

Differentiating Dyes: A Spectroscopic Investigation into the Composition of Scarlet Bloodroot (Haemodorum coccineum R.Br.) Rhizome

 , , and
, , and
Abstract
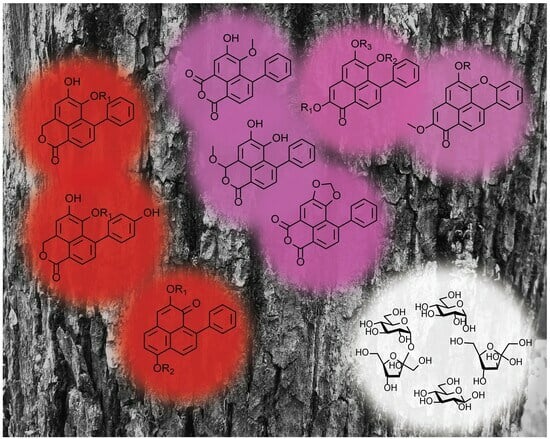
:
1. Introduction
2. Results and Discussion
2.1. Extration and Generation of Extract
2.2. Fractionation of Methanolic Extract
2.3. Malonate or Allophanyl?
2.4. Two Series of Glycosilated Phenylphenalenone
2.5. Pressurised Hot Water Extraction (PHWE)
2.6. Hydrolysis of PHW Extract
2.7. Preliminary Antimicrobial Activity
2.8. Rhizosheath Phytochemical Analysis
3. Conclusions
4. Materials and Methods
4.1. General
4.2. Plant Extraction and Isolation
- Solid-Phase Extraction (SPE)
- HPLC of MeOH(aq)
- Column of CHCl3
- PHWE
- Hydrolysis
- HPLC of organic soluble hydrolysis products.
4.3. Characterisation
4.4. Soil Soxhlet Extraction
4.5. Antimicrobial Assay
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cooke, R.G.; Segal, W. Coloring matters of Australian plants. IV. Haemocorin: A unique glycoside from Haemodorum corymbosum Vahl. Aust. J. Chem. 1955, 8, 107–113. [Google Scholar] [CrossRef]
- Thomas, R. Biosynthesis of the plant phenalenone hemocorin. J. Chem. Soc. D 1971, 14, 739–740. [Google Scholar] [CrossRef]
- Thomas, R. Biosynthesis of phenalenones. Pure Appl. Chem. 1973, 34, 515–528. [Google Scholar] [CrossRef]
- Bick, I.R.C.; Blackman, A.J. Haemodorin. a phenalenone pigment. Aust. J. Chem. 1973, 26, 1377–1380. [Google Scholar] [CrossRef]
- Edwards, J.M. Phenylphenalenones from Wachendorfia species. Phytochemistry 1974, 13, 290–291. [Google Scholar] [CrossRef]
- Dias, D.A.; Goble, D.J.; Silva, C.A.; Urban, S. Phenylphenalenones from the Australian Plant Haemodorum simplex. J. Nat. Prod. 2009, 72, 1075–1080. [Google Scholar] [CrossRef]
- Urban, S.; Brkljaca, R.; White, J.M.; Timmers, M.A. Phenylphenalenones and oxabenzochrysenones from the Australian plant Haemodorum simulans. Phytochemistry 2013, 95, 351–359. [Google Scholar] [CrossRef]
- Brkljaca, R.; Urban, S. HPLC-NMR and HPLC-MS profiling and bioassay-guided identification of secondary metabolites from the Australian plant Haemodorum spicatum. J. Nat. Prod. 2015, 78, 1486–1494. [Google Scholar] [CrossRef]
- Brock, J. Top End Native Plants; John Brock: Darwin, Australia, 1988. [Google Scholar]
- Aboriginal Communities of the Northern Territory. Traditional Aboriginal Medicines in the Northern Territory of Australia, 1st ed.; Conservation Commission of the Northern Territory: Darwin, Australia, 1993. [Google Scholar]
- Macfarlane, T.D. Haemodorum. In Flora of Australia; George, A.S., Ed.; Australian Government Publishing Service: Canberra, Australia, 1987; Volume 45, p. 134. [Google Scholar]
- Just, J.; Deans, B.J.; Olivier, W.J.; Paull, B.; Bissember, A.C.; Smith, J.A. New Method for Rapid Extraction of Natural Products: Efficient Isolation of Shikimic Acid from Star Anise. Org. Lett. 2015, 17, 2428–2430. [Google Scholar] [CrossRef]
- Ali, S.E.; El Gedaily, R.A.; Mocan, A.; Farag, M.A.; El-Seedi, H.R. Profiling Metabolites and Biological Activities of Sugarcane (Saccharum officinarum Linn.) Juice and its Product Molasses via a Multiplex Metabolomics Approach. Molecules 2019, 24, 934. [Google Scholar] [CrossRef]
- Fang, J.-J.; Paetz, C.; Hoelscher, D.; Munde, T.; Schneider, B. Phenylphenalenones and related natural products from Wachendorfia thyrsiflora L. Phytochem. Lett. 2011, 4, 203–208. [Google Scholar] [CrossRef]
- Fang, J.; Kai, M.; Schneider, B. Phytochemical profile of aerial parts and roots of Wachendorfia thyrsiflora L. studied by LC-DAD-SPE-NMR. Phytochemistry 2012, 81, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Opitz, S.; Hoelscher, D.; Oldham, N.J.; Bartram, S.; Schneider, B. Phenylphenalenone-related compounds: Chemotaxonomic markers of the Haemodoraceae from Xiphidium caeruleum. J. Nat. Prod. 2002, 65, 1122–1130. [Google Scholar] [CrossRef] [PubMed]
- Rajabi, F.; Wilhelm, C.; Thiel, W.R. A Bronsted acidic, ionic liquid containing, heteropolyacid functionalized polysiloxane network as a highly sensitive catalyst for the esterification of dicarboxylic acids. Green Chem. 2020, 22, 4438. [Google Scholar] [CrossRef]
- Macholl, S.; Borner, F.; Buntkowsky, G. Revealing the configuration and crytal packing of organic compounds by solid-state NMR Spectroscopy: Methoxycarbonylurea, a Case Study. Chem. Eur. J. 2004, 10, 4808–4816. [Google Scholar] [CrossRef] [PubMed]
- Opitz, S.; Schneider, B. Organ-specific analysis of phenylphenalenone-related compounds in Xiphidium caeruleum. Phytochemistry 2002, 61, 819–825. [Google Scholar] [CrossRef]
- Cooke, R.G.; Merrett, B.K.; O’Loughlin, G.J.; Pietersz, G.A. Colouring matters of Australian plants. XXIII A new synthesis of arylphenalenones and naphthoxanthenones. Aust. J. Chem. 1980, 33, 2317–2324. [Google Scholar] [CrossRef]
- Cooke, R.G.; Dagley, I.J. Colouring matters of Australian plants. XXI naphthoxanthenones in the Haemodoraceae. Aust. J. Chem. 1979, 32, 1841–1847. [Google Scholar] [CrossRef]
- DellaGreca, M.; Previtera, L.; Zarrelli, A. Structures of new phenylphenalene-related compounds from Eichhornia crassipes (water hyacinth). Tetrahedron 2009, 65, 8206–8208. [Google Scholar] [CrossRef]
- Hidalgo, W.; Cano, M.; Arbelaez, M.; Zarrazola, E.; Gil, J.; Schneider, B.; Otalvaro, F. 4-phenylphenalenones as a template for new photodynamic compounds against Mycosphaerella fijiensis. Pest Manag. Sci. 2016, 72, 796–800. [Google Scholar] [CrossRef]
- Norman, E.O.; Lever, J.; Brkljaca, R.; Urban, S. Distribution, biosynthesis, and biological activity of phenylphenalenone-type compounds derived from the family of plants, Haemodoraceae. Nat. Prod. Rep. 2019, 36, 753–768. [Google Scholar] [CrossRef] [PubMed]
- Dora, G.; Xie, X.-Q.; Edwards, J.M. Two novel phenalenones from Dilatris viscosa. J. Nat. Prod. 1993, 56, 2029–2033. [Google Scholar] [CrossRef]
- Carpinelli de Jesus, M.; Wapling, J.; Stockdale, T.; Leach, D.; Church, T.; Collins, R.; Leach, G.; Hewitt, D.; De Voss, J.J.; Blanchfield, J.T. Phytochemical composition of Denhamia obscura (A. Rich.) Meisn. Ex Walp. root bark, seeds and leaves. Arckivoc 2022, 4, 123–142. [Google Scholar] [CrossRef]
- CLSI. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria that Grow Aerobically, 9th ed.; CLSI standard M07; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2012. [Google Scholar]
- CLSI. Methods for Dilution Antifungal Susceptibility Testing of Yeasts, 4th ed.; CLSI standard M27; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2016. [Google Scholar]
- European Pharmacopoeia. Determination of Bactericidal, Fungicidal or Yeasticidal Activity of Antiseptic Medicinal Products; EDQM: Strasbourg, France, 2017; Version 9.2, p. Chapter 5.1.11; pp. 4348–4350. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formula | Accurate Mass | |
|---|---|---|
| Allophanyl | C2H4N2O3 | 104.022193 |
| Malonate | C3H4O4 | 104.010960 |
 | ||||||
| Compound | 1″ | Δurea | Δmal | 2″ | Δurea | Δmal |
| 4′ | 168.2 | 14.7 | 2.0 | 169.9 | 15.0 | 0.1 |
| 5′ | 169.3 | 15.8 | 3.1 | 171.5 | 16.6 | 1.7 |
| 19′’ | 170.0 | 16.5 | 3.8 | 172.4 | 17.5 | 2.6 |
| Formula | Accurate Mass | δ | |
|---|---|---|---|
| Reported | Compound 4′ [M + H] | 567.1486 | - |
| Allophanyl | C28H27N2O11 | 567.1615 | 0.0129 |
| Malonate | C29H27O12 | 567.1503 | 0.0017 |
| Reported | Compound 5′ [M + H] | 553.1278 | - |
| Allophanyl | C27H25N2O11 | 553.1458 | 0.0180 |
| Malonate | C28H25O12 | 553.1346 | 0.0068 |
| Reported | Compound 19′ [M + H] | 541.1378 | - |
| Allophanyl | C26H25N2O11 | 541.1458 | 0.0080 |
| Malonate | C27H25O12 | 541.1346 | 0.0032 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carpinelli de Jesus, M.; Church, T.; Wapling, J.A.; Collins, R.; Leach, G.J.; Leach, D.; De Voss, J.J.; Blanchfield, J.T. Differentiating Dyes: A Spectroscopic Investigation into the Composition of Scarlet Bloodroot (Haemodorum coccineum R.Br.) Rhizome. Molecules 2023, 28, 7422. https://doi.org/10.3390/molecules28217422
Carpinelli de Jesus M, Church T, Wapling JA, Collins R, Leach GJ, Leach D, De Voss JJ, Blanchfield JT. Differentiating Dyes: A Spectroscopic Investigation into the Composition of Scarlet Bloodroot (Haemodorum coccineum R.Br.) Rhizome. Molecules. 2023; 28(21):7422. https://doi.org/10.3390/molecules28217422
Chicago/Turabian StyleCarpinelli de Jesus, Matheus, Taylah Church, Johanna A. Wapling, Raelene Collins, Gregory J. Leach, David Leach, James J. De Voss, and Joanne T. Blanchfield. 2023. "Differentiating Dyes: A Spectroscopic Investigation into the Composition of Scarlet Bloodroot (Haemodorum coccineum R.Br.) Rhizome" Molecules 28, no. 21: 7422. https://doi.org/10.3390/molecules28217422
APA StyleCarpinelli de Jesus, M., Church, T., Wapling, J. A., Collins, R., Leach, G. J., Leach, D., De Voss, J. J., & Blanchfield, J. T. (2023). Differentiating Dyes: A Spectroscopic Investigation into the Composition of Scarlet Bloodroot (Haemodorum coccineum R.Br.) Rhizome. Molecules, 28(21), 7422. https://doi.org/10.3390/molecules28217422