
Deep Eutectic Solvent Based Reversed-Phase Dispersive Liquid–Liquid Microextraction and High-Performance Liquid Chromatography for the Determination of Free Tryptophan in Cold-Pressed Oils




Abstract
:1. Introduction
2. Results and Discussion
2.1. Selection of DESs
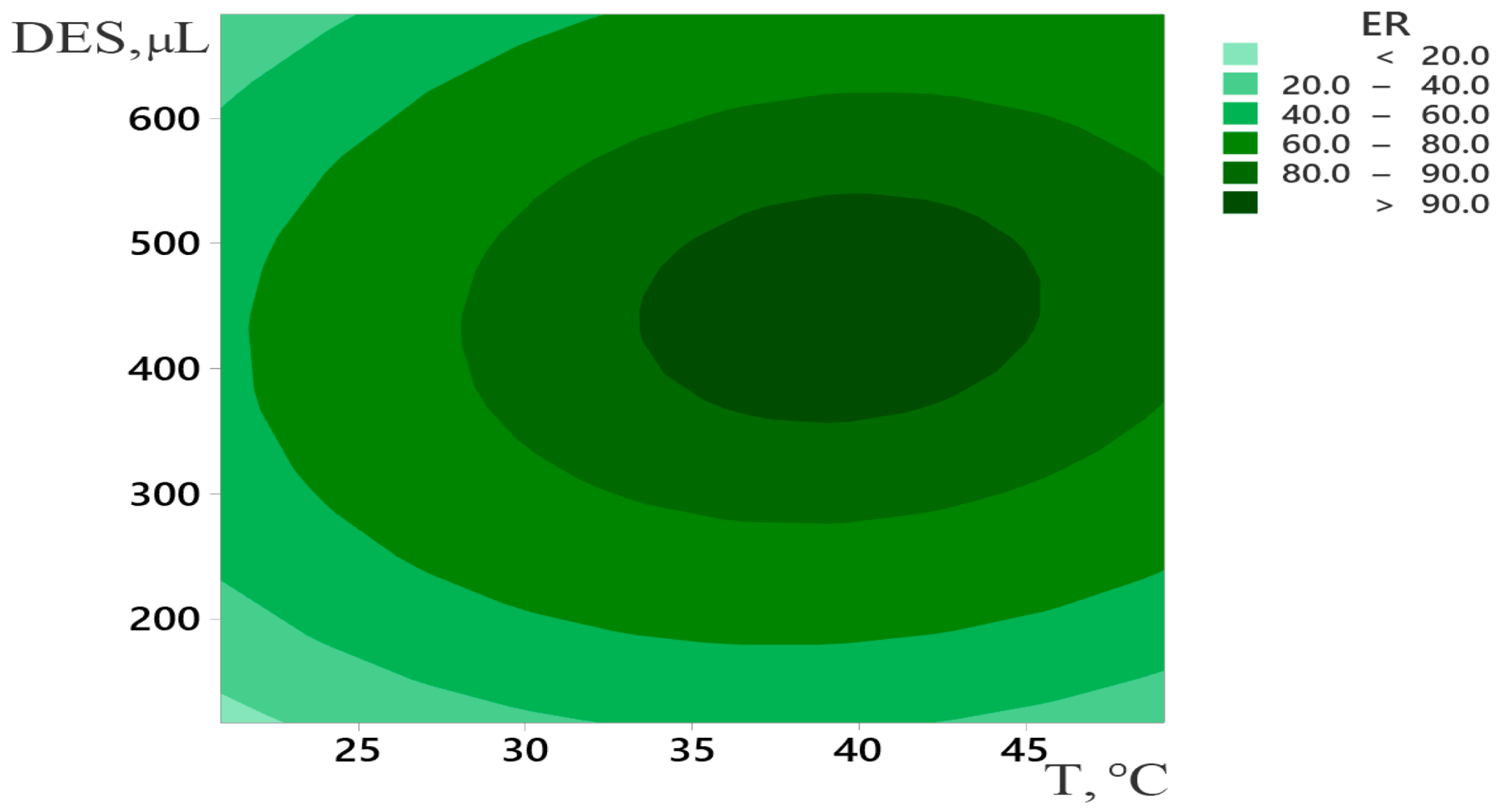
2.2. Optimization of DLLME Procedure
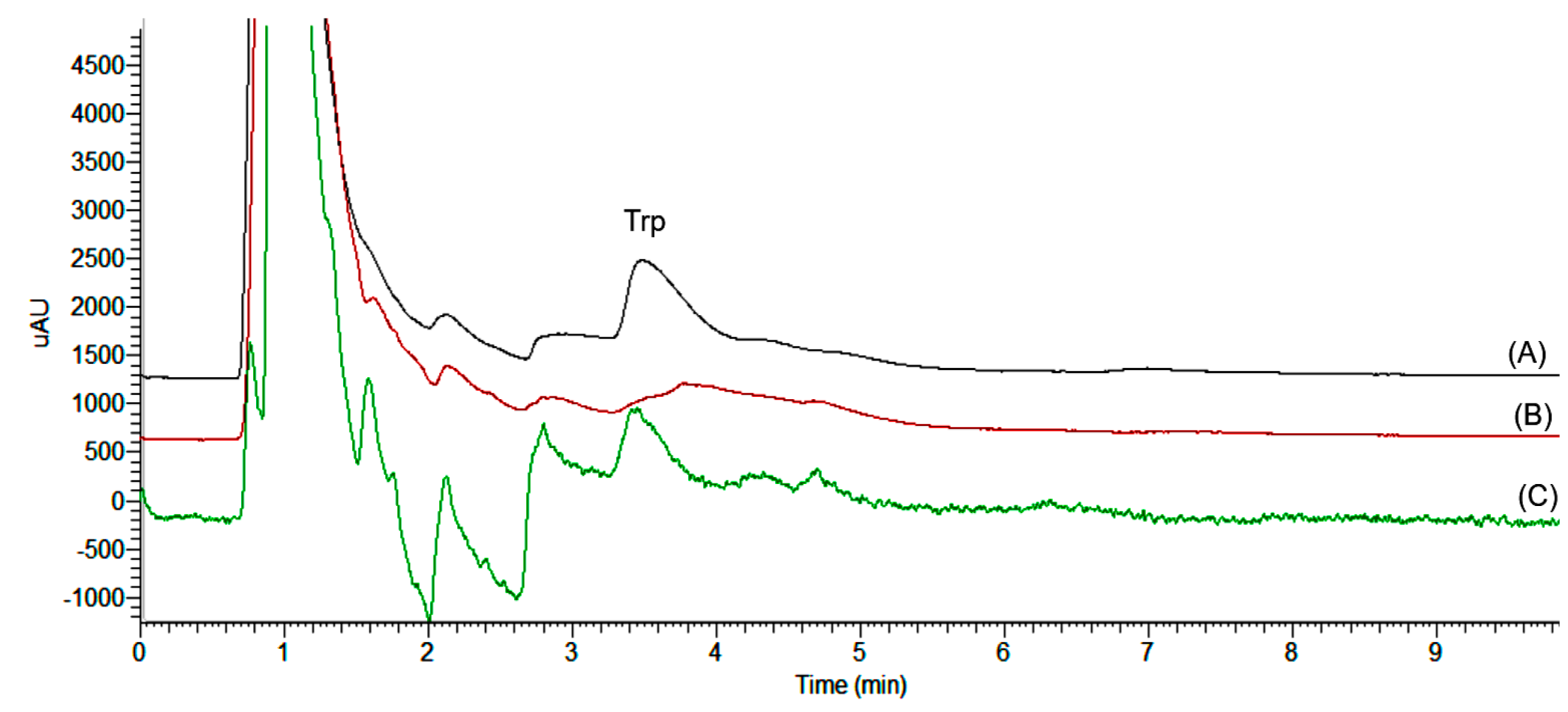
2.3. Validation of RP-DLLME-HPLC Method
2.4. Critical Analysis of the Method Performances
2.5. Analysis of Real Samples
3. Materials and Methods
3.1. Reagents and Chemicals
3.2. Sample Preparation
3.3. RP-DLLME Optimization
3.4. HPLC Measurements
3.5. Method Validation
3.6. Software
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Friedman, M. Analysis, Nutrition, and Health Benefits of Tryptophan. Int. J. Tryptophan Res. 2018, 11, 1178646918802282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Comai, S.; Bertazzo, A.; Brughera, M.; Crotti, S. Tryptophan in Health and Disease. In Advances in Clinical Chemistry; Elsevier: Amsterdam, The Netherlands, 2020; Volume 95, pp. 165–218. ISBN 978-0-12-821165-6. [Google Scholar]
- Strasser, B.; Gostner, J.M.; Fuchs, D. Mood, Food, and Cognition: Role of Tryptophan and Serotonin. Curr. Opin. Clin. Nutr. Metab. Care 2016, 19, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Venkatachalam, M.; Sathe, S.K. Chemical Composition of Selected Edible Nut Seeds. J. Agric. Food Chem. 2006, 54, 4705–4714. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Aloy, M.; Hulshof, P.J.M.; Estruel-Amades, S.; Osté, M.C.J.; Lankinen, M.; Geleijnse, J.M.; de Goede, J.; Ulaszewska, M.; Mattivi, F.; Bakker, S.J.L.; et al. Biomarkers of Food Intake for Nuts and Vegetable Oils: An Extensive Literature Search. Genes Nutr. 2019, 14, 7. [Google Scholar] [CrossRef] [Green Version]
- Pinter-Szakacs, M.; Molnar-Perl, I. Determination of Tryptophan in Unhydrolyzed Food and Feedstuffs by the Acid Ninhydrin Method. J. Agric. Food Chem. 1990, 38, 720–726. [Google Scholar] [CrossRef]
- Wu, Y.; Wang, T.; Zhang, C.; Xing, X.-H. A Rapid and Specific Colorimetric Method for Free Tryptophan Quantification. Talanta 2018, 176, 604–609. [Google Scholar] [CrossRef]
- Hosokawa, S.; Morinishi, T.; Ohara, K.; Yamaguchi, K.; Tada, S.; Tokuhara, Y. A Spectrophotometric Method for the Determination of Tryptophan Following Oxidation by the Addition of Sodium Hypochlorite Pentahydrate. PLoS ONE 2023, 18, e0279547. [Google Scholar] [CrossRef]
- Li, L.; Luo, Y.; Jia, L. Genetically Engineered Bacterium-Modified Magnetic Particles Assisted Chiral Recognition and Colorimetric Determination of D/L-Tryptophan in Millets. Food Chem. 2023, 407, 135125. [Google Scholar] [CrossRef]
- Fernández, L.G.; Vera-López, S.; Díez-Pascual, A.M.; San Andrés, M.P. Easy, Fast, and Clean Fluorescence Analysis of Tryptophan with Clays and Graphene/Clay Mixtures. J. Food Compos. Anal. 2022, 114, 104858. [Google Scholar] [CrossRef]
- Lima, D.; Andrade Pessôa, C.; Wohnrath, K.; Humberto Marcolino-Junior, L.; Fernando Bergamini, M. A Feasible and Efficient Voltammetric Sensor Based on Electropolymerized L-Arginine for the Detection of L-Tryptophan in Dietary Supplements. Microchem. J. 2022, 181, 107709. [Google Scholar] [CrossRef]
- Khoshsafar, H.; Bagheri, H.; Hashemi, P.; Bordbar, M.M.; Madrakian, T.; Afkhami, A. Combination of an Aptamer-Based Immunochromatography Assay with Nanocomposite-Modified Screen-Printed Electrodes for Discrimination and Simultaneous Determination of Tryptophan Enantiomers. Talanta 2023, 253, 124090. [Google Scholar] [CrossRef]
- Tasić, Ž.Z.; Mihajlović, M.B.P.; Radovanović, M.B.; Simonović, A.T.; Medić, D.V.; Antonijević, M.M. Electrochemical Determination of L-Tryptophan in Food Samples on Graphite Electrode Prepared from Waste Batteries. Sci. Rep. 2022, 12, 5469. [Google Scholar] [CrossRef]
- Rezaei, F.; Ashraf, N.; Zohuri, G.H. A Smart Electrochemical Sensor Based upon Hydrophilic Core–Shell Molecularly Imprinted Polymer for Determination of L-Tryptophan. Microchem. J. 2023, 185, 108260. [Google Scholar] [CrossRef]
- Alam, I.; Lertanantawong, B.; Sutthibutpong, T.; Punnakitikashem, P.; Asanithi, P. Molecularly Imprinted Polymer-Amyloid Fibril-Based Electrochemical Biosensor for Ultrasensitive Detection of Tryptophan. Biosensors 2022, 12, 291. [Google Scholar] [CrossRef]
- Forteschi, M.; Sotgia, S.; Assaretti, S.; Arru, D.; Cambedda, D.; Sotgiu, E.; Zinellu, A.; Carru, C. Simultaneous Determination of Aromatic Amino Acids in Human Blood Plasma by Capillary Electrophoresis with UV-Absorption Detection: Other Techniques. J. Sep. Sci. 2015, 38, 1794–1799. [Google Scholar] [CrossRef]
- Jastrzębska, A.; Kowalska, S.; Szłyk, E. Determination of Free Tryptophan in Beer Samples by Capillary Isotachophoretic Method. Food Anal. Methods 2020, 13, 850–862. [Google Scholar] [CrossRef] [Green Version]
- Michalczuk, L.; Bialek, K.; Cohen, J.D. Rapid Determination of Free Tryptophan in Plant Samples by Gas Chromatography-Selected Ion Monitoring Mass Spectrometry. J. Chromatogr. A 1992, 596, 294–298. [Google Scholar] [CrossRef]
- Zhu, Y.; Yang, Y.; Zhou, Z.; Li, G.; Jiang, M.; Zhang, C.; Chen, S. Direct Determination of Free Tryptophan Contents in Soy Sauces and Its Application as an Index of Soy Sauce Adulteration. Food Chem. 2010, 118, 159–162. [Google Scholar] [CrossRef]
- Lomenova, A.; Hroboňová, K. Application of Achiral–Chiral Two-dimensional HPLC for Separation of Phenylalanine and Tryptophan Enantiomers in Dietary Supplement. Biomed. Chromatogr. 2021, 35, e4972. [Google Scholar] [CrossRef]
- Draher, J.; White, N. HPLC Determination of Total Tryptophan in Infant Formula and Adult/Pediatric Nutritional Formula Following Enzymatic Hydrolysis: Single-Laboratory Validation, First Action 2017.03. J. AOAC Int. 2018, 101, 824–830. [Google Scholar] [CrossRef]
- Yust, M.M.; Pedroche, J.; Girón-Calle, J.; Vioque, J.; Millán, F.; Alaiz, M. Determination of Tryptophan by High-Performance Liquid Chromatography of Alkaline Hydrolysates with Spectrophotometric Detection. Food Chem. 2004, 85, 317–320. [Google Scholar] [CrossRef]
- ISO 13904; Animal Feeding Stuffs—Determination of Tryptophan Content. 2005. Available online: https://www.iso.org/standard/37259.html (accessed on 2 March 2023).
- Ravindran, G.; Ravindran, V.; Bryden, W.L. Total and Ileal Digestible Tryptophan Contents of Feedstuffs for Broiler Chickens. J. Sci. Food Agric. 2006, 86, 1132–1137. [Google Scholar] [CrossRef]
- Allred, M.C.; Macdonald, J.L. Determination of Sulfur Amino Acids and Tryptophan in Foods and Food and Feed Ingredients: Collaborative Study. J. AOAC Int. 1988, 71, 603–606. [Google Scholar] [CrossRef]
- Ravindran, G.; Bryden, W.L. Tryptophan Determination in Proteins and Feedstuffs by Ion Exchange Chromatography. Food Chem. 2005, 89, 309–314. [Google Scholar] [CrossRef]
- Ritota, M.; Manzi, P. Rapid Determination of Total Tryptophan in Yoghurt by Ultra High Performance Liquid Chromatography with Fluorescence Detection. Molecules 2020, 25, 5025. [Google Scholar] [CrossRef]
- Zhang, J.; Xue, X.; Zhou, J.; Chen, F.; Wu, L.; Li, Y.; Zhao, J. Determination of Tryptophan in Bee Pollen and Royal Jelly by High-Performance Liquid Chromatography with Fluorescence Detection. Biomed. Chromatogr. 2009, 23, 994–998. [Google Scholar] [CrossRef]
- la Cour, R.; Jørgensen, H.; Schjoerring, J.K. Improvement of Tryptophan Analysis by Liquid Chromatography-Single Quadrupole Mass Spectrometry Through the Evaluation of Multiple Parameters. Front. Chem. 2019, 7, 797. [Google Scholar] [CrossRef]
- Protti, M.; Cirrincione, M.; Mandrioli, R.; Rudge, J.; Regazzoni, L.; Valsecchi, V.; Volpi, C.; Mercolini, L. Volumetric Absorptive Microsampling (VAMS) for Targeted LC-MS/MS Determination of Tryptophan-Related Biomarkers. Molecules 2022, 27, 5652. [Google Scholar] [CrossRef]
- Boulet, L.; Faure, P.; Flore, P.; Montérémal, J.; Ducros, V. Simultaneous Determination of Tryptophan and 8 Metabolites in Human Plasma by Liquid Chromatography/Tandem Mass Spectrometry. J. Chromatogr. B 2017, 1054, 36–43. [Google Scholar] [CrossRef]
- Soto, M.E.; Ares, A.M.; Bernal, J.; Nozal, M.J.; Bernal, J.L. Simultaneous Determination of Tryptophan, Kynurenine, Kynurenic and Xanthurenic Acids in Honey by Liquid Chromatography with Diode Array, Fluorescence and Tandem Mass Spectrometry Detection. J. Chromatogr. A 2011, 1218, 7592–7600. [Google Scholar] [CrossRef]
- Su, M.; Cheng, Y.; Zhang, C.; Zhu, D.; Jia, M.; Zhang, Q.; Wu, H.; Chen, G. Determination of the Levels of Tryptophan and 12 Metabolites in Milk by Liquid Chromatography-Tandem Mass Spectrometry with the QuEChERS Method. J. Dairy Sci. 2020, 103, 9851–9859. [Google Scholar] [CrossRef]
- Vitalini, S.; Dei Cas, M.; Rubino, F.M.; Vigentini, I.; Foschino, R.; Iriti, M.; Paroni, R. LC-MS/MS-Based Profiling of Tryptophan-Related Metabolites in Healthy Plant Foods. Molecules 2020, 25, 311. [Google Scholar] [CrossRef] [Green Version]
- Weber, P. Determination of Amino Acids in Food and Feed by Microwave Hydrolysis and UHPLC-MS/MS. J. Chromatogr. B 2022, 1209, 123429. [Google Scholar] [CrossRef]
- Cooper, C.; Packer, N.; Williams, K. Amino Acid Analysis Protocols; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2000; Volume 159, ISBN 978-1-59259-047-6. [Google Scholar]
- Taş, N.G.; Yılmaz, C.; Gökmen, V. Investigation of Serotonin, Free and Protein-Bound Tryptophan in Turkish Hazelnut Varieties and Effect of Roasting on Serotonin Content. Food Res. Int. 2019, 120, 865–871. [Google Scholar] [CrossRef]
- Faraji, M.; Yamini, Y.; Gholami, M. Recent Advances and Trends in Applications of Solid-Phase Extraction Techniques in Food and Environmental Analysis. Chromatographia 2019, 82, 1207–1249. [Google Scholar] [CrossRef]
- Lukić, J.; Radulović, J.; Lučić, M.; Đurkić, T.; Onjia, A. Chemometric Optimization of Solid-Phase Extraction Followed by Liquid Chromatography-Tandem Mass Spectrometry and Probabilistic Risk Assessment of Ultraviolet Filters in an Urban Recreational Lake. Front. Environ. Sci. 2022, 10, 916916. [Google Scholar] [CrossRef]
- Rutkowska, M.; Płotka-Wasylka, J.; Sajid, M.; Andruch, V. Liquid–Phase Microextraction: A Review of Reviews. Microchem. J. 2019, 149, 103989. [Google Scholar] [CrossRef]
- Lukić, J.; Đurkić, T.; Onjia, A. Dispersive Liquid–Liquid Microextraction and Monte Carlo Simulation of Margin of Safety for Octocrylene, EHMC, 2ES, and Homosalate in Sunscreens. Biomed. Chromatogr. 2023, e5590. [Google Scholar] [CrossRef]
- Mohammadi, A.; Barzegar, F.; Kamankesh, M.; Mousavi Khaneghah, A. Heterocyclic Aromatic Amines in Doner Kebab: Quantitation Using an Efficient Microextraction Technique Coupled with Reversed-phase High-performance Liquid Chromatography. Food Sci. Nutr. 2020, 8, 88–96. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Li, J.; Ji, L.; Chen, L. Simultaneous Determination of Sulfonamides Antibiotics in Environmental Water and Seafood Samples Using Ultrasonic-Assisted Dispersive Liquid-Liquid Microextraction Coupled with High Performance Liquid Chromatography. Molecules 2022, 27, 2160. [Google Scholar] [CrossRef]
- Han, Q.; Liu, Y.; Huo, Y.; Li, D.; Yang, X. Determination of Ultra-Trace Cobalt in Water Samples Using Dispersive Liquid-Liquid Microextraction Followed by Graphite Furnace Atomic Absorption Spectrometry. Molecules 2022, 27, 2694. [Google Scholar] [CrossRef] [PubMed]
- Slavković-Beškoski, L.; Ignjatović, L.; Bolognesi, G.; Maksin, D.; Savić, A.; Vladisavljević, G.; Onjia, A. Dispersive Solid–Liquid Microextraction Based on the Poly(HDDA)/Graphene Sorbent Followed by ICP-MS for the Determination of Rare Earth Elements in Coal Fly Ash Leachate. Metals 2022, 12, 791. [Google Scholar] [CrossRef]
- Tadić, T.; Marković, B.; Radulović, J.; Lukić, J.; Suručić, L.; Nastasović, A.; Onjia, A. A Core-Shell Amino-Functionalized Magnetic Molecularly Imprinted Polymer Based on Glycidyl Methacrylate for Dispersive Solid-Phase Microextraction of Aniline. Sustainability 2022, 14, 9222. [Google Scholar] [CrossRef]
- Sereshti, H.; Karimi, M.; Samadi, S. Application of Response Surface Method for Optimization of Dispersive Liquid–Liquid Microextraction of Water-Soluble Components of Rosa damascena Mill. Essential Oil. J. Chromatogr. A 2009, 1216, 198–204. [Google Scholar] [CrossRef] [PubMed]
- Hashemi, P.; Raeisi, F.; Ghiasvand, A.R.; Rahimi, A. Reversed-Phase Dispersive Liquid–Liquid Microextraction with Central Composite Design Optimization for Preconcentration and HPLC Determination of Oleuropein. Talanta 2010, 80, 1926–1931. [Google Scholar] [CrossRef]
- Wang, Q.F.; Liang, L.J.; Sun, J.B.; Zhou, J. Application of a Reversed-Phase Ionic Liquid Dispersive Liquid-Liquid Microextraction Method for the Extraction and Preconcentration of Domoic Acid from Urine Samples. Heliyon 2022, 8, e10152. [Google Scholar] [CrossRef]
- Schettino, L.; García-Juan, A.; Fernández-Lozano, L.; Benedé, J.L.; Chisvert, A. Trace Determination of Prohibited Acrylamide in Cosmetic Products by Vortex-Assisted Reversed-Phase Dispersive Liquid-Liquid Microextraction and Liquid Chromatography-Tandem Mass Spectrometry. J. Chromatogr. A 2023, 1687, 463651. [Google Scholar] [CrossRef]
- Hassan, M.; Erbas, Z.; Alshana, U.; Soylak, M. Ligandless Reversed-Phase Switchable-Hydrophilicity Solvent Liquid–Liquid Microextraction Combined with Flame-Atomic Absorption Spectrometry for the Determination of Copper in Oil Samples. Microchem. J. 2020, 156, 104868. [Google Scholar] [CrossRef]
- Shishov, A.; Volodina, N.; Semenova, E.; Navolotskaya, D.; Ermakov, S.; Bulatov, A. Reversed-Phase Dispersive Liquid-Liquid Microextraction Based on Decomposition of Deep Eutectic Solvent for the Determination of Lead and Cadmium in Vegetable Oil. Food Chem. 2022, 373, 131456. [Google Scholar] [CrossRef]
- Ferreira, V.J.; Lemos, V.A.; Teixeira, L.S.G. Dynamic Reversed-Phase Liquid-Liquid Microextraction for the Determination of Cd, Cr, Mn, and Ni in Vegetable Oils by Energy Dispersive X-Ray Fluorescence Spectrometry. J. Food Compos. Anal. 2023, 117, 105098. [Google Scholar] [CrossRef]
- Ma, S.; Li, F.; Liu, L.; Liao, L.; Chang, L.; Tan, Z. Deep-Eutectic Solvents Simultaneously Used as the Phase-Forming Components and Chiral Selectors for Enantioselective Liquid-Liquid Extraction of Tryptophan Enantiomers. J. Mol. Liq. 2020, 319, 114106. [Google Scholar] [CrossRef]
- Santos, L.B.; Assis, R.S.; Barreto, J.A.; Bezerra, M.A.; Novaes, C.G.; Lemos, V.A. Deep Eutectic Solvents in Liquid-Phase Microextraction: Contribution to Green Chemistry. TrAC Trends Anal. Chem. 2022, 146, 116478. [Google Scholar] [CrossRef]
- Wang, H.; Huang, X.; Qian, H.; Lu, R.; Zhang, S.; Zhou, W.; Gao, H.; Xu, D. Vortex-Assisted Deep Eutectic Solvent Reversed-Phase Liquid–Liquid Microextraction of Triazine Herbicides in Edible Vegetable Oils. J. Chromatogr. A 2019, 1589, 10–17. [Google Scholar] [CrossRef]
- Hansen, B.B.; Spittle, S.; Chen, B.; Poe, D.; Zhang, Y.; Klein, J.M.; Horton, A.; Adhikari, L.; Zelovich, T.; Doherty, B.W.; et al. Deep Eutectic Solvents: A Review of Fundamentals and Applications. Chem. Rev. 2021, 121, 1232–1285. [Google Scholar] [CrossRef]
- Cao, J.; Wang, C.; Shi, L.; Cheng, Y.; Hu, H.; Zeng, B.; Zhao, F. Water Based-Deep Eutectic Solvent for Ultrasound-Assisted Liquid–Liquid Microextraction of Parabens in Edible Oil. Food Chem. 2022, 383, 132586. [Google Scholar] [CrossRef]
- Xie, Q.; Xia, M.; Lu, H.; Shi, H.; Sun, D.; Hou, B.; Jia, L.; Li, D. Deep Eutectic Solvent-Based Liquid-Liquid Microextraction for the HPLC-DAD Analysis of Bisphenol A in Edible Oils. J. Mol. Liq. 2020, 306, 112881. [Google Scholar] [CrossRef]
- Heidari, H.; Ghanbari-Rad, S.; Habibi, E. Optimization Deep Eutectic Solvent-Based Ultrasound-Assisted Liquid-Liquid Microextraction by Using the Desirability Function Approach for Extraction and Preconcentration of Organophosphorus Pesticides from Fruit Juice Samples. J. Food Compos. Anal. 2020, 87, 103389. [Google Scholar] [CrossRef]
- Lučić, M.; Sredović Ignjatović, I.; Lević, S.; Pećinar, I.; Antić, M.; Đurđić, S.; Onjia, A. Ultrasound-assisted Extraction of Essential and Toxic Elements from Pepper in Different Ripening Stages Using Box–Behnken Design. Food Process. Preserv. 2022, 46, e16493. [Google Scholar] [CrossRef]
- Du, C.; Zhao, B.; Chen, X.-B.; Birbilis, N.; Yang, H. Effect of Water Presence on Choline Chloride-2urea Ionic Liquid and Coating Platings from the Hydrated Ionic Liquid. Sci. Rep. 2016, 6, 29225. [Google Scholar] [CrossRef] [Green Version]
- Banjare, M.K.; Behera, K.; Satnami, M.L.; Pandey, S.; Ghosh, K.K. Self-Assembly of a Short-Chain Ionic Liquid within Deep Eutectic Solvents. RSC Adv. 2018, 8, 7969–7979. [Google Scholar] [CrossRef]
- Khezeli, T.; Daneshfar, A.; Sahraei, R. A Green Ultrasonic-Assisted Liquid–Liquid Microextraction Based on Deep Eutectic Solvent for the HPLC-UV Determination of Ferulic, Caffeic and Cinnamic Acid from Olive, Almond, Sesame and Cinnamon Oil. Talanta 2016, 150, 577–585. [Google Scholar] [CrossRef] [PubMed]
- dos Santos, C.; Padilha, C.; Damasceno, K.; Leite, P.; de Araújo, A.; Freitas, P.; Vieira, É.; Cordeiro, A.; de Sousa, F., Jr.; de Assis, C. Astaxanthin Recovery from Shrimp Residue by Solvent Ethanol Extraction Using Choline Chloride:Glycerol Deep Eutectic Solvent as Adjuvant. J. Braz. Chem. Soc. 2021, 32, 1030–1039. [Google Scholar] [CrossRef]
- Ijardar, S.P.; Singh, V.; Gardas, R.L. Revisiting the Physicochemical Properties and Applications of Deep Eutectic Solvents. Molecules 2022, 27, 1368. [Google Scholar] [CrossRef] [PubMed]
- FDA. FDA Guidelines for the Validation of Chemical Methods for the FDA Foods Program, 3rd ed.; FDA: Silver Spring, MD, USA, 2019.
- Karimi, M.; Shabani, A.M.H.; Dadfarnia, S. Deep Eutectic Solvent-Mediated Extraction for Ligand-Less Preconcentration of Lead and Cadmium from Environmental Samples Using Magnetic Nanoparticles. Microchim. Acta 2016, 183, 563–571. [Google Scholar] [CrossRef]
- Liu, W.; Zong, B.; Wang, X.; Yang, G.; Yu, J. Deep Eutectic Solvents as Switchable Solvents for Highly Efficient Liquid–Liquid Microextraction of Phenolic Antioxidant: Easily Tracking the Original TBHQ in Edible Oils. Food Chem. 2022, 377, 131946. [Google Scholar] [CrossRef]
- Alañón, M.E.; Ivanović, M.; Gómez-Caravaca, A.M.; Arráez-Román, D.; Segura-Carretero, A. Choline Chloride Derivative-Based Deep Eutectic Liquids as Novel Green Alternative Solvents for Extraction of Phenolic Compounds from Olive Leaf. Arab. J. Chem. 2020, 13, 1685–1701. [Google Scholar] [CrossRef]
- Zhang, K.; Guo, R.; Wang, Y.; Wang, J.; Nie, Q.; Li, B.; Zhu, G. Temperature-Controlled Air-Assisted Liquid–Liquid Microextraction Based on the Solidification of Floating Deep Eutectic Solvents for the Determination of Triclosan and Alkylphenols in Water Samples via HPLC. Microchem. J. 2022, 182, 107864. [Google Scholar] [CrossRef]
- Moema, D.; Makwakwa, T.A.; Gebreyohannes, B.E.; Dube, S.; Nindi, M.M. Hollow Fiber Liquid Phase Microextraction of Fluoroquinolones in Chicken Livers Followed by High Pressure Liquid Chromatography: Greenness Assessment Using National Environmental Methods Index Label (NEMI), Green Analytical Procedure Index (GAPI), Analytical GREEnness Metric (AGREE), and Eco Scale. J. Food Compos. Anal. 2023, 117, 105131. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Variable | Symbol | Level | ||
|---|---|---|---|---|---|
| −1 | 0 | +1 | |||
| 1. | Initial sample dilution ratio (:) | dil | 1:1 | 1:5 | 1:9 |
| 2. | DES amount (μL) | DES | 100 | 200 | 300 |
| 3. | Extraction time (min) | tex | 1 | 3 | 5 |
| 4. | Extraction temperature (°C) | T | 25 | 35 | 45 |
| 5. | Salt (NaCl) addition (%) | salt | 0 | 5 | 10 |
| 6. | Stirring type (vortex or ultrasonic) | stir | Vor | - | Us |
| 7. | Centrifuge speed (rpm) | w | 2000 | 6000 | 10,000 |
| 8. | Centrifuge time (min) | tcfg | 2 | 6 | 10 |
| Source | DF | Adj SS | Adj MS | F-Value | p-Value |
|---|---|---|---|---|---|
| Model | 8 | 2222 | 277.7 | 8.97 | 0.049 |
| Linear | 8 | 2222 | 277.7 | 8.97 | 0.049 |
| dil | 1 | 184.0 | 184.1 | 5.94 | 0.093 |
| DES | 1 | 374.0 | 374.1 | 12.1 | 0.040 |
| tex | 1 | 24.08 | 24.08 | 0.78 | 0.443 |
| T | 1 | 954.0 | 954.0 | 30.8 | 0.012 |
| salt | 1 | 270.7 | 270.7 | 8.78 | 0.060 |
| stir | 1 | 310.1 | 310.1 | 10.01 | 0.051 |
| W | 1 | 90.75 | 90.75 | 2.93 | 0.185 |
| tcfg | 1 | 14.08 | 14.08 | 0.45 | 0.548 |
| Error | 3 | 92.92 | 30.97 | ||
| Total | 11 | 2314 |
| No. | Matrix | Concentration Range | Sample Preparation Method | Reagents/ Extractant | Analytical Technique | Limit of Detection | Linearity (R2) | Recovery (%) | RSD (%) | Reference |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Protein | 10-100 μg | acid hydrolysis | HCl/ninhydrin | ViS | n.a. | n.a. | 98.3 | 2.7 | [6] |
| 2 | Yeast extract * | 100–600 μM | enzymatic hydrolysis | hydroxylamine | ViS | 100 μM | 0.6969 | 86 | n.a. | [7] |
| 3 | Solution * | 10–100 mg/L | oxidation | NaOCl | ViS | 10 mg/L | 0.9996 | 90.5 | 1.19 | [8] |
| 4 | Millets * | 9–36 mg/L | biorecognition | MP@PDA-E. coli | ViS | 5.6 μM | 0.98 | 106 | 7.3 | [9] |
| 5 | Beer * | 0.02–0.12 mg/L | dSPE | graphene/clay/ Brij L23 | FL | 0.01 mg/L | 0.9991 | 90 | 5.0 | [10] |
| 6 | Dietary supplements * | 1.0–7.0 μmol/L | dilution | GCE/p-ARG | SWV | 0.30 μmol/L | 0.990 | 97.6 | 2.1 | [11] |
| 7 | Plasma * | 0.08–20.0 μM | screen-printed electrode | PdCuCo/RGO | DPV | 0.03 μM | 0.997 | 103.7 | 2.8 | [12] |
| 8 | Milk * | 5.0–150 µM | electrochemical sensor | graphite electrode | DPV | 5.78 µM | 0.9841 | 99.3 | 8.6 | [13] |
| 9 | Pharmaceutics * | 1–350 μM | MIP electrochemical sensor | AuNPs@PVP@SiO2MIP | LSV | 1 μM | 0.995 | 105 | 4 | [14] |
| 10 | Milk * | 0.01–80 μM | MIP biosensor | MIP-AF | EIS | 0.008 μM | 0.99 | 98.2 | 1.8 | [15] |
| 11 | Plasma * | 0.005–0.1 mol/L | dilution | HBP/SSA | CE | 5 μmol/L | 0.998 | 101.9 | 5.4 | [16] |
| 12 | Beer * | n.d.–40.7 mg/L | acid hydrolysis | HCl/HEC//BTP/EACA/AMPD | cITP | 4.35 mg/L | 0.9993 | 95.9 | 4.3 | [17] |
| 13 | Leaf tissue * | n.a. | SPE | acetic anhydride deriv. | GC-MS | n.a. | n.a. | 60 | 2 | [18] |
| 14 | Soy sauces * | 136–262 mg/L | precipitation | ethanol | HPLC | 1 mg/L | 0.995 | 108 | 4.9 | [19] |
| 15 | Dietary supplements * | 5.0–500 μg/m | HILIC | 1-octane sulfonate | HPLC | 1.2 mg/mL | 0.979 | 96.5 | 2.3 | [20] |
| 16 | Infant formula | 0.018–30 mg/kg | enzymatic hydrolysis | pronase enzyme | HPLC | 18 μg/kg | 0.9999 | 93.8 | 6.9 | [21] |
| 17 | Rapeseed | 10–400 ng | alkaline hydrolysis | NaOH | HPLC | 10 ng | 0.998 | 98.6 | 1.6 | [22] |
| 18 | Pig feed * | n.a. | dilution | HCl | HPLC | n.a. | n.a. | n.a. | 5.0 | [23] |
| 19 | Chicken feed | 59–130 g/kg | alkaline hydrolysis | NaOH/o-phthalaldehyde | HPLC-FLD | n.a. | n.a. | 86 | 4.0 | [24] |
| 20 | White bread | alkaline hydrolysis | NaOH | HPLC | n.a. | n.a. | 85 | 16.1 | [25] | |
| 21 | Wheat | 1.3–14.8 g/kg | alkaline hydrolysis | NaOH/O-phthalaldehyde | HPLC-FLD | n.a. | n.a. | 91.6 | 1.9 | [26] |
| 22 | Yogurt | 352–1220 mg/kg | alkaline hydrolysis | NaOH/5-methyl-l-tryptophan | HPLC-FLD | 11 μg/kg | 0.9995 | 93 | 1.1 | [27] |
| 23 | Bee pollen * | 0.069 mg/g | ultrasonic extraction | ACN | HPLC-FLD | 0.003 mg/L | 0.9998 | 93.8 | 3.82 | [28] |
| 24 | Ryegrass shoot | 0.5–40 µM | alkaline hydrolysis | NaOH | LC-MS | 0.02 µM | 0.99 | 89.9 | 8.5 | [29] |
| 25 | Whole blood | 0.1–25 ng/mL | VAMS | ACN/H2O | LC-MS/MS | 25 ng/mL | 0.9987 | 85 | 9.6 | [30] |
| 26 | Plasma | 0–160 μM | acid hydrolysis | MeOH/ZnSO4/TFA | LC-MS/MS | 83 nM/L | 0.995 | 88 | 11 | [31] |
| 27 | Honey * | 0.7–9.94 mg/kg | SPE | Oasis MCX 30 µm | LC-MS/MS | 1.0 μg/kg | n.a. | 60 | 4.3 | [32] |
| 28 | Milk * | 89.6–117 | QuEChERS | CAN | LC-MS/MS | 2 ng/mL | 0.99 | 103.7 | 2.6 | [33] |
| 29 | Plant material * | 1–50 ng/mL | SPE | Hybrid SPE–phospholipids | LC-MS/MS | 4 ng/mL | 0.996 | 87.8 | 15 | [34] |
| 30 | Chicken feed | n.a. | microwave hydrolysis | AQC-derivatization | LC-MS/MS | 1 fmol | n.a. | 99 | 4.2 | [35] |
| 31 | Hazelnut * | 42–127 μg/g | water extraction | water | UPLC-MS/MS | n.a. | n.a. | 117 | 30 | [37] |
| 32 | Nuts and seed oils * | 10–400 mg/kg | RP-DLLME | DES (ChCl:U) | HPLC | 11 mg/kg | 0.996 | 91 | 9.0 | This study |
| No. | Oils Made from | Trp Content (mg/100 g) | ± | Variation of Trp Content between Samples (%) |
|---|---|---|---|---|
| 1. | Almonds | 16 | ± | 16 |
| 2. | Brazil nuts | 14 | ± | 18 |
| 3. | Cashews | 11 | ± | 15 |
| 4. | Hazelnuts | 17 | ± | 14 |
| 5. | Peanuts | 18 | ± | 13 |
| 6. | Pumpkin seeds | 32 | ± | 26 |
| 7. | Sesame seeds | 33 | ± | 27 |
| 8. | Sunflower seeds | 38 | ± | 21 |
| 9. | Walnuts | 12 | ± | 14 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ražić, S.; Bakić, T.; Topić, A.; Lukić, J.; Onjia, A. Deep Eutectic Solvent Based Reversed-Phase Dispersive Liquid–Liquid Microextraction and High-Performance Liquid Chromatography for the Determination of Free Tryptophan in Cold-Pressed Oils. Molecules 2023, 28, 2395. https://doi.org/10.3390/molecules28052395
Ražić S, Bakić T, Topić A, Lukić J, Onjia A. Deep Eutectic Solvent Based Reversed-Phase Dispersive Liquid–Liquid Microextraction and High-Performance Liquid Chromatography for the Determination of Free Tryptophan in Cold-Pressed Oils. Molecules. 2023; 28(5):2395. https://doi.org/10.3390/molecules28052395
Chicago/Turabian StyleRažić, Slavica, Tamara Bakić, Aleksandra Topić, Jelena Lukić, and Antonije Onjia. 2023. "Deep Eutectic Solvent Based Reversed-Phase Dispersive Liquid–Liquid Microextraction and High-Performance Liquid Chromatography for the Determination of Free Tryptophan in Cold-Pressed Oils" Molecules 28, no. 5: 2395. https://doi.org/10.3390/molecules28052395
APA StyleRažić, S., Bakić, T., Topić, A., Lukić, J., & Onjia, A. (2023). Deep Eutectic Solvent Based Reversed-Phase Dispersive Liquid–Liquid Microextraction and High-Performance Liquid Chromatography for the Determination of Free Tryptophan in Cold-Pressed Oils. Molecules, 28(5), 2395. https://doi.org/10.3390/molecules28052395