State Physics; John Wiley & Sons: Hoboken, NJ, USA, 2005. [Google Scholar]
- Tang, D.; Zhang, J. Theoretical investigation on CO oxidation catalyzed by a copper nanocluster. RSC Adv. 2013, 3, 15225–15236. [Google Scholar] [CrossRef]
- Akça, A.; Karaman, O.; Karaman, C. Mechanistic insights into catalytic reduction of N2O by CO over Cu-embedded graphene: A density functional theory perspective. ECS J. Solid State Sci. Technol. 2021, 10, 041003. [Google Scholar] [CrossRef]
- Wannakao, S.; Nongnual, T.; Khongpracha, P.; Maihom, T.; Limtrakul, J. Reaction mechanisms for CO catalytic oxidation by N2O on Fe-embedded graphene. J. Phys. Chem. C 2012, 116, 16992–16998. [Google Scholar] [CrossRef]
- Junkaew, A.; Namuangruk, S.; Maitarad, P.; Ehara, M. Silicon-coordinated nitrogen-doped graphene as a promising metal-free catalyst for N2O reduction by CO: A theoretical study. RSC Adv. 2018, 8, 22322–22330. [Google Scholar] [CrossRef]
- Zhou, S.; Pei, W.; Du, Q.; Zhao, J. Foreign atom encapsulated Au 12 golden cages for catalysis of CO oxidation. Phys. Chem. Chem. Phys. 2019, 21, 10587–10593. [Google Scholar] [CrossRef]
- Chase, M.W. NIST–JANAF thermochemical tables for the bromine oxides. J. Phys. Chem. Ref. Data 1996, 25, 1069–1111. [Google Scholar] [CrossRef]
- Wongnongwa, Y.; Namuangruk, S.; Kungwan, N.; Jungsuttiwong, S. Mechanistic study of CO oxidation by N2O over Ag7Au6 cluster investigated by DFT methods. Appl. Catal. A Gen. 2017, 538, 99–106. [Google Scholar] [CrossRef]
- Piskorz, W.; Zasada, F.; Stelmachowski, P.; Kotarba, A.; Sojka, Z. DFT modeling of reaction mechanism and ab initio microkinetics of catalytic N2O decomposition over alkaline earth oxides: From molecular orbital picture account to simulation of transient and stationary rate profiles. J. Phys. Chem. C 2013, 117, 18488–18501. [Google Scholar] [CrossRef]
- Pei, W.; Zhou, S.; Bai, Y.; Zhao, J. N-doped graphitic carbon materials hybridized with transition metals (compounds) for hydrogen evolution reaction: Understanding the synergistic effect from atomistic level. Carbon 2018, 133, 260–266. [Google Scholar] [CrossRef]
- Zhou, S.; Yang, X.; Pei, W.; Liu, N.; Zhao, J. Heterostructures of MXenes and N-doped graphene as highly active bifunctional electrocatalysts. Nanoscale 2018, 10, 10876–10883. [Google Scholar] [CrossRef] [PubMed]
- Xin, H.; Linic, S. Communications: Exceptions to the d-band model of chemisorption on metal surfaces: The dominant role of repulsion between adsorbate states and metal d-states. J. Chem. Phys. 2010, 132, 221101. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| M | dCu-M (Å) | dM-M (Å) | Eb (kcal/mol) | Mag (μB) | CTM (e) | CTCu (e) | εd (eV) | Eg (eV) |
|---|---|---|---|---|---|---|---|---|
| Cu | 2.42 | 2.49 | −70.10 | 3 | +0.06 | −0.017 | 2.42 | 0.1 |
| Co | 2.42 | 2.45 | −77.48 | 21 | −0.05 | 0.017 | 2.21 | 0.01 |
| Ni | 2.43 | 2.45 | −77.48 | 8 | −0.01 | 0.003 | 2.07 | 0.01 |
| Zn | 2.44 | 2.59 | −55.81 | 2 | −0.16 | 0.052 | 3.51 | 0.59 |
| Ru | 2.62 | 2.61 | −90.40 | 4 | +0.01 | −0.002 | 2.45 | 0.02 |
| Rh | 2.48 | 2.69 | −81.86 | 9 | +0.12 | −0.035 | 2.22 | 0.02 |
| Pd | 2.48 | 2.70 | −70.79 | 2 | +0.18 | −0.054 | 2.11 | 0.17 |
| Pt | 2.65 | 2.82 | −80.71 | 0 | +0.28 | −0.086 | 2.26 | 0.16 |
| E-R | L-H | |||||||
|---|---|---|---|---|---|---|---|---|
| M13@Cu42 | ΔEN2O (kcal/mol) | ΔECO (kcal/mol) | CT (e) | TS (kcal/mol) | ΔEN2O-CO (kcal/mol) | ΔEO-CO (kcal/mol) | CT (e) | TS (kcal/mol) |
| Cu | −53.27 | −27.44 | 0.97 | 11.64 | −72.87 | −7.61 | 1.09 | 10.58 |
| Co | −50.04 | −28.59 | 1.07 | 14.75 | −77.48 | −2.77 | 1.10 | 9.80 |
| Ni | −46.12 | −32.51 | 1.08 | 10.97 | −74.02 | −5.53 | 1.08 | 9.68 |
| Zn | −50.27 | −28.36 | 0.97 | 17.35 | −78.40 | −2.31 | 1.09 | 19.58 |
| Ru | −50.04 | −28.36 | 1.11 | 12.90 | −86.71 | −5.77 | 1.12 | 11.99 |
| Rh | −50.96 | −27.67 | 1.10 | 13.99 | −84.40 | −2.54 | 1.13 | 12.53 |
| Pd | −70.33 | −20.06 | 1.09 | 22.37 | −97.77 | −0.92 | 1.18 | 18.22 |
| Pt | −57.88 | −20.75 | 1.11 | 14.93 | −75.41 | −4.84 | 1.12 | 12.47 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Z.; Wang, H.; Gao, Y.; Zhao, J. Mechanisms in the Catalytic Reduction of N2O by CO over the M13@Cu42 Clusters of Aromatic-like Inorganic and Metal Compounds. Molecules 2023, 28, 4485. https://doi.org/10.3390/molecules28114485
Liu Z, Wang H, Gao Y, Zhao J. Mechanisms in the Catalytic Reduction of N2O by CO over the M13@Cu42 Clusters of Aromatic-like Inorganic and Metal Compounds. Molecules. 2023; 28(11):4485. https://doi.org/10.3390/molecules28114485
Chicago/Turabian StyleLiu, Ziyang, Haifeng Wang, Yan Gao, and Jijun Zhao. 2023. "Mechanisms in the Catalytic Reduction of N2O by CO over the M13@Cu42 Clusters of Aromatic-like Inorganic and Metal Compounds" Molecules 28, no. 11: 4485. https://doi.org/10.3390/molecules28114485
APA StyleLiu, Z., Wang, H., Gao, Y., & Zhao, J. (2023). Mechanisms in the Catalytic Reduction of N2O by CO over the M13@Cu42 Clusters of Aromatic-like Inorganic and Metal Compounds. Molecules, 28(11), 4485. https://doi.org/10.3390/molecules28114485