
Quantitative Structure-Antioxidant Activity Models of Isoflavonoids: A Theoretical Study


Abstract
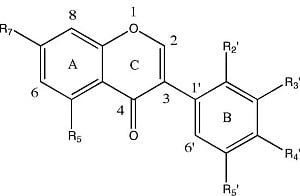
:
1. Introduction
2. Results and Discussion

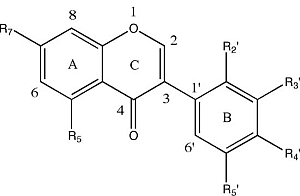{kind=link}
| Molecular Structure | Entry | Isoflavones | R5 | R7 | R2′ | R3′ | R4′ | R5′ |
 | 1 | Khrinone C | OH | OH | OMe | OH | OMe | H |
| 2 | Calycosin | H | OH | H | OH | OMe | H | |
| 3 | Genistein | OH | OH | H | H | OH | H | |
| 4 | 3′-O-Methylorobol | OH | OH | H | OMe | OH | H | |
| 5 | Cajanin | OH | OMe | OH | H | OH | H | |
| 6 | Khrinone B | OH | OH | OH | H | OMe | OH | |
| 7 | Biochanin A | OH | OH | H | H | OMe | H | |
| 8 | Formononetin | H | OH | H | H | OMe | H | |
| Molecular Structure | Entry | Isoflavanones | R5 | R7 | R2′ | R3′ | R4′ | R5′ |
 | 9 | 3(R,S)-Violanone | H | OH | OMe | OH | OMe | H |
| 10 | 3(S)-Secundiflorol H | OH | OH | OMe | OH | OMe | H | |
| 11 | 3(R,S)-Dalparvin | H | OH | OMe | H | OMe | OH | |
| 12 | 3(R,S)-Onogenin | H | OH | OMe | H | OCH2O | ||
| 13 | 3(S)-Sativanone | H | OH | OMe | H | OMe | H | |
| 14 | 3(R,S)-3′-O-Methylviolanone | H | OH | OMe | OMe | OMe | H | |
| Molecular Structure | Entry | Isoflavans | R7 | R8 | R2′ | R3′ | R4′ | R5′ |
 | 15 | 3(R)-Vestitol | OH | H | OH | H | OMe | H |
| 16 | 3(R)(+)-Mucronulatol | OH | H | OMe | OH | OMe | H | |
| 17 | 3(S)-8-Demethylduartin | OH | OH | OMe | OH | OMe | H | |
| Entry | X/XO Assay SC50 [μM] a | ORAC Assay TE [μM] b | DPPH Assay SC50 [μM] a | Log X/XO | Log ORAC | Mw [Da] c | ALog P d | ACD Log P e | ACD Log D f | ACD pKa g | RBN h | QEDw i | PSA [Å2] j | NH + OH | HBD k | N + O | HBA l |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 0.64 | 43.5 | 61.7 | −0.194 | 1.638 | 330 | 2.11 | 2.25 | 0.82 | 6.32 | 3 | 0.79 | 105 | 3 | 3 | 7 | 7 |
| 2 | 0.25 | 37.8 | 96.2 | −0.602 | 1.577 | 284 | 2.37 | 1.33 | 0.75 | 6.95 | 2 | 0.89 | 76 | 2 | 2 | 5 | 5 |
| 3 | 9.0 | 37.8 | 300 | 0.954 | 1.577 | 270 | 2.14 | 3.11 | 1.93 | 6.51 | 1 | 0.74 | 87 | 3 | 3 | 5 | 5 |
| 4 | 36.7 | 35.7 | 81.2 | 1.565 | 1.553 | 300 | 2.12 | 2.63 | 1.25 | 6.35 | 2 | 0.79 | 96.2 | 3 | 3 | 6 | 6 |
| 5 | 54.3 | 34.7 | 70.8 | 1.735 | 1.540 | 369 | 3.52 | 3.88 | 3.86 | 8.93 | 8 | 0.54 | 96.2 | 3 | 3 | 6 | 6 |
| 6 | 0.60 | 34.2 | 133.6 | −0.222 | 1.534 | 316 | 1.88 | 1.71 | 0.37 | 6.38 | 2 | 0.63 | 116 | 4 | 4 | 7 | 7 |
| 7 | 203.3 | 26.6 | 300 | 2.308 | 1.425 | 284 | 2.37 | 3.34 | 2.11 | 6.5 | 2 | 0.89 | 76 | 2 | 2 | 5 | 5 |
| 8 | 116.92 | 2.8 | 300 | 2.068 | 0.447 | 268 | 2.61 | 6.99 | 2.86 | 2.31 | 2 | 0.91 | 55.8 | 1 | 1 | 4 | 4 |
| 9 | 43.7 | 31.1 | 89.7 | 1.640 | 1.493 | 286 | 2.48 | 7.69 | 2.63 | 2.44 | 2 | 0.89 | 76 | 2 | 2 | 6 | 5 |
| 10 | 247.2 | 27.4 | 74.3 | 2.393 | 1.438 | 302 | 2.24 | 2.76 | 2.34 | 7.5 | 2 | 0.79 | 96.2 | 3 | 3 | 7 | 6 |
| 11 | 48.2 | 21.8 | 80.4 | 1.683 | 1.338 | 332 | 2.22 | 7.48 | 3.01 | 2.58 | 3 | 0.79 | 105 | 2 | 3 | 6 | 7 |
| 12 | 56.9 | 0.0 | 300 | 1.755 | 0.0 | 330 | 2.25 | 4.52 | 4.1 | 7.48 | 2 | 0.87 | 94.4 | 1 | 2 | 6 | 7 |
| 13 | 59.3 | 0.0 | 300 | 1.773 | 0.0 | 270 | 2.72 | 3.48 | 3.31 | 7.7 | 2 | 0.91 | 55.8 | 1 | 1 | 5 | 4 |
| 14 | 300 | 0.0 | 300 | 2.477 | 0.0 | 330 | 2.69 | 2.93 | 2.74 | 7.67 | 7 | 0.93 | 74.2 | 1 | 1 | 6 | 6 |
| 15 | 6.4 | 40.1 | 204.1 | 0.806 | 1.603 | 272 | 3.2 | 3.26 | 3.25 | 9.53 | 2 | 0.88 | 58.9 | 2 | 2 | 4 | 4 |
| 16 | 10.0 | 39.8 | 75.41 | 1.000 | 1.600 | 302 | 3.18 | 2.84 | 2.84 | 9.87 | 3 | 0.91 | 68.2 | 2 | 2 | 5 | 5 |
| 17 | 13.4 | 27.0 | 115.4 | 1.127 | 1.431 | 318 | 2.94 | 1.65 | 1.65 | 9.75 | 3 | 0.75 | 88.4 | 3 | 3 | 6 | 6 |
2.1. Correlations between the Different Methods, and Physicochemical and Physiochemical Properties
2.1.1. Xanthine/Xanthine Oxidase Assay
2.1.2. Oxygen Radical Absorbance Capacity Assay
2.1.3. 2,2-Diphenyl-1-picrylhydrazyl Assay
2.1.4. Comparison between the Three Methods
| m | −Log X/XO Equation (3) | −Log X/XO Equation (4) | Log ORAC Equation (5) | Log ORAC Equation (6) | −DPPH Equation (7) | −DPPH Equation (8) | DPPH Equation (11) | DPPH Equation (12) | ALog P Equation (13) |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 0.553 | 0.726 | 0.758 | 0.870 | 0.400 | 0.655 | 0.638 | 0.748 | 0.824 |
| 2 | 0.552 | 0.725 | 0.757 | 0.870 | 0.405 | 0.653 | 0.638 | 0.748 | 0.828 |
| 3 | 0.550 | 0.724 | 0.756 | 0.869 | 0.409 | 0.651 | 0.638 | 0.747 | 0.832 |
| 4 | 0.549 | 0.722 | 0.755 | 0.867 | 0.415 | 0.648 | 0.638 | 0.746 | 0.836 |
| 5 | 0.546 | 0.720 | 0.753 | - | 0.422 | 0.645 | 0.637 | 0.745 | 0.839 |
| 6 | 0.544 | 0.717 | 0.751 | - | 0.431 | 0.641 | 0.637 | 0.744 | - |
| 7 | 0.540 | 0.713 | 0.749 | - | 0.442 | 0.636 | 0.635 | 0.743 | - |
| 8 | 0.537 | 0.707 | 0.746 | - | - | - | 0.633 | 0.742 | - |
| 9 | 0.532 | 0.698 | - | - | - | - | 0.629 | 0.741 | - |
| 10 | 0.527 | 0.682 | - | - | - | - | 0.624 | 0.740 | - |
| 11 | 0.521 | 0.650 | - | - | - | - | 0.615 | 0.741 | - |
| 12 | 0.516 | - | - | - | - | - | 0.600 | - | - |
| 13 | 0.509 | - | - | - | - | - | 0.565 | - | - |
| 14 | 0.436 | - | - | - | - | - | - | - | - |
| Property | Mw [Da] a | CLog P b | RBN c | QEDw d | N + O | CLog P − (N + O) | NH + OH | PSA [Å2] e | HBD f | HBA g | Others |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Tractability | 200–800 i | ≤8 i | ≤16 | >0.2 | ≤16 | >−8 | ≤8 | ≤140 | ≤8 | ≤15 | No metal, sugar, carbohydrates |
| Drug-likeness | 100–500 i | ≤5 i | ≤10 | >0.5 | ≤10 | >−5 | ≤5 | ≤105 | ≤5 | ≤10 | No metal, sugar, carbohydrates |
| Stringent drug-likeness | 100–450 i | ≤4 i | ≤10 | >0.5 | ≤8–9 | >−4.5 | ≤3 | ≤105 | ≤3 | ≤8–9 | No metal, sugar, carbohydrates |
| CNS-active h | 100–400 | ≤3.5 i,j | ≤7 | >0.7 | ≤5 | >0 | ≤4 | ≤70j | ≤4 | ≤5 | No metal, sugar, carbohydrates |
2.2. Discussion
3. Experimental Section
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Williams, R.J.; Spencer, J.P.; Rice-Evans, C. Flavonoids: Antioxidants or signalling molecules? Free Radic. Biol. Med. 2014, 36, 838–849. [Google Scholar] [CrossRef] [PubMed]
- Stahl, W.; Ale-Agha, N.; Polidori, M.C. Non-antioxidant properties of carotenoids. Biol. Chem. 2002, 383, 553–558. [Google Scholar] [CrossRef] [PubMed]
- Jäger, A.K.; Saaby, L. Flavonoids and the CNS. Molecules 2011, 16, 1471–1485. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.G.; Liu, B.G.; Liang, G.Z.; Ning, Z.X. Structure-activity relationship of flavonoids active against lard oil oxidation based on quantum chemical analysis. Molecules 2009, 14, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Harsa, A.M.; Harsa, T.E.; Bolboacâ, S.D.; Diudea, M.V. QSAR in flavonoids by similarity cluster prediction. Curr. Comput. Aided Drug Des. 2014, 10, 115–128. [Google Scholar] [CrossRef] [PubMed]
- Goto, H.; Terao, Y.; Akai, S. Synthesis of various kinds of isoflavones, isoflavanes, and biphenyl-ketones and their 1,1-diphenyl-2-picrylhydrazyl radical-scavenging activities. Chem. Pharm. Bull. 2009, 57, 346–360. [Google Scholar] [CrossRef] [PubMed]
- Mitra, I.; Saha, A.; Roy, K. Chemometric modeling of free radical scavenging activity of flavone derivatives. Eur. J. Med. Chem. 2010, 45, 5071–5079. [Google Scholar] [CrossRef] [PubMed]
- Umehara, K.; Nemoto, K.; Kimijima, K.; Matsushita, A.; Terada, E.; Monthakantirat, O.; De-Eknamkul, W.; Miyase, T.; Warashina, T.; Degawa, M.; et al. Estrogenic constituents of the heartwood of Dalbergia parviflora. Phytochemistry 2008, 69, 546–552. [Google Scholar] [CrossRef] [PubMed]
- Umehara, K.; Nemoto, K.; Matsushita, A.; Terada, E.; Monthakantirat, O.; de-Eknamkul, W.; Miyase, T.; Warashina, T.; Degawa, M.; Noguchi, H. Flavonoids from the heartwood of the Thai medicinal plant Dalbergia parviflora and their effects on estrogenic-responsive human breast cancer cells. J. Nat. Prod. 2009, 72, 2163–2168. [Google Scholar] [CrossRef] [PubMed]
- Promden, W.; Monthakantirat, O.; Umehara, K.; Noguchi, H.; de-Eknamkul, W. Structure and antioxidant activity relationships of isoflavonoids from Dalbergia parviflora. Molecules 2014, 19, 2226–2237. [Google Scholar] [CrossRef] [PubMed]
- McCord, J.M.; Fridovich, I. The reduction of cytochrome c by milk xanthine oxidase. J. Biol. Chem. 1968, 243, 5753–5760. [Google Scholar] [PubMed]
- Prior, R.L.; Hoang, H.; Gu, L.; Wu, X.; Bacchiocca, M.; Howard, L.; Hampsch-Woodill, M.; Huang, D.; Ou, B.; Jacob, R. Assays for hydrophilic and lipophilic antioxidant capacity (oxygen radical absorbance capacity (ORAC(FL))) of plasma and other biological and food samples. J. Agric. Food Chem. 2003, 51, 3273–3329. [Google Scholar] [CrossRef] [PubMed]
- Blois, M.S. Antioxidant determinations by the use of a stable free radical. Nature 1958, 181, 1199–1200. [Google Scholar] [CrossRef]
- Walker, J.D.; Jaworska, J.; Comber, J.H.I.; Schultz, T.W.; Dearden, J.C. Guidelines for developing and using quantitative structure-activity relationships. Environ. Toxicol. Chem. 2003, 22, 1653–1665. [Google Scholar] [CrossRef] [PubMed]
- Perkins, R.; Fang, H.; Tong, W.; Welsh, W.J. Quantitative structure-activity relationship methods: Perspectives on drug discovery and toxicology. Environ. Toxicol. Chem. 2003, 22, 1666–1679. [Google Scholar] [CrossRef] [PubMed]
- Benigni, R.; Giuliani, A. Putting the predictive toxicology challenge into perspective: Reflections on the results. Bioinformatics 2013, 19, 1194–1200. [Google Scholar] [CrossRef]
- Cherkasov, A.; Muratov, E.N.; Fourches, D.; Varnek, A.; Baskin, I.I.; Cronin, M.; Dearden, J.; Gramatica, P.; Martin, Y.C.; Todeschini, R.; et al. QSAR modeling: Where have you been? Where are you going to? J. Med. Chem. 2014, 57, 4977–5010. [Google Scholar] [CrossRef] [PubMed]
- Torrens, F.; Castellano, G. QSPR prediction of retention times of phenylurea herbicides by biological plastic evolution. Curr. Drug Saf. 2012, 7, 262–268. [Google Scholar] [CrossRef] [PubMed]
- Torrens, F.; Castellano, G. QSPR prediction of chromatographic retention times of pesticides: Partition and fractal indices. J. Environ. Sci. Health Part B 2014, 49, 400–407. [Google Scholar] [CrossRef] [PubMed]
- Wright, J.S.; Johnson, E.R.; DiLabio, G.A. Predicting the activity of phenolic antioxidants: Theoretical methods, analysis of substituent effects, and application to major families of antioxidants. J. Am. Chem. Soc. 2001, 123, 1173–1183. [Google Scholar] [CrossRef] [PubMed]
- Fujita, T.; Ban, T. Structure–activity study of phenethylamines as substrates of biosynthetic enzymes of sympathetic transmitters. J. Med. Chem. 1971, 14, 148–152. [Google Scholar] [CrossRef] [PubMed]
- Gaulton, A.; Bellis, L.J.; Bento, A.P.; Chambers, J.; Davies, M.; Hersey, A.; Light, Y.; McGlinchey, S.; Michalovich, D.; Al-Lazikani, B.; et al. ChEMBL: A large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012, 40, D1100–D1107. [Google Scholar] [CrossRef] [PubMed]
- López, A.; Faus, V.; Díez-Sales, O.; Herráez, M. Skin permeation model of phenyl alcohols: Comparison of experimental conditions. Int. J. Pharm. 1998, 173, 183–191. [Google Scholar] [CrossRef]
- Cramer, R.D., III. BC(DEF) parameters. 1. The intrinsic dimensionality of intermolecular interactions in the liquid state. J. Am. Chem. Soc. 1980, 102, 1837–1849. [Google Scholar] [CrossRef]
- Leeson, P.D.; Davis, A.M. Time-related differences in the physical property profiles of oral drugs. J. Med. Chem. 2004, 47, 6338–6348. [Google Scholar] [CrossRef] [PubMed]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. Prediction of hydrophobic (lipophilic) properties of small organic molecules using fragmental methods: An analysis of ALog P and CLog P methods. J. Phys. Chem. A 1998, 102, 3762–3772. [Google Scholar] [CrossRef]
- Leo, A.; Jow, P.Y.C.; Silipo, C.; Hansch, C. Calculation of hydrophobic constant (log P) from π and f constants. J. Med. Chem. 1975, 18, 865–868. [Google Scholar] [CrossRef] [PubMed]
- Fichert, T.; Yazdanian, M.; Proudfoot, J.R. A structure-permeability study of small drug-like molecules. Bioorg. Med. Chem. Lett. 2003, 13, 719–722. [Google Scholar] [CrossRef]
- Valentin, J.P.; Hammond, T. Safety and secondary pharmacology: Successes, threats, challenges and opportunities. J. Pharmacol. Toxicol. Methods 2008, 58, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Campbell, N.A.; Williamson, B.; Heyden, R.J. Biology: Exploring Life; Pearson Prentice Hall: Boston, MA, USA, 2006. [Google Scholar]
- Kamlet, M.J.; Abboud, J.L.M.; Abraham, M.H.; Taft, R.W. Linear solvation energy relationships. 23. A comprehensive collection of the solvatochromic parameters, π*, α, and β, and some methods for simplifying the generalized solvatochromic equation. J. Org. Chem. 1983, 48, 2877–2887. [Google Scholar] [CrossRef]
- Ertl, P.; Rohde, B.; Selzer, P. Fast calculation of molecular polar surface area as a sum of fragment-based contributions and its application to the prediction of drug transport properties. J. Med. Chem. 2000, 43, 3714–3717. [Google Scholar] [CrossRef] [PubMed]
- Prasanna, S.; Doerksen, R.J. Topological polar surface area: A useful descriptor in 2D-QSAR. Curr. Med. Chem. 2009, 16, 21–41. [Google Scholar] [CrossRef] [PubMed]
- Hughes, J.D.; Blagg, J.; Price, D.A.; Bailey, S.; DeCrescenzo, G.A.; Devraj, R.V.; Ellsworth, E.; Fobian, Y.M.; Gibbs, M.E.; Gilles, R.W.; et al. Physicochemical drug properties associated with in vivo toxicological outcomes. Bioorg. Med. Chem. Lett. 2008, 18, 4872–4875. [Google Scholar] [CrossRef] [PubMed]
- Gleeson, M.P. Generation of a set of simple, interpretable ADMET rules of thumb. J. Med. Chem. 2008, 51, 817–834. [Google Scholar] [CrossRef] [PubMed]
- Bickerton, G.R.; Paolini, G.V.; Besnard, J.; Muresan, S.; Hopkins, A.L. Quantifying the chemical beauty of drugs. Nat. Chem. 2012, 4, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Segall, M. Advances in multiparameter optimization methods for de novo drug design. Expert Opin. Drug Discov. 2014, 9, 803–817. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2012, 64, 4–17. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Norinder, U.; Haeberlein, M. Computational approaches to the prediction of the blood-brain distribution. Adv. Drug Deliv. Rev. 2002, 54, 291–313. [Google Scholar] [CrossRef]
- Wenlock, M.C.; Austin, R.P.; Barton, P.; Davis, A.M.; Leeson, P.D. A comparison of physiochemical property profiles of development and marketed oral drugs. J. Med. Chem. 2003, 46, 1250–1256. [Google Scholar] [CrossRef] [PubMed]
- Besalú, E. Fast computation of cross-validated properties in full linear leave-many-out procedures. J. Math. Chem. 2001, 29, 191–203. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castellano, G.; Torrens, F. Quantitative Structure-Antioxidant Activity Models of Isoflavonoids: A Theoretical Study. Int. J. Mol. Sci. 2015, 16, 12891-12906. https://doi.org/10.3390/ijms160612891
Castellano G, Torrens F. Quantitative Structure-Antioxidant Activity Models of Isoflavonoids: A Theoretical Study. International Journal of Molecular Sciences. 2015; 16(6):12891-12906. https://doi.org/10.3390/ijms160612891
Chicago/Turabian StyleCastellano, Gloria, and Francisco Torrens. 2015. "Quantitative Structure-Antioxidant Activity Models of Isoflavonoids: A Theoretical Study" International Journal of Molecular Sciences 16, no. 6: 12891-12906. https://doi.org/10.3390/ijms160612891
APA StyleCastellano, G., & Torrens, F. (2015). Quantitative Structure-Antioxidant Activity Models of Isoflavonoids: A Theoretical Study. International Journal of Molecular Sciences, 16(6), 12891-12906. https://doi.org/10.3390/ijms160612891