

Electrochemical Monitoring in Anticoagulation Therapy



Abstract
:1. Introduction
2. Overview of Haemostasis
2.1. Platelets
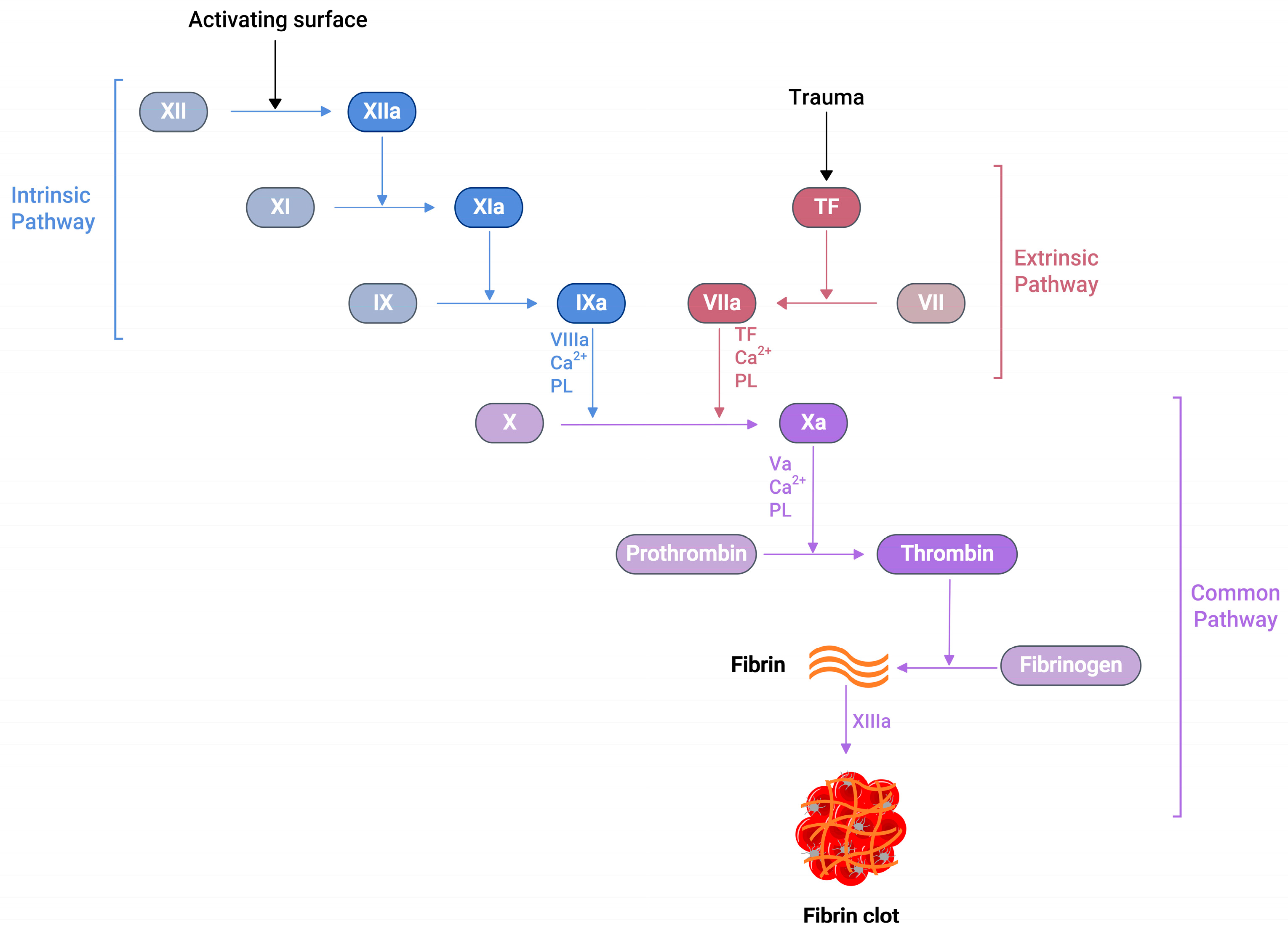
2.2. Coagulation Cascade
2.3. Thrombosis
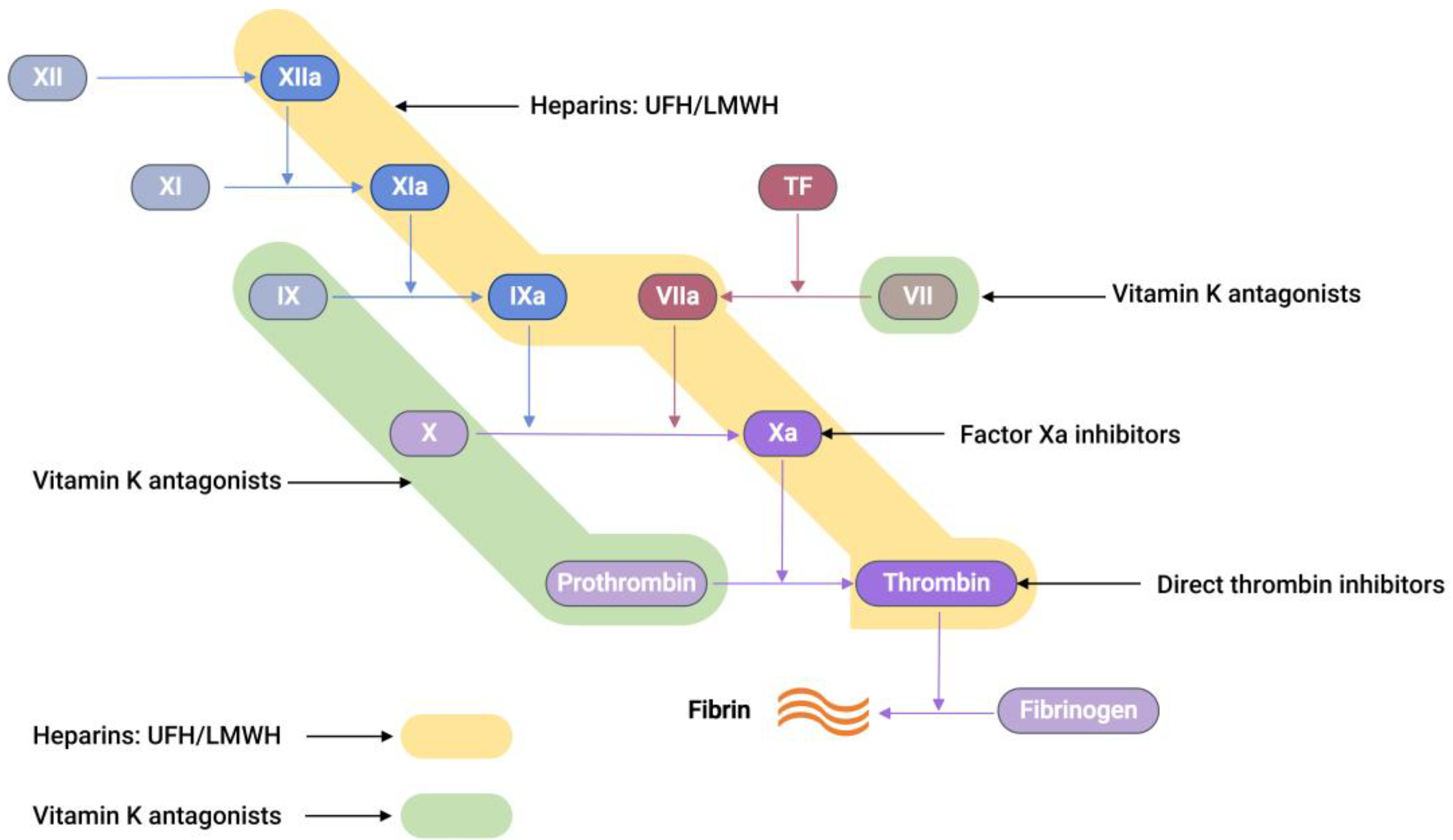
3. Drugs Used in Anticoagulation Therapy

4. Laboratory Testing of Coagulation
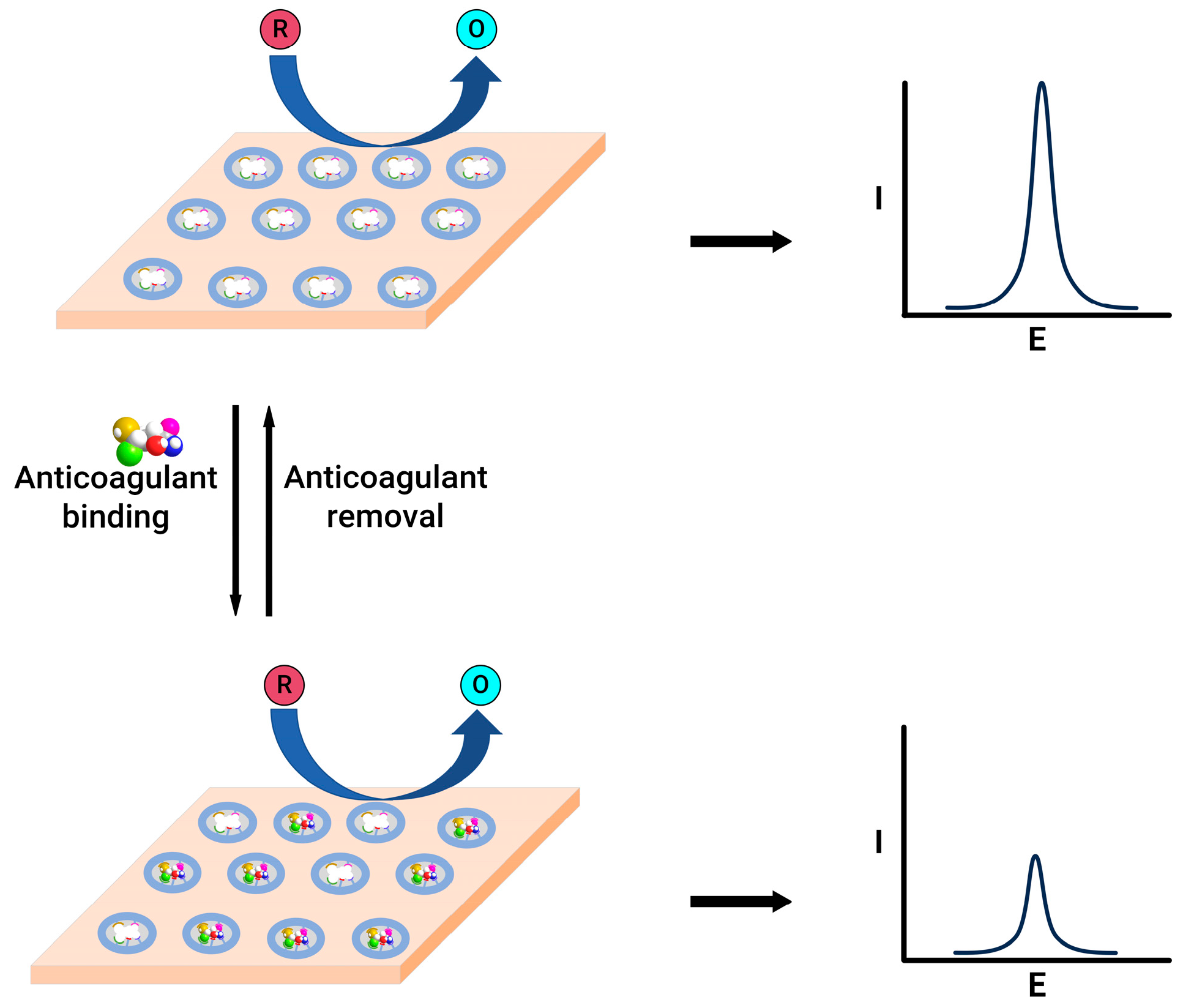
5. Electrochemical Sensors for Anticoagulants Based on Coagulation Factors
- (1)
- When a thrombin-specific peptide containing an electroactive group is added to the sample, the hydrolysis of the peptide by thrombin releases an electroactive group, which can then be quantified by applying a respective redox potential.
- (2)
- Thrombin can be directly quantified with the help of electrode modification by thrombin-specific aptamers; a change in the current response is a direct indication of the quantity of thrombin in the sample.
5.1. Electrochemical Sensors for Anticoagulants Using Thrombin-Specific Peptides
5.2. Electrochemical Sensors for Anticoagulants Using Modified Electrode Surfaces
6. Common Calibration Methods
7. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Levine, S.R. Hypercoagulable states and stroke: A selective review. CNS Spectr. 2005, 10, 567–578. [Google Scholar] [CrossRef]
- Kahn, M.J. Hypercoagulability as a cause of stroke in adults. (Featured CME Topic: Stroke). South. Med. J. 2003, 96, 350–354. [Google Scholar] [CrossRef]
- Falanga, A.; Marchetti, M.; Vignoli, A. Coagulation and cancer: Biological and clinical aspects. J. Thromb. Haemost. 2013, 11, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Lijfering, W.M.; Sprenger, H.G.; Georg, R.R.; van der Meulen, P.A.; van der Meer, J. Relationship between progression to AIDS and thrombophilic abnormalities in HIV infection. Clin. Chem. 2008, 54, 1226–1233. [Google Scholar] [CrossRef] [PubMed]
- Van Gorp, E.C.M.; Suharti, C.; ten Cate, H.; Dolmans, W.M.V.; van der Meer, J.W.M.; ten Cate, J.W.; Brandjes, D.P.M. Review: Infectious Diseases and Coagulation Disorders. J. Infect. Dis. 1999, 180, 176–186. [Google Scholar] [CrossRef] [PubMed]
- Levi, M.; Keller, T.T.; van Gorp, E.; ten Cate, H. Infection and inflammation and the coagulation system. Cardiovasc. Res. 2003, 60, 26–39. [Google Scholar] [CrossRef] [PubMed]
- Martini, W.Z. Coagulation complications following trauma. Mil. Med. Res. 2016, 3, 35. [Google Scholar] [CrossRef] [PubMed]
- Ceriello, A. Coagulation activation in diabetes mellitus: The role of hyperglycaemia and therapeutic prospects. Diabetologia 1993, 36, 1119–1125. [Google Scholar] [CrossRef] [PubMed]
- Alzahrani, S.H.; Ajjan, R.A. Coagulation and fibrinolysis in diabetes. Diabetes Vasc. Dis. Res. 2010, 7, 260–273. [Google Scholar] [CrossRef]
- Rugeri, L.; Levrat, A.; David, J.S.; Delecroix, E.; Floccard, B.; Gros, A.; Allaouchiche, B.; Negrier, C. Diagnosis of early coagulation abnormalities in trauma patients by rotation thrombelastography. J. Thromb. Haemost. 2007, 5, 289–295. [Google Scholar] [CrossRef]
- Kuhli-Hattenbach, C.; Scharrer, I.; Luchtenberg, M.; Hattenbach, L.O. Coagulation disorders and the risk of retinal vein occlusion. Thromb. Haemost. 2010, 103, 299–305. [Google Scholar]
- Fegan, C.D. Central retinal vein occlusion and thrombophilia. Eye 2002, 16, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Ganter, M.T.; Hofer, C.K. Coagulation monitoring: Current techniques and clinical use of viscoelastic point-of-care coagulation devices. Anesth. Analg. 2008, 106, 1366–1375. [Google Scholar] [CrossRef] [PubMed]
- Harris, L.F.; Castro-López, V.; Killard, A.J. Coagulation monitoring devices: Past, present, and future at the point of care. TrAC Trends Anal. Chem. 2013, 50, 85–95. [Google Scholar] [CrossRef]
- Mohammadi Aria, M.; Erten, A.; Yalcin, O. Technology Advancements in Blood Coagulation Measurements for Point-of-Care Diagnostic Testing. Front. Bioeng. Biotechnol. 2019, 7, 395. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, A.; Green, L. Physiology of haemostasis. Anaesth. Intensive Care Med. 2019, 20, 152–158. [Google Scholar] [CrossRef]
- Tynngård, N.; Lindahl, T.L.; Ramström, S. Assays of different aspects of haemostasis—What do they measure? Thromb. J. 2015, 13, 8. [Google Scholar] [CrossRef] [PubMed]
- Van der Meijden, P.E.J.; Heemskerk, J.W.M. Platelet biology and functions: New concepts and clinical perspectives. Nat. Rev. Cardiol. 2019, 16, 166–179. [Google Scholar] [CrossRef]
- Vinik, A.I.; Erbas, T.; Park, T.S.; Nolan, R.; Pittenger, G.L. Platelet Dysfunction in Type 2 Diabetes. Diabetes Care 2001, 24, 1476–1485. [Google Scholar] [CrossRef]
- Yan, H.; Naadiya, C.; Yiming, W.; Reid, C.G.; Alexandra, M.; Heyu, N. Platelets in hemostasis and thrombosis: Novel mechanisms of fibrinogen-independent platelet aggregation and fibronectin mediated protein wave of hemostasis. J. Biomed. Res. 2015, 29, 437–444. [Google Scholar] [CrossRef]
- Bryckaert, M.; Rosa, J.P.; Denis, C.V.; Lenting, P.J. Of von Willebrand factor and platelets. Cell. Mol. Life Sci. 2015, 72, 307–326. [Google Scholar] [CrossRef]
- Yun, S.H.; Sim, E.H.; Goh, R.Y.; Park, J.I.; Han, J.Y. Platelet Activation: The Mechanisms and Potential Biomarkers. BioMed Res. Int. 2016, 2016, 9060143. [Google Scholar] [CrossRef]
- Lentz, B.R. Exposure of platelet membrane phosphatidylserine regulates blood coagulation. Prog. Lipid Res. 2003, 42, 423–438. [Google Scholar] [CrossRef]
- Smith, S.A.; Travers, R.J.; Morrissey, J.H. How it all starts: Initiation of the clotting cascade. Crit. Rev. Biochem. Mol. Biol. 2015, 50, 326–336. [Google Scholar] [CrossRef] [PubMed]
- Adams, R.L.C.; Bird, R.J. Review article: Coagulation cascade and therapeutics update: Relevance to nephrology. Part 1: Overview of coagulation, thrombophilias and history of anticoagulants. Nephrology 2009, 14, 462–470. [Google Scholar] [CrossRef] [PubMed]
- Davie, E.W.; Fujikawa, K.; Kisiel, W. The coagulation cascade: Initiation, maintenance, and regulation. Biochemistry 1991, 30, 10363–10370. [Google Scholar] [CrossRef] [PubMed]
- Mackman, N. Triggers, targets and treatments for thrombosis. Nature 2008, 451, 914–918. [Google Scholar] [CrossRef]
- Oklu, R. Thrombosis. Cardiovasc. Diagn. Ther. 2017, 7 (Suppl. S3), S131–S133. [Google Scholar] [CrossRef]
- Rosenberg, R.D.; Rosenberg, J.S. Natural anticoagulant mechanisms. J. Clin. Investig. 1984, 74, 1–6. [Google Scholar] [CrossRef]
- Tollefsen, D.M. Heparin Cofactor II Modulates the Response to Vascular Injury. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 454–460. [Google Scholar] [CrossRef]
- Braune, S.; Küpper, J.-H.; Jung, F. Effect of Prostanoids on Human Platelet Function: An Overview. Int. J. Mol. Sci. 2020, 21, 9020. [Google Scholar] [CrossRef] [PubMed]
- Cesarman-Maus, G.; Hajjar, K.A. Molecular mechanisms of fibrinolysis. Br. J. Haematol. 2005, 129, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Liu, L.; Voglmeir, J. Chemoenzymatic synthesis of ultralow and low-molecular weight heparins. Biochim. Biophys. Acta (BBA)—Proteins Proteom. 2020, 1868, 140301. [Google Scholar] [CrossRef] [PubMed]
- Hirsh, J. Heparin. N. Engl. J. Med. 1991, 324, 1565–1574. [Google Scholar] [PubMed]
- Kumano, O.; Akatsuchi, K.; Amiral, J. Updates on Anticoagulation and Laboratory Tools for Therapy Monitoring of Heparin, Vitamin K Antagonists and Direct Oral Anticoagulants. Biomedicines 2021, 9, 264. [Google Scholar] [CrossRef] [PubMed]
- Jay, R.M.; Lui, P. How anticoagulants work. Tech. Reg. Anesth. Pain Manag. 2006, 10, 30–39. [Google Scholar] [CrossRef]
- Qiu, M.; Huang, S.; Luo, C.; Wu, Z.; Liang, B.; Huang, H.; Ci, Z.; Zhang, D.; Han, L.; Lin, J. Pharmacological and clinical application of heparin progress: An essential drug for modern medicine. Biomed. Pharmacother. 2021, 139, 111561. [Google Scholar] [CrossRef]
- Schein, J.R.; White, C.M.; Nelson, W.W.; Kluger, J.; Mearns, E.S.; Coleman, C.I. Vitamin K antagonist use: Evidence of the difficulty of achieving and maintaining target INR range and subsequent consequences. Thromb. J. 2016, 14, 14. [Google Scholar] [CrossRef]
- Harter, K.; Levine, M.; Henderson, S.O. Anticoagulation drug therapy: A review. West. J. Emerg. Med. 2015, 16, 11–17. [Google Scholar] [CrossRef]
- Ebner, M.; Birschmann, I.; Peter, A.; Härtig, F.; Spencer, C.; Kuhn, J.; Blumenstock, G.; Zuern, C.S.; Ziemann, U.; Poli, S. Emergency Coagulation Assessment During Treatment with Direct Oral Anticoagulants. Stroke 2017, 48, 2457–2463. [Google Scholar] [CrossRef]
- Dias, J.D.; Lopez-Espina, C.G.; Ippolito, J.; Hsiao, L.H.; Zaman, F.; Muresan, A.A.; Thomas, S.G.; Walsh, M.; Jones, A.J.; Grisoli, A.; et al. Rapid point-of-care detection and classification of direct-acting oral anticoagulants with the TEG 6s: Implications for trauma and acute care surgery. J. Trauma Acute Care Surg. 2019, 87, 364–370. [Google Scholar] [CrossRef] [PubMed]
- Pai, M. Chapter 129—Laboratory Evaluation of Hemostatic and Thrombotic Disorders. In Hematology, 7th ed.; Hoffman, R., Benz, E.J., Silberstein, L.E., Heslop, H.E., Weitz, J.I., Anastasi, J., Salama, M.E., Abutalib, S.A., Eds.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 1922–1931. [Google Scholar]
- Campbell, S. Chapter 26—Hemostasis. In Contemporary Practice in Clinical Chemistry, 4th ed.; Clarke, W., Marzinke, M.A., Eds.; Academic Press: Cambridge, MA, USA, 2020; pp. 445–467. [Google Scholar]
- Tripodi, A.; Caldwell, S.H.; Hoffman, M.; Trotter, J.F.; Sanyal, A.J. Review article: The prothrombin time test as a measure of bleeding risk and prognosis in liver disease. Aliment. Pharmacol. Ther. 2007, 26, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Perry, D.J.; Fitzmaurice, D.A.; Kitchen, S.; Mackie, I.J.; Mallett, S. Point-of-care testing in haemostasis. Br. J. Haematol. 2010, 150, 501–514. [Google Scholar] [CrossRef] [PubMed]
- Alserr, A.H.K.; Menshawey, R.; Kotb, A.; Hussein, N.; Kotp, N.; Ashraf-Taha, M.; Anwar, N.; Abdalla, A.; Abdullah, M.; Ela, S.A.; et al. A Comparison of International Normalized Ratio Results by Point-of-Care Device and Clinical Laboratory Analyzers in a Vascular Surgery Department. Point Care 2020, 19, 106–111. [Google Scholar] [CrossRef]
- Bardakci, H.; Altıntaş, G.; Çiçek, O.F.; Kervan, U.; Yilmaz, S.; Kaplan, S.; Birincioglu, C.L. Comparison of the CoaguChek XS Handheld Coagulation Analyzer and Conventional Laboratory Methods Measuring International Normalised Ratio (INR) Values during the Time to Therapeutic Range after Mechanical Valve Surgery. J. Card. Surg. 2013, 28, 254–257. [Google Scholar] [CrossRef] [PubMed]
- Luke, R.B.; Shane, L.J.; Gregory, M.P.; Ella, C.J.; Katherine, A.M.; David, M.J. Accuracy and clinical utility of the CoaguChek XS portable international normalised ratio monitor in a pilot study of warfarin home-monitoring. J. Clin. Pathol. 2007, 60, 311. [Google Scholar]
- Wieloch, M.; Hillarp, A.; Strandberg, K.; Nilsson, C.; Svensson, P.J. Comparison and evaluation of a Point-of-care device (CoaguChek XS) to Owren-type prothrombin time assay for monitoring of oral anticoagulant therapy with warfarin. Thromb. Res. 2009, 124, 344–348. [Google Scholar] [CrossRef] [PubMed]
- Piacenza, F.; Galeazzi, R.; Cardelli, M.; Moroni, F.; Provinciali, M.; Pierpaoli, E.; Giovagnetti, S.; Appolloni, S.; Marchegiani, F. Precision and accuracy of the new XPrecia Stride mobile coagulometer. Thromb. Res. 2017, 156, 51–53. [Google Scholar] [CrossRef]
- Spielmann, N.; Mauch, J.Y.; Madjdpour, C.; Schmugge, M.; Albisetti, M.; Weiss, M.; Haas, T. Comparison of point-of-care testing (POCT): I-STAT® international normalized ratio (INR) vs reference laboratory INR in pediatric patients undergoing major surgery. Pediatr. Anesth. 2011, 21, 1041–1045. [Google Scholar] [CrossRef]
- Deborah, M.; Andrea, R.; David, A.F. An evaluation of a coagulation system (Xprecia Stride) for utilisation in anticoagulation management. J. Clin. Pathol. 2018, 71, 20. [Google Scholar]
- Schober, P.; Bossers, S.M.; Koolwijk, J.; Terra, M.; Schwarte, L.A. Prehospital coagulation measurement by a portable blood analyzer in a helicopter emergency medical service (HEMS). Am. J. Emerg. Med. 2021, 46, 137–140. [Google Scholar] [CrossRef]
- Thuerlemann, C.; Haeberli, A.; Alberio, L. Monitoring thrombin generation by electrochemistry: Development of an amperometric biosensor screening test for plasma and whole blood. Clin. Chem. 2009, 55, 505–512. [Google Scholar] [CrossRef]
- Ebner, M.; Peter, A.; Spencer, C.; Härtig, F.; Birschmann, I.; Kuhn, J.; Wolf, M.; Winter, N.; Russo, F.; Zuern, C.S.; et al. Point-of-Care Testing of Coagulation in Patients Treated with Non–Vitamin K Antagonist Oral Anticoagulants. Stroke 2015, 46, 2741–2747. [Google Scholar] [CrossRef] [PubMed]
- Mruthunjaya, A.K.V.; Chatelier, R.C.; Torriero, A.A.J. Electrochemical Disposable Biosensor to Monitor Dabigatran in Point-of-Care Anticoagulation Therapy. Molecules 2023, 28, 4953. [Google Scholar] [CrossRef] [PubMed]
- Mruthunjaya, A.K.V.; Hodges, A.M.; Chatelier, R.C.; Torriero, A.A.J. Calibration-free disposable electrochemical sensor with co-facing electrodes: Theory and characterisation with fixed and changing mediator concentration. Electrochim. Acta 2023, 460, 142596. [Google Scholar] [CrossRef]
- Mruthunjaya, A.K.V.; Chatelier, R.C.; Torriero, A.A.J. Calibration-free electrochemical sensor to monitor factor-Xa inhibitors at the point-of-care anticoagulation therapy. Talanta 2024, 270, 125593. [Google Scholar] [CrossRef]
- Deng, B.; Lin, Y.; Wang, C.; Li, F.; Wang, Z.; Zhang, H.; Li, X.-F.; Le, X.C. Aptamer binding assays for proteins: The thrombin example—A review. Anal. Chim. Acta 2014, 837, 1–15. [Google Scholar] [CrossRef]
- Sun, H.; Wang, N.; Zhang, L.; Meng, H.; Li, Z. Aptamer-Based Sensors for Thrombin Detection Application. Chemosensors 2022, 10, 255. [Google Scholar] [CrossRef]
- Eivazzadeh-Keihan, R.; Saadatidizaji, Z.; Maleki, A.; de la Guardia, M.; Mahdavi, M.; Barzegar, S.; Ahadian, S. Recent Progresses in Development of Biosensors for Thrombin Detection. Biosensors 2022, 12, 767. [Google Scholar] [CrossRef]
- Wolberg, A.S.; Campbell, R.A. Thrombin generation, fibrin clot formation and hemostasis. Transfus. Apher. Sci. 2008, 38, 15–23. [Google Scholar] [CrossRef]
- Butenas, S.; Mann, K.G.; Butenas. Blood Coagulation. Biochemistry 2002, 67, 3–12. [Google Scholar] [PubMed]
- Xiao, Y.; Lubin, A.A.; Heeger, A.J.; Plaxco, K.W. Label-Free Electronic Detection of Thrombin in Blood Serum by Using an Aptamer-Based Sensor. Angew. Chem. 2005, 117, 5592–5595. [Google Scholar] [CrossRef]
- Suprun, E.; Shumyantseva, V.; Bulko, T.; Rachmetova, S.; Rad’ko, S.; Bodoev, N.; Archakov, A. Au-nanoparticles as an electrochemical sensing platform for aptamer–thrombin interaction. Biosens. Bioelectron. 2008, 24, 825–830. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.; Moon, J.M.; Choi, J.; Hwang, H.; Shim, Y.B. Magnetic force assisted electrochemical sensor for the detection of thrombin with aptamer-antibody sandwich formation. Biosens. Bioelectron. 2018, 117, 480–486. [Google Scholar] [CrossRef]
- Chen, H.J.; Chen, R.L.C.; Hsieh, B.C.; Hsiao, H.Y.; Kung, Y.; Hou, Y.T.; Cheng, T.J. Label-free and reagentless capacitive aptasensor for thrombin. Biosens. Bioelectron. 2019, 131, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Xu, F.; Miao, S.; Xie, C.; Li, H.; Li, S.; Xia, F. Incorporation of a Multi-Valent Aptamer into Electrochemical Biosensors to Achieve an Improved Performance for Thrombin Analysis in Blood Serum. Chempluschem 2022, 87, e202200325. [Google Scholar] [CrossRef]
- Yagati, A.K.; Behrent, A.; Tomanek, V.; Chavan, S.G.; Go, A.; Park, S.R.; Jin, Z.; Baeumner, A.J.; Lee, M.H. Polypyrrole-palladium nanocomposite as a high-efficiency transducer for thrombin detection with liposomes as a label. Anal. Bioanal. Chem. 2022, 414, 3205–3217. [Google Scholar] [CrossRef]
- Dhara, K.; Mahapatra, D.R. Recent advances in electrochemical nonenzymatic hydrogen peroxide sensors based on nanomaterials: A review. J. Mater. Sci. 2019, 54, 12319–12357. [Google Scholar] [CrossRef]
- Liu, X.; Huang, L.; Qian, K. Nanomaterial-Based Electrochemical Sensors: Mechanism, Preparation, and Application in Biomedicine. Adv. NanoBiomed Res. 2021, 1, 2000104. [Google Scholar] [CrossRef]
- Chen, A.; Chatterjee, S. Nanomaterials based electrochemical sensors for biomedical applications. Chem. Soc. Rev. 2013, 42, 5425–5438. [Google Scholar] [CrossRef]
- Rezaei, B.; Rahmanian, O.; Ensafi, A.A. An electrochemical sensor based on multiwall carbon nanotubes and molecular imprinting strategy for warfarin recognition and determination. Sens. Actuators B Chem. 2014, 196, 539–545. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, Y.; Jiang, M.; Tian, L.; Sun, S.; Zhao, N.; Zhao, F.; Li, Y. Electrochemical microfluidic chip based on molecular imprinting technique applied for therapeutic drug monitoring. Biosens. Bioelectron. 2017, 91, 714–720. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, L.; Liu, J.; Zhou, S.-F.; Al-Ghanim, K.A.; Mahboob, S.; Ye, B.-C.; Zhang, X. A novel sensitive and selective electrochemical sensor based on molecularly imprinted polymer on a nanoporous gold leaf modified electrode for warfarin sodium determination. RSC Adv. 2016, 6, 43724–43731. [Google Scholar] [CrossRef]
- Gholivand, M.B.; Mohammadi-Behzad, L. An electrochemical sensor for warfarin determination based on covalent immobilization of quantum dots onto carboxylated multiwalled carbon nanotubes and chitosan composite film modified electrode. Mater. Sci. Eng. C 2015, 57, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Maheshwaran, S.; Akilarasan, M.; Chen, S.-M.; Tamilalagan, E.; Keerthiga, E.; Alothman, A.A.; Alqahtani, K.N.; Ganesh, P.S. Synthesis of nickel-doped ceria nanospheres for in situ profiling of Warfarin sodium in biological media. Bioelectrochemistry 2022, 146, 108166. [Google Scholar] [CrossRef] [PubMed]
- Hashemi, M.; Rahimnejad, M.; Ezoji, H. Facile and sensitive electrochemical sensing device based on carbon paste electrode for warfarin determination. Monatshefte Chem.—Chem. Mon. 2023, 155, 29–35. [Google Scholar] [CrossRef]
- Belal, F.; Anderson, J.L. Flow injection analysis of warfarin sodium with amperometric detection. Microchim. Acta 1985, 86, 145–151. [Google Scholar] [CrossRef]
- Saeedi, I.; Ahmadi, S.; Thompson, M.; Hashemi, P.; Ramezani, Z. Electrochemical Sensor for the Direct Determination of Warfarin in Blood. Chemosensors 2022, 10, 44. [Google Scholar] [CrossRef]
- Huo, H.Y.; Luo, H.Q.; Li, N.B. Electrochemical sensor for heparin based on a poly(thionine) modified glassy carbon electrode. Microchim. Acta 2009, 167, 195–199. [Google Scholar] [CrossRef]
- Tian, L.; Zhao, H.; Zhao, Z.; Zhai, J.; Zhang, Z. A facile voltammetric method for detection of heparin in plasma based on the polyethylenimine modified electrode. Anal. Methods 2019, 11, 1324–1330. [Google Scholar] [CrossRef]
- 8Rengaraj, A.; Haldorai, Y.; Hwang, S.K.; Lee, E.; Oh, M.H.; Jeon, T.J.; Han, Y.K.; Huh, Y.S. A protamine-conjugated gold decorated graphene oxide composite as an electrochemical platform for heparin detection. Bioelectrochemistry 2019, 128, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Aljohani, M.M.; Chinnappan, R.; Eissa, S.; Alsager, O.A.; Weber, K.; Cialla-May, D.; Popp, J.; Zourob, M. In Vitro Selection of Specific DNA Aptamers Against the Anti-Coagulant Dabigatran Etexilate. Sci. Rep. 2018, 8, 13290. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.; Sinha, A. Electrocatalytic quantification of thrombin inhibitor dabigatran etexilate in solubilized system. Ionics 2015, 21, 1445–1452. [Google Scholar] [CrossRef]
- Festinger, N.; Smarzewska, S.; Ciesielski, W. Comparative study of boron-doped diamond, basal-plane pyrolytic graphite, and graphite flake paste electrodes for the voltammetric determination of rivaroxaban and dabigatran etexilate in pharmaceuticals and urine samples. Diam. Relat. Mater. 2021, 118, 108539. [Google Scholar] [CrossRef]
- Abou El-Alamin, M.M.; Mohamed, D.A.; Toubar, S.S. New disposable ion-selective sensors for the determination of dabigatran etexilate: The oral anticoagulant of choice in patients with non-valvular atrial fibrillation and COVID-19 infection. Measurement 2022, 198, 111406. [Google Scholar] [CrossRef]
- Ebrahimi, R.; Barzegari, A.; Teimuri-Mofrad, R.; Kordasht, H.K.; Hasanzadeh, M.; Khoubnasabjafari, M.; Jouyban-Gharamaleki, V.; Rad, A.A.; Shadjou, N.; Rashidi, M.R.; et al. Selection of Specific Aptamer against Rivaroxaban and Utilization for Label-Free Electrochemical Aptasensing Using Gold Nanoparticles: First Announcement and Application for Clinical Sample Analysis. Biosensors 2022, 12, 773. [Google Scholar] [CrossRef]
- Ebrahimi, R.; Hasanzadeh, M.; Rashidi, M.-R.; Jouyban, A. Low fouling aptasensing of rivaroxaban in real samples using poly (toluidine blue) decorated by silver nanoparticle: A new platform for the cardiovascular disease analysis. Microchem. J. 2023, 189, 108529. [Google Scholar] [CrossRef]
- Abdallah, A.B.; Saher, A.; Molouk, A.F.S.; Mortada, W.I.; Khalifa, M.E. Applications of electrochemical techniques for determination of anticoagulant drug (Rivaroxaban) in real samples. Biosens. Bioelectron. 2022, 208, 114213. [Google Scholar] [CrossRef]
- Ayish, N.S.; Marzouk, H.M.; El-Zeany, B.A.; Fayed, A.S. A Novel Nanoparticles-based Electrochemical Sensing Platform for Sensitive Detection of Oral Anticoagulant; Edoxaban in Human Plasma. Electroanalysis 2022, 34, 1266–1272. [Google Scholar] [CrossRef]
- Kiliç, A.; Aslan, M.; Önal, G.; Levent, A. Firstly electrochemical investigetions and determination of anticoagulant drug edoxaban at single-use pencil graphite electrode: An eco-friendly and cost effective voltammetric method. DARU J. Pharm. Sci. 2023, 31, 233–241. [Google Scholar] [CrossRef]
- Kiliç, A.; Aslan, M.; Levent, A. Investigation of the electrochemical properties of edoxaban using glassy carbon and boron-doped diamond electrodes and development of an eco-friendly and cost effective voltammetric method for its determination. Anal. Biochem. 2024, 685, 115386. [Google Scholar] [CrossRef]
- Rizk, M.; Sultan, M.A.; Taha, E.A.; Attia, A.K.; Abdallah, Y.M. Sensitive validated voltammetric determination of apixaban using a multi-walled carbon nanotube-modified carbon paste electrode: Application to a drug product and biological sample. Anal. Methods 2017, 9, 2523–2534. [Google Scholar] [CrossRef]
- Hoffman, M. Heparins: Clinical Use and Laboratory Monitoring. Lab. Med. 2010, 41, 621–626. [Google Scholar] [CrossRef]
- Hirsh, J.; Anand, S.S.; Halperin, J.L.; Fuster, V. Guide to Anticoagulant Therapy: Heparin. Circulation 2001, 103, 2994–3018. [Google Scholar] [CrossRef]
- Bromfield, S.M.; Wilde, E.; Smith, D.K. Heparin sensing and binding—Taking supramolecular chemistry towards clinical applications. Chem. Soc. Rev. 2013, 42, 9184–9195. [Google Scholar] [CrossRef]
- Amemiya, S.; Kim, Y.; Ishimatsu, R.; Kabagambe, B. Electrochemical heparin sensing at liquid/liquid interfaces and polymeric membranes. Anal. Bioanal. Chem. 2011, 399, 571–579. [Google Scholar] [CrossRef]
- Vishenkova, D.A.; Korotkova, E.I. Electrochemical methods for the determination of heparin. J. Anal. Chem. 2017, 72, 349–353. [Google Scholar] [CrossRef]
- Shishkanova, T.V.; Briza, T.; Rezanka, P.; Kejik, Z.; Jakubek, M. Pentamethinium Salts Nanocomposite for Electrochemical Detection of Heparin. Materials 2021, 14, 5357. [Google Scholar] [CrossRef] [PubMed]
- Yoshimi, Y.; Yagisawa, Y.; Yamaguchi, R.; Seki, M. Blood heparin sensor made from a paste electrode of graphite particles grafted with molecularly imprinted polymer. Sens. Actuators B Chem. 2018, 259, 455–462. [Google Scholar] [CrossRef]
- Yoshimi, Y.; Kani, S.; Aaryashree. A disposable edoxaban sensor chip using carbon paste electrode grafted with molecularly imprinted polymer. J. Artif. Organs 2023, 27, 77–81. [Google Scholar] [CrossRef] [PubMed]
- Shahbazi-Derakhshi, P.; Abbasi, M.; Akbarzadeh, A.; Mokhtarzadeh, A.; Hosseinpour, H.; Soleymani, J. A ratiometric electrochemical probe for the quantification of apixaban in unprocessed plasma samples using carbon aerogel/BFO modified glassy carbon electrodes. RSC Adv. 2023, 13, 21432–21440. [Google Scholar] [CrossRef]
- Van Pelt, L.J.; Lukens, M.V.; Testa, S.; Chatelain, B.; Douxfils, J.; Mullier, F. The DaXa-inhibition assay: A concept for a readily available, universal aXa assay that measures the direct inhibitory effect of all anti-Xa drugs. Thromb. Res. 2018, 168, 63–66. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sensor Composition | Anticoagulant Detected | Detection Limit | Quantification Limit | Ref. |
|---|---|---|---|---|
| AuNP/MIP/f-MWCNT/GCE | warfarin | 0.078 nM | 0.101 nM | [73] |
| MIP/NPAMW | warfarin | 8.00 pM | 20 pM | [74] |
| MIP/NPGL/GE | warfarin | 41.0 pM | NA | [75] |
| CdS-QDs/CS/MWCNTs/GCE | warfarin | 8.50 nM | 28 nM | [76] |
| CeO2@Ni/GCE | warfarin | 6.30 nM | 10 nM | [77] |
| CPE | warfarin | 0.315 µM | 1.05 µM | [78] |
| GCE | warfarin | 16.0 nM | 3.24 µM | [79] |
| TDDA/ISE | warfarin | 14 µM (blood) | NA | [80] |
| 0.125 µM (buffer) | NA | |||
| poly(thionine)/GCE | heparin | 18.7 nM | 0.27 µM | [81] |
| [Fe(CN)6]3−/PEI/SWCNT-GCE | heparin | 0.088 µM | 0.133 µM | [82] |
| GO/Au-protamine/GCE | heparin | 0.9 nM | 1.59 nM | [83] |
| Aptamer/GE | dabigatran | 15.9 pM | NA | [84] |
| GR/CeO2/GCE | dabigatran | 19.9 nM | 66.3 nM | [85] |
| BDDE | dabigatran | 2.78 nM | 9.25 nM | [86] |
| BPPGE | 16.7 nM | 55.8 nM | [86] | |
| ISCPEs | dabigatran | 4.36 µM | NA | [87] |
| ISPGEs | 0.426 µM | |||
| Aptamer-AuNPs-PET-OH-ITO | rivaroxaban | 14.1 nM (plasma) 6.03 nM (EBC) | NA | [88] |
| ITO-PET-OH-p(TB-Ag)-Aptamer-PEG | rivaroxaban | 6.65 nM (plasma) 4.13 nM (EBC) | NA | [89] |
| MIP/PVC/GCE | rivaroxaban | 2.29 µM | 6.88 µM | [90] |
| GCE/MWCNT-ISM | edoxaban | 3.39 µM | NA | [91] |
| PGE | edoxaban | 0.073 µM | 0.243 µM | [92] |
| GCE | edoxaban | 0.24 µM | 0.84 µM | [93] |
| BDDE | 0.57 µM | 1.90 µM | ||
| MWCNT/CPE | apixaban | 0.618 µM | 1.99 µM | [94] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mruthunjaya, A.K.V.; Torriero, A.A.J. Electrochemical Monitoring in Anticoagulation Therapy. Molecules 2024, 29, 1453. https://doi.org/10.3390/molecules29071453
Mruthunjaya AKV, Torriero AAJ. Electrochemical Monitoring in Anticoagulation Therapy. Molecules. 2024; 29(7):1453. https://doi.org/10.3390/molecules29071453
Chicago/Turabian StyleMruthunjaya, Ashwin K. V., and Angel A. J. Torriero. 2024. "Electrochemical Monitoring in Anticoagulation Therapy" Molecules 29, no. 7: 1453. https://doi.org/10.3390/molecules29071453
APA StyleMruthunjaya, A. K. V., & Torriero, A. A. J. (2024). Electrochemical Monitoring in Anticoagulation Therapy. Molecules, 29(7), 1453. https://doi.org/10.3390/molecules29071453