
Condensation of Benzyl Carbamate with Glyoxal in Polar Protic and Aprotic Solvents


Abstract
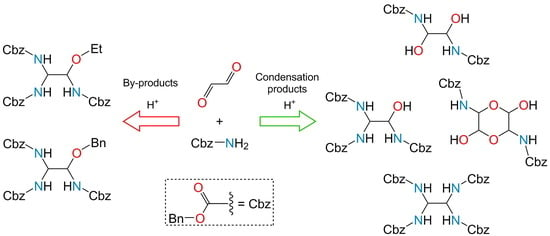
:
1. Introduction
2. Results and Discussion
3. Materials and Methods
4. Experimental
4.1. N,N′-Bis(carbobenzoxy)-3,6-diamino-1,4-dioxane-2,5-diol (2)
4.2. N,N′-Bis(carbobenzoxy)ethan-1,2-diol (3)
4.3. N,N′,N″-Tris(carbobenzoxy)ethanol (4)
4.4. N,N′,N″,N‴-Tetrakis(carbobenzoxy)ethan (5)
4.5. N,N′,N″-Tris(carbobenzoxy)-2-ethoxyethan (7)
4.6. N,N′,N″-Tris(carbobenzoxy)-2-benzoxyethan (8)
5. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Xue, Q.; Bi, F.; Zhang, J.; Zhang, J.; Wang, B.; Wu, M. Synthesis and characterization of two 1,2,4-oxadiazole-furazan-based nitrate ester compounds as potential energetic plasticizers. FirePhysChem 2023, 3, 16–22. [Google Scholar] [CrossRef]
- Parakhin, V.V.; Smirnov, G.A. Research progress on design, synthesis and performance of energetic polynitro hexaazaisowurtzitane derivatives: Towards improved CL-20 analogues. FirePhysChem 2023, in press. [Google Scholar] [CrossRef]
- Yadav, A.K.; Jujam, M.; Ghule, V.D.; Dharavath, S. High-performing, insensitive and thermally stable energetic materials from zwitterionic gem-dinitromethyl substituted C-C bonded 1,2,4-triazole and 1,3,4-oxadiazole. Chem. Commun. 2023, 59, 4324–4327. [Google Scholar] [CrossRef]
- Zhang, W.; Yang, Y.; Wang, Y.; Fei, T.; Wang, Y.; Sun, C.; Pang, S. Challenging the limits of the oxygen balance of a pyrazole ring. Chem. Eng. J. 2023, 451, 138609. [Google Scholar] [CrossRef]
- Wang, T.; Lu, Z.; Bu, S.; Kuang, B.; Zhang, L.; Yi, Z.; Wang, K.; Zhu, S.; Zhang, J. Combination of nitrogen-rich skeleton and coordination group: Synthesis of a high-energy primary explosive based on 1H-tetrazole-5-carbohydrazide. Def. Technol. 2023, in press. [Google Scholar] [CrossRef]
- Wang, K.; Li, X.; Yang, K.; Huo, H.; Xue, Q.; Wang, B.; Bi, F. A novel synthetic method of 1,1,4,4-tetramethyl-2-tetrazene (TMTZ) via photocatalytic reaction. FirePhysChem 2022, 2, 267–271. [Google Scholar] [CrossRef]
- Xiong, J.; Cai, J.; Lai, Q.; Yin, P.; Pang, S. Asymmetric assembly of pyrazole and 1,2,3-triazole with a methylene bridge: Regioisomerism and energetic properties. Chem. Commun. 2022, 58, 10647–10650. [Google Scholar] [CrossRef]
- Ma, W.; Zhang, Z.Q.; Ma, Q.; Tang, J.; Yang, W.; Yang, H.; Cheng, G.; Fan, G.J. Bicyclic high-energy and low-sensitivity regioisomeric energetic compounds based on polynitrobenzene and pyrazoles. Cryst. Growth Des. 2023, 23, 1127–1132. [Google Scholar] [CrossRef]
- Konnov, A.A.; Klenov, M.S.; Churakov, A.M.; Dalinger, I.L.; Strelenko, Y.A.; Fedyanin, I.V.; Lempert, D.B.; Pivkina, A.N.; Kon’kova, T.S.; Matyushin, Y.N.; et al. Novel energetic furazans containing isomeric N-(azoxy)-dinitropyrazole moieties: Synthesis, characterization and comparison of properties. Energetic Mater. Front. 2023, 4, 1–9. [Google Scholar] [CrossRef]
- Pandey, K.; Bhatia, P.; Dolui, P.; Ghule, V.D.; Kumar, D. Connecting energetic nitropyrazole and nitrobenzene moieties with C-C bonds using suzuki cross-coupling reaction: A novel route to thermally stable energetic materials. Asian J. Org. Chem. 2022, 11, e202200543. [Google Scholar] [CrossRef]
- Leonov, N.E.; Emel’yanov, A.E.; Klenov, M.S.; Churakov, A.M.; Strelenko, Y.A.; Pivkina, A.N.; Fedyanin, I.V.; Lempert, D.B.; Kon’kova, T.S.; Matyushin, Y.N.; et al. Novel (1H-tetrazol-5-yl-NNO-azoxy)furazans and their energetic salts: Synthesis, characterization and energetic properties. Mendeleev Commun. 2022, 32, 714–716. [Google Scholar] [CrossRef]
- Sheremetev, A.B.; Mel’nikova, S.F.; Kokareva, E.S.; Nekrutenko, R.E.; Strizhenko, K.V.; Suponitsky, K.Y.; Pham, T.D.; Pivkina, A.N.; Sinditskii, V.P. Nitroxy- and azidomethyl azofurazans as advanced energetic materials. Def. Technol. 2022, 18, 1369–1381. [Google Scholar] [CrossRef]
- Larin, A.A.; Ananyev, I.V.; Dubasova, E.V.; Teslenko, F.E.; Monogarov, K.A.; Khakimov, D.V.; He, C.; Pang, S.; Gazieva, G.A.; Fershtat, L.L. Simple and energetic: Novel combination of furoxan and 1,2,4-triazole rings in the synthesis of energetic materials. Energ. Mater. Front. 2022, 3, 146–153. [Google Scholar] [CrossRef]
- Paromov, A.E.; Sysolyatin, S.V. Oxaazatetracyclo[5.5.0.03,11.05,9]dodecanes—A promising foundation for the design of ther-mally stable, high-density energetic compounds. Chem. Heterocycl. Compd. 2017, 53, 630–637. [Google Scholar] [CrossRef]
- Gong, X.; Sun, C.; Pang, S.; Zhang, J.; Li, Y.; Zhao, X. Research Progress in Study of Isowurtzitane Derivatives. Chin. J. Org. Chem. 2012, 3, 486–496. [Google Scholar] [CrossRef]
- Kodama, T.; Tojo, M.; Ikeda, M. Hexaazaisowurtzitane Derivatives and Process for Producing the Same. WO Patent 9623792 A1, 17 May 2000. [Google Scholar]
- Nielsen, A.T.; Nissan, R.A.; Vanderah, D.J. Polyazapolycyclics by condensation of aldehydes with amines. 2. Formation of 2,4,6,8,10,12-hexabenzyl-2,4,6,8,10,12-hexaazatetracyclo[5.5.0.05.9.03,11]dodecanes from glyoxal and benzylamines. J. Org. Chem. 1990, 55, 1459–1466. [Google Scholar] [CrossRef]
- Nielsen, A.T. Caged Polynitramine Compound. U.S. Patent 5693794, 2 December 1997. [Google Scholar]
- Nielsen, A.T.; Chafin, A.P.; Christian, S.L.; Moore, D.W.; Nadler, M.P.; Nissan, R.A.; Vanderah, D.J.; Flippen-Anderson, J.L. Synthesis of polyazapolycyclic caged polynitramines. Tetrahedron 1998, 54, 11793–11812. [Google Scholar] [CrossRef]
- Sakovich, G.V.; Sysolyatin, S.V.; Kozyrev, N.V.; Makarovets, N.A. Explosive Composition. RU Patent 2252925, 27 May 2005. [Google Scholar]
- Viswanath, D.S.; Ghosh, T.K.; Boddu, V.M. Hexanitrohexaazaisowurtzitane (HNIW, CL-20). In Emerging Energetic Materials: Synthesis, Physicochemical, and Detonation Properties; Viswanath, D.S., Ghosh, T.K., Boddu, V.M., Eds.; Springer: Dordrecht, The Netherlands, 2018; pp. 59–100. [Google Scholar] [CrossRef]
- Sysolyatin, S.V.; Lobanova, A.A.; Chernikova, Y.T.; Sakovich, G.V. Methods of synthesis and properties of hexanitrohexaazaisowurtzitane. Russ. Chem. Rev. 2005, 74, 757–764. [Google Scholar] [CrossRef]
- Venkata Viswanath, J.; Venugopal, K.J.; Srinivasa Rao, N.V.; Venkataraman, A. An overview on importance, synthetic strategies and studies of 2,4,6,8,10,12-hexanitro-2,4,6,8,10,12-hexaazaisowurtzitane (HNIW). Def. Technol. 2016, 12, 401–418. [Google Scholar] [CrossRef]
- Nair, U.R.; Sivabalan, R.; Gore, G.M.; Geetha, M.; Asthana, S.N.; Singh, H. Hexanitrohexaazaisowurtzitane (CL-20) and CL-20-based formulations (review). Combust. Explos. Shock. Waves 2005, 41, 121–132. [Google Scholar] [CrossRef]
- Bumpus, J.A. A Theoretical Investigation of the Ring Strain Energy, Destabilization Energy, and Heat of Formation of CL-20. Adv. Phys. Chem. 2012, 2012, 175146. [Google Scholar] [CrossRef]
- Krause, H.H. New Energetic Materials. In Energetic Materials: Particle Processing and Characterization; Teipel, U., Ed.; Wiley-VCH: Weinheim, Germany, 2005; pp. 1–25. [Google Scholar]
- Mandal, A.K.; Pant, C.S.; Kasar, S.M.; Soman, T. Process Optimization for Synthesis of CL-20. J. Energ. Mater 2009, 27, 231–246. [Google Scholar] [CrossRef]
- Talawar, M.B.; Sivabalan, R.; Anniyappan, M.; Gore, G.M.; Asthana, S.N.; Gandhe, B.R. Emerging trends in advanced high energy materials. Combust. Explos. Shock Waves 2007, 43, 62–72. [Google Scholar] [CrossRef]
- Singh, H. Survey of New Energetic and Eco-friendly. Materials for Propulsion of Space Vehicles. In Chemical Rocket Propulsion. A Comprehensive Survey of Energetic Materials; De Luca, L.T., Shimada, T., Sinditskii, V.P., Calabro, M., Eds.; Springer: Dordrecht, The Netherlands, 2017; pp. 127–138. [Google Scholar]
- Aldoshin, S.M.; Lempert, D.B.; Goncharov, T.K.; Kazakov, A.I.; Soglasnova, S.I.; Dorofeenko, E.M.; Plishkin, N.A. Energetic potential of solid composite propellants based on CL-20-containing bimolecular crystals. Russ. Chem. Bull. 2016, 65, 2018–2024. [Google Scholar] [CrossRef]
- Wu, Z.; Liu, N.; Zheng, W.; Chen, J.; Song, X.; Wang, J.; Cui, C.; Zhang, D.; Zhao, F. Application and Properties of CL-20/HMX Cocrystal in Composite Modified Double Base Propellants. Propellants Explos. Pyrotech. 2020, 45, 92–100. [Google Scholar] [CrossRef]
- Wang, J.; Yang, L.; Zheng, W.; Zhang, J. Study on Comparative Performance of CL-20/RDX-based CMDB Propellants. Propellants Explos. Pyrotech. 2019, 44, 1175–1182. [Google Scholar] [CrossRef]
- Sergienko, A.V.; Popenko, E.M.; Slyusarsky, K.V.; Larionov, K.B.; Dzidziguri, E.L.; Kondratyeva, E.S.; Gromov, A.A. Burning Characteristics of the HMX/CL-20/AP/Polyvinyltetrazole Binder/Al Solid Propellants Loaded with Nanometals. Propellants Explos. Pyrotech. 2019, 44, 217–223. [Google Scholar] [CrossRef]
- Sinditskii, V.P.; Chernyi, A.N.; Egorshev, V.Y.; Dashko, D.V.; Goncharov, T.K.; Shisho, N.I. Combustion of CL-20 cocrystals. Combust. Flame 2019, 207, 51–62. [Google Scholar] [CrossRef]
- Zhou, S.; Wu, F.; Tang, G.; Wang, Y.; Pang, A. Effects of 2CL-20/HMX cocrystals on the thermal decomposition behavior and combustion properties of polyether solid propellants. Energetic Mater. Front. 2021, 2, 96–104. [Google Scholar] [CrossRef]
- Wang, Y.; Wen, J.; Yang, J.; Zhang, G.; Wang, N.; Wu, Y. Investigations on the thermal response of a solid rocket motor with complex charge structure using CL-20/GAP propellant. Case Stud. Therm. Eng. 2022, 37, 102257. [Google Scholar] [CrossRef]
- Li, M.; Hu, R.; Xu, M.; Wang, Q.; Yang, W. Burning characteristics of high density foamed GAP/CL-20 propellants. Def. Technol. 2021, 18, 1914–1921. [Google Scholar] [CrossRef]
- Yang, L.-F.; Shi, X.-R.; Li, C.-Z.; Wu, B.; Pei, C.-H. Microfluidic assisted 90% loading CL-20 spherical particles: Enhancing self-sustaining combustion performance. Def. Technol. 2023, 22, 176–184. [Google Scholar] [CrossRef]
- Shi, Y.; Bai, L.; Gong, J.; Ju, X. Theoretical calculation into the structures, stability, sensitivity, and mechanical properties of 2,4,6,8,10,12-hexanitro-2,4,6,8,10,12 hexaazaisowurtzitane (CL-20)/1-amino-3-methyl-1,2,3-triazoliumnitrate (1-AMTN) cocrystal and its mixture. Struct. Chem. 2020, 31, 647–655. [Google Scholar] [CrossRef]
- Zhu, Y.; Luo, J.; Lu, Y.; Li, H.; Gao, B.; Wang, D.; Zhang, X.; Guo, C. Emulsion synthesis of CL-20/DNA composite with excellent superfine spherical improved sensitivity performance via a combined ultrasonic–microwave irradiation approach. J. Mater. Sci. 2018, 53, 14231–14240. [Google Scholar] [CrossRef]
- Chen, T.; Zhang, Y.; Guo, S.-F.; Zhao, L.-m.; Chen, W.; Hao, G.-Z.; Xiao, L.; Ke, X.; Jiang, W. Preparation and property of CL-20/BAMO-THF energetic nanocomposites. Def. Technol. 2019, 15, 306–312. [Google Scholar] [CrossRef]
- Chapman, C.J.; Groven, L.J. Evaluation of a CL-20/TATB Energetic Co-crystal. Propellants Explos. Pyrotech. 2019, 44, 293–300. [Google Scholar] [CrossRef]
- Liu, N.; Duan, B.; Lu, X.; Mo, H.; Xu, M.; Zhanga, Q.; Wang, B. Preparation of CL-20/DNDAP cocrystals by a rapid and continuous spray drying method: An alternative to cocrystal formation. CrystEngComm 2018, 20, 2060–2067. [Google Scholar] [CrossRef]
- Herrmannsdorfer, D.; Gerber, P.; Heintz, T.; Herrmann, M.J.; Klapotke, T.M. Investigation of Crystallisation Conditions to Produce CL-20/HMX Cocrystal for Polymer-bonded Explosives. Propellants Explos. Pyrotech. 2019, 44, 668–678. [Google Scholar] [CrossRef]
- Tan, Y.; Yang, Z.; Wang, H.; Li, H.; Nie, F.; Liu, Y.; Yu, Y. High Energy Explosive with Low Sensitivity: A New Energetic Cocrystal Based on CL-20 and 1,4-DNI. Cryst. Growth Des. 2019, 19, 4476–4482. [Google Scholar] [CrossRef]
- Li, P.; Liu, K.; Ao, D.; Liu, X.; Xu, H.; Duan, X.; Pei, C. A Low-Sensitivity Nanocomposite of CL-20 and TATB. Cryst. Res. Technol. 2018, 53, 1800189. [Google Scholar] [CrossRef]
- Wu, C.-L.; Zhang, S.-H.; Gou, R.-J.; Ren, F.-D.; Han, G.; Zhu, S.-F. Theoretical insight into the effect of solvent polarity on the formation and morphology of 2,4,6,8,10,12-hexanitrohexaazaisowurtzitane (CL-20)/2,4,6-trinitro-toluene(TNT) cocrystal explosive. Comput. Theor. Chem. 2018, 1127, 22–30. [Google Scholar] [CrossRef]
- Vuppuluri, V.S.; Samuels, P.J.; Caflin, K.C.; Gunduz, I.E.; Son, S.F. Detonation Performance Characterization of a Novel CL-20 Cocrystal Using Microwave Interferometry. Propellants Explos. Pyrotech. 2018, 43, 38–47. [Google Scholar] [CrossRef]
- Hai, L.; Yi, L.; Zhaoxia, M.; Zhixuan, Z.; Junling, L.; Yuanhang, H. Study on the Initial Decomposition Mechanism of Energetic Co-Crystal 2,4,6,8,10,12-Hexanitro-2,4,6,8,10,12-Hexaazaisowurtzitane (CL-20)/1,3,5,7-Tetranitro-1,3,5,7-Tetrazacyclooctane (HMX) under a Steady Shock Wave. Acta Phys.-Chim. Sin. 2019, 35, 858–867. [Google Scholar] [CrossRef]
- Sun, S.; Zhang, H.; Xu, J.; Wang, H.; Wang, S.; Yu, Z.; Zhua, C.; Suna, J. Design, preparation, characterization and formation mechanism of a novel kinetic CL-20-based cocrystal. Acta Cryst. 2019, B75, 310–317. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Gou, R.-j.; Zhang, S.-h.; Chen, Y.-H.; Chen, M.-H.; Liu, Y.-B. Effect of solvent mixture on the formation of CL-20/HMX cocrystal explosives. J. Mol. Model. 2020, 26, 8. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Duan, B.; Lu, X.; Zhang, Q.; Xu, M.; Moa, H.; Wang, B. Preparation of CL-20/TFAZ cocrystals under aqueous conditions: Balancing high performance and low sensitivity. CrystEngComm 2019, 21, 7271–7279. [Google Scholar] [CrossRef]
- Viswanath, J.V.; Shanigaram, B.; Vijayadarshan, P.; Chowadary, T.V.; Gupta, A.; Bhanuprakash, K.; Niranjana, S.R.; Venkataraman, A. Studies and Theoretical Optimization of CL-20: RDX Cocrystal. Propellants Explos. Pyrotech. 2019, 44, 1570–1582. [Google Scholar] [CrossRef]
- Stepanov, V.; Patel, R.B.; Mudryy, R.; Qiu, H. Investigation of Nitramine-Based Amorphous Energetics. Propellants Explos. Pyrotech. 2016, 41, 142–147. [Google Scholar] [CrossRef]
- Chiquete, C.; Jackson, S.I. Detonation performance of the CL-20-based explosive LX-19. Proc. Combust. Inst. 2021, 38, 3661–3669. [Google Scholar] [CrossRef]
- Sysolyatin, S.V.; Chernikova, Y.T.; Lobanova, A.A.; Sakovich, G.V.; Surmachev, V.N.; Kadulin, V.V.; Kalashnikov, A.I.; Gudkova, N.I. Nitrolysis of tetraacetyl derivatives of hexaazaisowurtzitanes. Khimicheskaya Tekhnologiya 2005, 11, 12–15. [Google Scholar]
- Bayat, Y.; Hajighasemali, F. Synthesis of CL-20 by a Greener Method Using Nitroguanidine/HNO3. Propellants Explos. Pyrotech. 2016, 41, 20–23. [Google Scholar] [CrossRef]
- Koskin, A.P.; Simakova, I.L.; Parmon, V.N. Reductive debenzylation of hexabenzylhexaazaisowurtzitane—The key step of the synthesis of polycyclic nitramine hexanitrohexaazaisowurtzitane. Russ. Chem. Bull. 2007, 56, 2370–2375. [Google Scholar] [CrossRef]
- Zhao, W.; Liu, S.; Wang, H.; Yang, J.; Chen, X. Ultrasmall Pd Nanoparticles Supported on TiO2 for Catalytic Debenzylation via Hydrogenative C–N Bond Cleavage. ACS Appl. Nano Mater. 2020, 4, 159–166. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, D.; Dong, K.; Lv, P.; Pang, S.; Sun, C. Kinetics Study of a Complex Reaction: Nitration of Caged 2,6,8,12-Tetraacetyl-4,10-dinitro-2,4,6,8,10,12-hexaazaisowurtzitane. Org. Process Res. Dev. 2016, 20, 1911–1916. [Google Scholar] [CrossRef]
- Latypov, N.V.; Wellmar, U.; Goede, P.; Bellamy, A.J. Synthesis and Scale-Up of 2,4,6,8,10,12-Hexanitro-2,4,6,8,10,12-hexaazaisowurtzitane from 2,6,8,12-Tetraacetyl-4,10-dibenzyl-2,4,6,8,10,12-hexaazaisowurtzitane (HNIW, CL-20). Org. Process Res. Dev. 2000, 4, 156–158. [Google Scholar] [CrossRef]
- Dong, K.; Sun, C.H.; Song, J.W.; Wei, G.X.; Pang, S.P. Synthesis of 2,6,8,12-Tetraacetyl-2,4,6,8,10,12-hexaazaisowurtzitane (TAIW) from 2,6,8,12-Tetraacetyl-4,10-dibenzyl-2,4,6,8,10,12-hexaazaisowurtzitane (TADBIW) by Catalytic Hydrogenolysis Using a Continuous Flow Process. Org. Process Res. Dev. 2014, 18, 1321–1325. [Google Scholar] [CrossRef]
- Bayat, Y.; Hajimirsadeghi, S.S.; Pourmortazavi, S.M. Statistical Optimization of Reaction Parameters for the Synthesis of 2,4,6,8,10,12-Hexanitro-2,4,6,8,10,12-hexaazaisowurtzitane. Org. Process Res. Dev. 2011, 15, 810–816. [Google Scholar] [CrossRef]
- Pang, S.-P.; Yu, Y.-Z.; Zhao, X.-Q. A Novel Synthetic Route to Hexanitrohexaazaisowurtzitane. Propellants Explos. Pyrotech. 2005, 30, 442–444. [Google Scholar] [CrossRef]
- Surmachev, V.N.; Kubasova, V.A.; Zimin, D.E. A Study on Nitration of 4,10-Dibenzyl-2,6,8,12-Tetraacetyl-2,4,6,8,10,12-Hexaazaisowurtzitane. Propellants Explos. Pyrotech. 2020, 45, 1841–1844. [Google Scholar] [CrossRef]
- Kalashnikov, A.I.; Sysolyatin, S.V.; Sakovich, G.V.; Dubkov, A.S.; Kulagina, D.A. Nitrolysis of 2,6,8,12-tetraacetyl-4,10-dibenzyl-2,4,6,8,10,12-hexaazatetracyclo[5.5.0.03,11.05,9]dodecane. Russ. Chem. Bull. 2017, 66, 531–536. [Google Scholar] [CrossRef]
- Chapman, R.D.; Hollins, R.A. Benzylamine-Free, Heavy-Metal-Free Synthesis of CL-20 via Hexa(1-propenyl)hexaazaisowurtzitane. J. Energ. Mater. 2008, 26, 246–273. [Google Scholar] [CrossRef]
- Lou, D.; Wang, H.; Liu, S.; Li, L.; Zhao, W.; Chen, X.; Yang, J. PdFe bimetallic catalysts for debenzylation of hexabenzylhexaazaisowurtzitane (HBIW) and tetraacetyldibenzylhexaazaisowurtzitane (TADBIW). Catal. Commun. 2018, 109, 28–32. [Google Scholar] [CrossRef]
- Qian, H.; Ye, Z.-W.; Lv, C.-X. An Efficient and Facile Synthesis of Hexanitrohexaazaisowurtzitane (HNIW). Lett. Org. Chem. 2007, 4, 482–485. [Google Scholar] [CrossRef]
- Kai, W.; Dong, B.; Yang, C.; Qian, H. Acidic ionic liquids and green and recyclable catalysts in the clean nitration of TAIW to CL-20 using HNO3 electrolyte. Can. J. Chem. 2017, 95, 190–193. [Google Scholar] [CrossRef]
- Chapman, R.D.; Hollins, R.A. Processes for Preparing Certain Hexaazaisowurtzitanes and Their Use in Preparing Hexanitrohexaazaisowurtzitane. U.S. Patent 8268991 B1, 18 September 2012. [Google Scholar]
- Wright, M.E. Three-Step Synthesis of CL-20. U.S. Patent 9056868 B1, 16 June 2015. [Google Scholar]
- Wardle, R.B.; Hinshaw, J.C. Polycyclic, Polyamides as Precursors for Energetic Polycyclic Polynitramine Oxidizers. U.S. Patent 7129348 B1, 31 October 2006. [Google Scholar]
- Lu, Y.; Lei, Q.; Ren, X.; Ye, D.; Guo, Y.; Ding, N.; He, J. A Kind of Method that Three-Step Reaction Prepares CL 20. CN Patent 107353293 A, 16 August 2019. [Google Scholar]
- Cagnon, G.; Eck, G.; Herve, G.; Jacob, G. Process for the Synthesis of Hexanitrohexaazaisowurtzitane in 2 Steps from a Primary Amine. EP Patent 1479683 A1, 26 October 2005. [Google Scholar]
- Wang, J.; Li, Y.; Xue, M.; Chen, L.; Cao, D.; Li, Y. Method for Preparing CL-20 through Two-Step Method. CN Patent 110117289 B, 4 January 2022. [Google Scholar]
- Kalashnikov, A.I.; Sysoljatin, S.V.; Lapina, J.T.; Lobanova, A.A.; Kadulin, V.V. Method for Preparation of 2,4,6,8,10,12-hexanitro-2,4,6,8,10,12-hexaazatetracyclo[5,5,0,03,11,05,9]dodecane. RU Patent 2360916 C1, 10 July 2009. [Google Scholar]
- Jin, Z.; Wang, M.; Li, J.; Tie, Z. Method for Efficiently Synthesizing CL-20 High-Energy Cage Compound Based on Monatomic Catalyst. CN Patent 115611905 A, 2023. Available online: https://patents.google.com/patent/CN115611905A/en?oq=CN+Patent+115611905+A (accessed on 12 October 2023).
- Badard, O.; Renouard, J.; Marc, S.; Tenaglia, A. Procede de Synthese de l’Hexanitrohexaazaisowurtzitane a Partir de l’Hexaallylhexaazaisowurtzitane; Intermediaires. FR Patent 2997697 A1, 17 June 2016. [Google Scholar]
- Parakhin, V.V.; Pokhvisneva, G.V.; Ternikova, T.V.; Nikitin, S.V.; Smirnov, G.A.; Kon’kova, A.S.; Lempert, D.B.; Pivkina, A.N. Energetic alkylnitramine-functionalized pentanitro hexaazaisowurtzitanes: Towards advanced less sensitive CL-20 analogues. J. Mater. Chem. A 2022, 10, 818–828. [Google Scholar] [CrossRef]
- Luk’yanov, O.A.; Shlykova, N.I. Pentanitro- and pentanitronitroso-2,4,6,8,10,12-hexaazaisowurtzitanes. Russ. Chem. Bull. 2004, 53, 566–568. [Google Scholar] [CrossRef]
- Bellamy, A.J.; MacCuish, A.; Golding, P.; Mahon, M.F. The Use of trifluoroacetyl as an N- and O-protecting group during the synthesis of energetic compounds containing nitramine and/or nitrate ester groups. Propellants Explos. Pyrotech. 2007, 32, 20–31. [Google Scholar] [CrossRef]
- Sun, C.H.; Zhao, X.Q.; Li, Y.C.; Pang, S.P. Synthesis of two new cage molecules containing trinitromethyl group. Chin. Chem. Lett. 2010, 21, 572–575. [Google Scholar] [CrossRef]
- Zhang, Y.; Hu, J.; Li, Y.; Xu, C.; Chen, S.; Ge, Z.; Sun, C.; Pang, S. Energetic properties, thermal behavior and thermal safety of 4-(2,2,2-trinitroethyl)-2,6,8,10,12-pentanitro-2,4,6,8,10,12-hexaazaisowurtzitane. J. Anal. Appl. Pyrolysis 2020, 152, 104924. [Google Scholar] [CrossRef]
- Yukai, W.; Yuxiang, O.; Jinquan, L.; Lihua, L.; Boren, C.; Zhiguo, L. Synthesis, crystal structure and theoretical study of tetranitrodiazidoacetylhexaazaisowurtzitane (TNDAIW). Propellants Explos. Pyrotech. 2004, 29, 155–159. [Google Scholar] [CrossRef]
- Duddu, R.; Dave, P.R.; Damavarapu, R.; Surapaneni, R.; Gilardi, R.; Parrish, D. Synthesis of Azido Heterocycles. Synth. Commun. 2008, 38, 767–774. [Google Scholar] [CrossRef]
- Meng, Z.; Ou, Y.; Liu, J.; Wang, Y. Synthesis mechanism of tetranitrodiazidopropionylhexaazaisowurtzitane. Chin. J. Explos. Propellants 2006, 29, 65–67. [Google Scholar]
- Arabian, R.; Ramazani, A.; Mohtat, B.; Azizkhani, V.; Joo, S.W.; Rouhani, M. A Convenient and Efficient Protocol for the Synthesis of HBIW Catalyzed by Silica Nanoparticles under Ultrasound Irradiation. J. Energ. Mater. 2014, 32, 300–305. [Google Scholar] [CrossRef]
- Azizkhani, V.; Montazeri, F.; Molashahi, E.; Ramazani, A. Magnetically Recyclable CuFe2O4 Nanoparticles as an Efficient and Reusable Catalyst for the Green Synthesis of 2,4,6,8,10,12-Hexabenzyl-2,4,6,8,10,12-hexaazaisowurtzitane as CL-20 Explosive Precursor. J. Energ. Mater 2016, 35, 314–320. [Google Scholar] [CrossRef]
- Wang, L.; Yu, Z. Method for Efficiently Preparing Hexabenzyl Hexaazaisowurtzitanes. CN Patent 115594685 A, 2023. Available online: https://patents.google.com/patent/CN115594685A/en?oq=CN+Patent+115594685+A (accessed on 12 October 2023).
- Liu, W.; She, C.C.; Chao, H.; Wang, N.; Chen, S.S.; Jin, S.H.; Wang, J.F.; Chen, K. Role of the Bromide on the Hydrodebenzylation of 2,4,6,8,10,12-Hexabenzyl-2,4,6,8,10,12-hexaazaisowurtzitane (HBIW). ChemistrySelect 2022, 7, e202104216. [Google Scholar] [CrossRef]
- Yang, J.; Liu, S. Application of the Pd Radicel Duplex Metal Catalyst in HBIW Catalytic Hydrogenolytic Cleavages. CN Patent 106946894A, 15 March 2019. [Google Scholar]
- Kozlov, A.I.; Zbarskij, V.L.; Grunskij, V.N.; Judin, N.V.; Kuznetsov, L.A.; Merkin, A.A.; Komarov, A.A.; Kozlov, I.A.; Rybin, V.E.; Mikhajlov, J.M.; et al. Method of Producing Substituted Hexaazaisowurtzitanes. RU Patent 2451020 C1, 20 May 2012. [Google Scholar]
- Koskin, A.P.; Simakova, I.L.; Parmon, V.N. Study of palladium catalyst deactivation in synthesis of 4,10-diformyl-2,6,8,12-tetraacetyl-2,4,6,8,10,12-hexaazaisowurtzitane. React. Kinet. Catal. Lett. 2007, 92, 293–302. [Google Scholar] [CrossRef]
- Tang, X.; Lian, P.; Zhang, Y.; Zhu, J.; Jang, S.-R.; Wang, X.; Li, W.; Chen, S. Method for Synthesizing TADB by Continuous Hydrogenolysis and Debenzylation of HBIW through Acoustic Resonance Enhancement. CN Patent 114634513 A, 12 September 2023. [Google Scholar]
- Zhang, Q.; Wang, M.; Qian, J.; Lou, S.; Jin, J.; Li, B.; Lu, C.; Feng, F.; Lv, J.; Wang, Q.; et al. Deactivation and Regeneration of Palladium Catalysts for Hydrogenation Debenzylation of 2,4,6,8,10,12-Hexabenzyl-2,4,6,8,10,12-Hexaazaisowurtzitane (HBIW). Catalysts 2022, 12, 1547. [Google Scholar] [CrossRef]
- Fotouhi-Far, F.; Bashiri, H.; Hamadanian, M. Study of Deactivation of Pd(OH)2/C Catalyst in Reductive Debenzylation of Hexabenzylhexaazaisowurtzitane. Propellants Explos. Pyrotech. 2016, 42, 213–219. [Google Scholar] [CrossRef]
- Aravindu, P.; Rani, K.D.; Shaik, A.M.; Kommu, N.; Rao, V.K. Synthesis of Novel Hexaazaisowurtzitane Cages to Access CL-20. Asian J. Org. Chem. 2022, 11, 333–337. [Google Scholar] [CrossRef]
- Shang, F.; Liu, R.; Lv, M.; Ma, Y.; Liu, J.; Zhou, P.; Zhang, C.; Han, K.L. Unraveling the Key Role of the Benzyl Group in the Synthesis of CL-20 Precursor HBIW. ACS Omega 2022, 7, 21912–21924. [Google Scholar] [CrossRef] [PubMed]
- Paromov, A.; Shchurova, I.; Rogova, A.; Bagryanskaya, I.; Polovyanenko, D. Acid-Catalyzed Condensation of Benzamide with Glyoxal, and Reaction Features. Molecules 2022, 27, 1094. [Google Scholar] [CrossRef] [PubMed]
- Paromov, A.E.; Sysolyatin, S.V.; Gatilov, Y.V. An acid-catalyzed cascade synthesis of oxaazatetracyclo[5.5.0.03,11.05,9]dodecane Derivatives. J. Energ. Mater. 2017, 35, 363–373. [Google Scholar] [CrossRef]
- Paromov, A.E.; Sysolyatin, S.V. Synthesis of new N-polysubstituted oxaazaisowurtzitanes by acid-catalyzed condensation of sulfonamides with glyoxal. Russ. J. Org. Chem. 2017, 53, 1717–1725. [Google Scholar] [CrossRef]
- Paromov, A.E.; Sysolyatin, S.V.; Shchurova, I.A.; Rogova, A.I.; Malykhin, V.V.; Gatilov, Y.V. Synthesis of oxaazaisowurtzitanes by condensation of 4-dimethylaminobenzenesulfonamide with glyoxal. Tetrahedron 2020, 76, 131298. [Google Scholar] [CrossRef]
- Paromov, A.E.; Sysolyatin, S.V.; Shchurova, I.A. Condensation of 4-Tert-butyl-2,6-dimethylbenzenesulfonamide with Glyoxal and Reaction Features: A New Process for Symmetric and Asymmetric Aromatic Sulfones. Molecules 2022, 27, 7793. [Google Scholar] [CrossRef] [PubMed]
- Kovalevsky, R.A.; Vasechkin, K.V.; Kucherenko, A.S.; Zlotin, S.G. Enantioselective Catalytic Synthesis of α-Stereogenic Chromen-4-one Amino Derivatives. Adv. Synth. Catal. 2023, 365, 3162–3166. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Solvent | ω(H2SO4 1),% 2 | Composition of Principal Reaction Products (HPLC),% | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 2 | 3 | 4 | 5 | 7 | 8 | 9 | ||||
| 1 | H2O | 40 | residue (0.339 g) 3: | 3.4 | 8.5 | 9.7 | 0.2 | - | 0.3 | - |
| 2 | 44 | residue (0.360 g) 3: | 0.3 | 4.3 | 18.3 | 1.8 | - | 4.1 | - | |
| 3 | 48 | residue (0.372 g) 3: | 0.2 | 4.2 | 18.5 | 2.7 | - | 7.3 | - | |
| 4 | 51 | residue (0.331 g) 3: | - | 2.5 | 12.9 | 3.0 | - | 15.0 | - | |
| 5 | 54 | residue (0.322 g) 3: | - | 1.4 | 3.6 | 3.0 | - | 25.1 | - | |
| Entry | Solvent | ω(H2SO4 1),% 2 | Composition of Principal Reaction Products (HPLC),% | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 2 | 3 | 4 | 5 | 7 | 8 | 9 | ||||
| 1 | Et2O | 0.3 | residue (0.033 g) 3: | 41.8 | 36.3 | 2.2 | - | traces | - | - |
| filtrate: | 3.0 | 12.1 | 1.5 | - | 0.2 | - | 2.7 | |||
| 2 | 2 | residue (0.630 g) 3: | - | 2.0 | 70.2 | - | 11.0 | 0.7 | - | |
| filtrate: | - | 4.4 | 5.3 | - | - | 0.6 | - | |||
| 3 | THF | 0.3 | reaction mass: | 6.7 | 20.6 | 3.8 | - | - | - | - |
| 4 | 2 | reaction mass: | 1.7 | 11.4 | 21.6 | traces | - | 0.4 | - | |
| 5 | 7 | residue (0.081 g) 3: | - | - | 10.4 | 82.1 | - | - | - | |
| filtrate: | 0.5 | 5.1 | 9.0 | - | - | 0.7 | - | |||
| 6 | AcOEt | 0.3 | residue (0.004 g) 3: | - | - | 1.6 | 43.4 | 44.9 | - | - |
| filtrate: | - | 2.8 | 1.8 | traces | 14.0 | - | 0.6 | |||
| 7 | 2 | residue (0.255 g) 3: | - | - | 1.6 | 17.1 | 61.1 | - | - | |
| filtrate: | - | 4.0 | 2.4 | 2.7 | 14.8 | 1.3 | - | |||
| 8 | (CH3)2CO | 0.3 | reaction mass: | 3.0 | 12.0 | 4.8 | - | - | - | - |
| 9 | 2 | residue (0.030 g) 3: | - | - | 15.4 | 72.5 | - | - | - | |
| filtrate: | 1.4 | 8.5 | 12.3 | traces | - | - | - | |||
| 10 | CH3CN | 0.3 | residue (0.131 g) 3: | - | - | 35.7 | 55.5 | - | - | - |
| filtrate: | 1.2 | 8.7 | 12.6 | - | - | - | - | |||
| 11 | 2 | residue (0.233 g) 3: | - | - | 14.5 | 75.2 | - | - | - | |
| filtrate: | 0.6 | 2.4 | 3.1 | - | - | - | - | |||
| 12 | 7 | residue (0.162 g) 3: | - | - | 4.5 | 74.0 | - | - | - | |
| filtrate: | - | - | - | - | - | - | - | |||
| 13 | CH2Cl2 | 0.3 | reaction mass: | 3.2 | 6.2 | 1.3 | 0.8 | - | - | - |
| 14 | 2 | residue (0.340 g) 3: | - | - | 7.7 | 81.0 | - | 2.8 | - | |
| filtrate: | - | 2.3 | 6.1 | - | - | 10.7 | - | |||
| 15 | DMSO | 0.3 | reaction mass: | traces | 1.9 | - | - | - | - | - |
| 16 | 2 | reaction mass: | 2.4 | 7.8 | 0.3 | - | - | - | - | |
| 17 | 7 | reaction mass: | 4.4 | 23.7 | 2.6 | 0.3 | - | - | - | |
| 18 | FoOH | - | residue (0.038 g) 3: | - | - | 15.5 | 67.0 | - | - | - |
| filtrate: | - | 0.7 | 5.2 | - | - | - | 23.0 | |||
| 19 | 2 | residue (0.034 g) 3: | - | - | 3.0 | 82.2 | - | - | - | |
| filtrate: | - | - | - | - | - | - | 74.9 | |||
| 20 | 7 | reaction mass: | - | - | - | - | - | - | 87.0 | |
| 21 | AcOH | - | residue (0.057 g) 3: | 70.0 | 16.0 | - | - | - | - | - |
| filtrate: | 0.7 | 6.1 | traces | - | - | - | - | |||
| 22 | 2 | residue (0.158 g) 3: | - | - | 9.9 | 72.5 | - | - | - | |
| filtrate: | - | - | 2.1 | - | - | - | - | |||
| 23 | 7 | residue (0.211 g) 3: | - | - | 5.8 | 81.8 | - | - | - | |
| filtrate: | - | - | - | - | - | - | - | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paromov, A.E. Condensation of Benzyl Carbamate with Glyoxal in Polar Protic and Aprotic Solvents. Molecules 2023, 28, 7648. https://doi.org/10.3390/molecules28227648
Paromov AE. Condensation of Benzyl Carbamate with Glyoxal in Polar Protic and Aprotic Solvents. Molecules. 2023; 28(22):7648. https://doi.org/10.3390/molecules28227648
Chicago/Turabian StyleParomov, Artyom E. 2023. "Condensation of Benzyl Carbamate with Glyoxal in Polar Protic and Aprotic Solvents" Molecules 28, no. 22: 7648. https://doi.org/10.3390/molecules28227648
APA StyleParomov, A. E. (2023). Condensation of Benzyl Carbamate with Glyoxal in Polar Protic and Aprotic Solvents. Molecules, 28(22), 7648. https://doi.org/10.3390/molecules28227648