New Insights into the Role of Inflammation in the Pathogenesis of Atherosclerosis




Abstract
: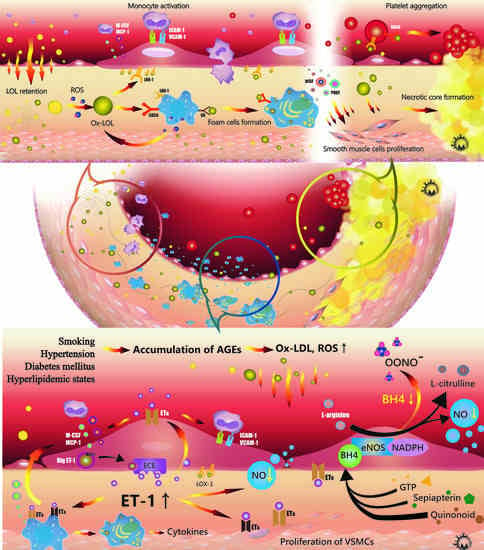
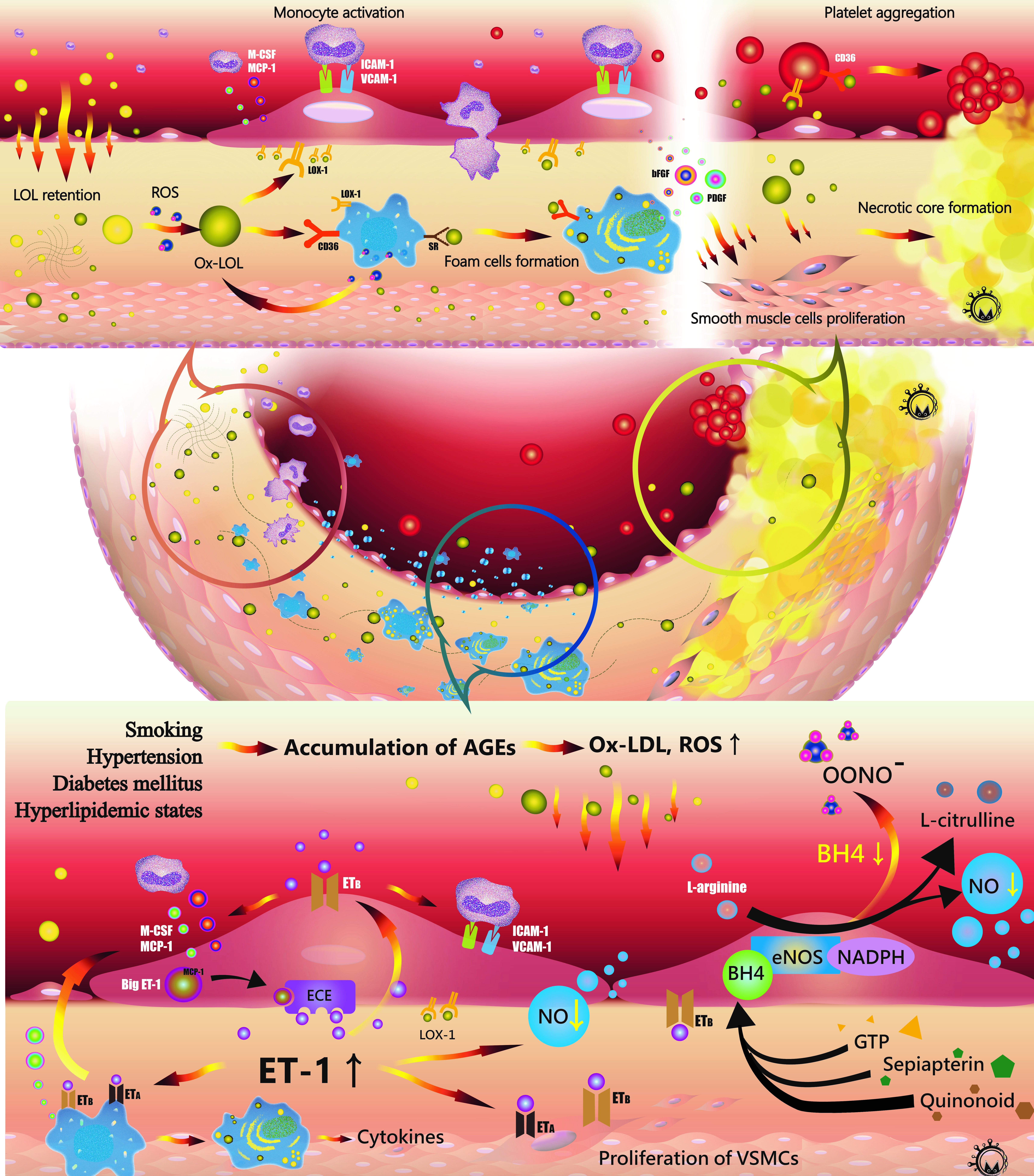{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Lipid Metabolism and Low-Density Lipoprotein (LDL) Modification
2.1. Roles of Lipid Metabolism in Atherosclerosis
2.2. LDL Modification and Oxidization in Atherosclerosis
3. Endothelial Function and Microenvironment
3.1. Roles of NO in Atherosclerosis
3.2. Role of ET-1 in Atherosclerosis
4. Roles of Immune-Mediator Regulation
4.1. Macrophages
4.2. Dendritic Cells
4.3. T Cells
4.4. Other Cells in Atherosclerosis
5. Plaque Rupture and Platelet Activation
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| AGEs | Advanced glycation end products |
| ALK5 | Transforming growth factor, beta receptor I |
| APCs | Antigen-presenting cells |
| Apo | Apolipoprotein |
| BCR | B cell receptor |
| bFGF | Basic fibroblast growth factor |
| BH4 | Tetrahydrobiopterin |
| CD36 | Cluster of differentiation 36 |
| DCs | Dendritic cells |
| ECEs | ET converting enzymes |
| ET-1 | Endothelin-1 |
| eNOS | Endothelial isoform of NOS |
| GTP | Guanosine 5′-triphosphate |
| GTPCH I | Guanosine 5′-triphosphate cyclohydrolase I |
| HSP | Heat shock proteins |
| ICAM-1 | Intercellular adhesion molecule-1 |
| JNK | c-Jun N-terminal kinase |
| KLF2 | Kruppel-like factor 2 |
| LDL | Low-density lipoprotein |
| LFA-1 | Lymphocyte function-associated antigen 1 |
| LOX-1 | Lectin-like oxidized LDL receptor-1 |
| LPL | Lipoprotein lipase |
| LSS | Laminar shear stress |
| Ox-LDL | Oxidized LDL |
| MAPK | Mitogen-activated protein kinase |
| MCP-1 | Monocyte chemoattractant protein-1 |
| M-CSF | Macrophage colony stimulating factors |
| MHC-II | Major histocompatibility complex class II |
| MMPs | Matrix metalloproteinases |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| NK T cells | Natural killer T cells |
| NO | Nitric oxide |
| NOS | NO synthase |
| PDGF | Platelet-derived growth factor |
| PGE2 | Prostaglandin E2 |
| PGI2 | Prostaglandin I2 |
| PRRs | Pattern recognition receptors |
| PSGL-1 | P-selectin glycoprotein 1 |
| ROS | Reactive oxygen species |
| SMAD3 | Mothers against decapentaplegic homolog 3 |
| SMC | Smooth muscle cell |
| SR | Scavenger receptor |
| Th1 | Type 1 T helper response |
| TG | Triacylglycerol |
| TLR | Toll-like receptor |
| TxA2 | Thromboxane A2 |
| VCAM-1 | Vascular-cell adhesion molecule 1 |
| VEGF | Vascular endothelial growth factor |
| VLA-4 | Very late antigen-4 |
| VLDL | Very low-density lipoproteins |
| VWF | Von Willebrand factor |
References
- Falk, E. Pathogenesis of Atherosclerosis. J. Am. Coll. Cardiol. 2006, 47, C7–C12. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.R.; Mengi, S.A.; Xu, Y.J.; Arneja, A.S.; Dhalla, N.S. Pathogenesis of Atherosclerosis: A Multifactorial Process. Exp. Clin. Cardiol. 2002, 7, 40–53. [Google Scholar]
- Albertini, R.; Moratti, R.; de Luca, G. Oxidation of Low-Density Lipoprotein in Atherosclerosis from Basic Biochemistry to Clinical Studies. Curr. Mol. Med. 2002, 2, 579–592. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, P.; Leray, V.; Diez, M.; Serisier, S.; le Bloc’h, J.; Siliart, B.; Dumon, H. Liver Lipid Metabolism. J. Anim. Physiol. Anim. Nutr. 2008, 92, 272–283. [Google Scholar] [CrossRef] [PubMed]
- Khalil, F.M.; Wagner, W.D.; Goldberg, I.J. Molecular Interactions Leading to Lipoprotein Retention and the Initiation of Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 2211–2218. [Google Scholar] [CrossRef] [PubMed]
- Kramer-Guth, A.; Greiber, S.; Pavenstadt, H.; Quaschning, T.; Winkler, K.; Schollmeyer, P.; Wanner, C. Interaction of Native and Oxidized Lipoprotein(a) with Human Mesangial Cells and Matrix. Kidney Int. 1996, 49, 1250–1261. [Google Scholar] [CrossRef] [PubMed]
- Williams, K.J. Arterial Wall Chondroitin Sulfate Proteoglycans: Diverse Molecules with Distinct Roles in Lipoprotein Retention and Atherogenesis. Curr. Opin. Lipidol. 2001, 12, 477–487. [Google Scholar] [CrossRef] [PubMed]
- Wiśniewska, A.; Olszanecki, R.; Totoń-Żurańska, J.; Kuś, K.; Stachowicz, A.; Suski, M.; Gębska, A.; Gajda, M.; Jawień, J.; Korbut, R. Anti-Atherosclerotic Action of Agmatine in ApoE-Knockout Mice. Int. J. Mol. Sci. 2017, 18, 1706. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.S.; Harkewicz, R.; Lee, J.H.; Boullier, A.; Almazan, F.; Li, A.C.; Witztum, J.L.; Bae, Y.S.; Miller, Y.I. Lipoprotein Accumulation in Macrophages Via Toll-Like Receptor-4-Dependent Fluid Phase Uptake. Circ. Res. 2009, 104, 1355–1363. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.M.; Chadipiralla, K.; Mendez, A.J.; Jaimes, E.A.; Silverstein, R.L.; Webster, K.; Raij, L. Nicotine Potentiates Proatherogenic Effects of OxLDL by Stimulating and Upregulating Macrophage CD36 Signaling. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H563–H574. [Google Scholar] [CrossRef] [PubMed]
- Bloomer, R.J. Decreased Blood Antioxidant Capacity and Increased Lipid Peroxidation in Young Cigarette Smokers Compared to Nonsmokers: Impact of Dietary Intake. Nutr. J. 2007, 6, 39. [Google Scholar] [CrossRef] [PubMed]
- Frostegård, J.; Ruihua, W.U.; Lemne, C.; Thulin, T.; Witztum, J.L.; de Faire, U. Circulating Oxidized Low-Density Lipoprotein Is Increased in Hypertension. Clin. Sci. 2003, 105, 615–620. [Google Scholar] [CrossRef] [PubMed]
- Levitan, I.; Volkov, S.; Subbaiah, P.V. Oxidized Ldl: Diversity, Patterns of Recognition, and Pathophysiology. Antioxid. Redox. Signal. 2010, 13, 39–75. [Google Scholar] [CrossRef] [PubMed]
- Parthasarathy, S.; Raghavamenon, A.; Garelnabi, M.O.; Santanam, N. Oxidized Low-Density Lipoprotein. Methods Mol. Biol. 2010, 610, 403–417. [Google Scholar] [PubMed]
- Badrnya, S.; Assinger, A.; Volf, I. Native High Density Lipoproteins (HDL) Interfere with Platelet Activation Induced by Oxidized Low Density Lipoproteins (OxLDL). Int. J. Mol. Sci. 2013, 14, 10107. [Google Scholar] [CrossRef] [PubMed]
- Cominacini, L.; Garbin, U.; Pasini, A.F.; Davoli, A.; Campagnola, M.; Contessi, G.B.; Pastorino, A.M.; Cascio, V.L. Antioxidants Inhibit the Expression of Intercellular Cell Adhesion Molecule-1 and Vascular Cell Adhesion Molecule-1 Induced by Oxidized LDL on Human Umbilical Vein Endothelial Cells. Free Radic. Biol. Med. 1997, 22, 117–127. [Google Scholar] [CrossRef]
- Frostegard, J.; Haegerstrand, A.; Gidlund, M.; Nilsson, J. Biologically Modified LDL Increases the Adhesive Properties of Endothelial Cells. Atherosclerosis 1991, 90, 119–126. [Google Scholar] [CrossRef]
- Quinn, M.T.; Parthasarathy, S.; Steinberg, D. Lysophosphatidylcholine: A Chemotactic Factor for Human Monocytes and Its Potential Role in Atherogenesis. Proc. Natl. Acad. Sci. USA 1988, 85, 2805–2809. [Google Scholar] [CrossRef] [PubMed]
- Barbieri, S.S.; Cavalca, V.; Eligini, S.; Brambilla, M.; Caiani, A.; Tremoli, E.; Colli, S. Apocynin Prevents Cyclooxygenase 2 Expression in Human Monocytes through Nadph Oxidase and Glutathione Redox-Dependent Mechanisms. Free Radic. Biol. Med. 2004, 37, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Carr, A.C.; McCall, M.R.; Frei, B. Oxidation of LDL by Myeloperoxidase and Reactive Nitrogen Species: Reaction Pathways and Antioxidant Protection. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1716–1723. [Google Scholar] [CrossRef] [PubMed]
- Cyrus, T.; Witztum, J.L.; Rader, D.J.; Tangirala, R.; Fazio, S.; Linton, M.F.; Funk, C.D. Disruption of the 12/15-Lipoxygenase Gene Diminishes Atherosclerosis in Apo E-Deficient Mice. J. Clin. Investig. 1999, 103, 1597–1604. [Google Scholar] [CrossRef] [PubMed]
- Nagy, L.; Tontonoz, P.; Alvarez, J.G.; Chen, H.; Evans, R.M. Oxidized LDL Regulates Macrophage Gene Expression through Ligand Activation of PPARγ. Cell 1998, 93, 229–240. [Google Scholar] [CrossRef]
- Park, Y.M. CD36 Modulates Migration of Mouse and Human Macrophages in Response to Oxidized LDL and May Contribute to Macrophage Trapping in the Arterial Intima. J. Clin. Investig. 2009, 119, 136–145. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.M.; Drazba, J.A.; Vasanji, A.; Egelhoff, T.; Febbraio, M.; Silverstein, R.L. Oxidized LDL/CD36 Interaction Induces Loss of Cell Polarity and Inhibits Macrophage Locomotion. Mol. Biol. Cell 2012, 23, 3057–3068. [Google Scholar] [CrossRef] [PubMed]
- Hansson, G.K.; Robertson, A.K.; Soderberg-Naucler, C. Inflammation and Atherosclerosis. Annu. Rev. Pathol. 2006, 1, 297–329. [Google Scholar] [CrossRef] [PubMed]
- Liao, F.; Andalibi, A.; deBeer, F.C.; Fogelman, A.M.; Lusis, A.J. Genetic Control of Inflammatory Gene Induction and NF-κB-Like Transcription Factor Activation in Response to an Atherogenic Diet in Mice. J. Clin. Investig. 1993, 91, 2572–2579. [Google Scholar] [CrossRef] [PubMed]
- Kohno, M.; Yokokawa, K.; Yasunari, K.; Minami, M.; Kano, H.; Hanehira, T.; Yoshikawa, J. Induction by Lysophosphatidylcholine, a Major Phospholipid Component of Atherogenic Lipoproteins, of Human Coronary Artery Smooth Muscle Cell Migration. Circulation 1998, 98, 353–359. [Google Scholar] [CrossRef] [PubMed]
- Lindner, V.; Lappi, D.A.; Baird, A.; Majack, R.A.; Reidy, M.A. Role of Basic Fibroblast Growth Factor in Vascular Lesion Formation. Circ. Res. 1991, 68, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Stiko-Rahm, A.; Hultgardh-Nilsson, A.; Regnstrom, J.; Hamsten, A.; Nilsson, J. Native and Oxidized LDL Enhances Production of PDGF AA and the Surface Expression of PDGF Receptors in Cultured Human Smooth Muscle Cells. Arterioscler. Thromb. 1992, 12, 1099–1109. [Google Scholar] [CrossRef] [PubMed]
- Maiolino, G.; Rossitto, G.; Caielli, P.; Bisogni, V.; Rossi, G.P.; Cala, L.A. The Role of Oxidized Low-Density Lipoproteins in Atherosclerosis: The Myths and the Facts. Mediat. Inflamm. 2013, 2013, 13. [Google Scholar] [CrossRef] [PubMed]
- Loidl, A.; Claus, R.; Ingolic, E.; Deigner, H.P.; Hermetter, A. Role of Ceramide in Activation of Stress-Associated MAP Kinases by Minimally Modified LDL in Vascular Smooth Muscle Cells. Biochim. Biophys. Acta 2004, 1690, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Rajavashisth, T.B.; Liao, J.K.; Galis, Z.S.; Tripathi, S.; Laufs, U.; Tripathi, J.; Chai, N.N.; Xu, X.P.; Jovinge, S.; Shah, P.K.; et al. Inflammatory Cytokines and Oxidized Low Density Lipoproteins Increase Endothelial Cell Expression of Membrane Type 1-Matrix Metalloproteinase. J. Biol. Chem. 1999, 274, 11924–11929. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.P.; Meisel, S.R.; Ong, J.M.; Kaul, S.; Cercek, B.; Rajavashisth, T.B.; Sharifi, B.; Shah, P.K. Oxidized Low-Density Lipoprotein Regulates Matrix Metalloproteinase-9 and Its Tissue Inhibitor in Human Monocyte-Derived Macrophages. Circulation 1999, 99, 993–998. [Google Scholar] [CrossRef] [PubMed]
- Podrez, E.A.; Byzova, T.V.; Febbraio, M.; Salomon, R.G.; Ma, Y.; Valiyaveettil, M.; Poliakov, E.; Sun, M.; Finton, P.J.; Curtis, B.R.; et al. Platelet CD36 Links Hyperlipidemia, Oxidant Stress and a Prothrombotic Phenotype. Nat. Med. 2007, 13, 1086–1095. [Google Scholar] [CrossRef] [PubMed]
- Wraith, K.S.; Magwenzi, S.; Aburima, A.; Wen, Y.; Leake, D.; Naseem, M.K. Oxidized Low-Density Lipoproteins Induce Rapid Platelet Activation and Shape Change through Tyrosine Kinase and Rho Kinase-Signaling Pathways. Blood 2013, 122, 580–589. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Kakutani, M.; Naruko, T.; Ueda, M.; Narumiya, S.; Masaki, T.; Sawamura, T. Activation-Dependent Surface Expression of LOX-1 in Human Platelets. Biochem. Biophys. Res. Commun. 2001, 282, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Pirillo, A.; Norata, G.D.; Catapano, A.L. LOX-1, OxLDL, and Atherosclerosis. Mediat. Inflamm. 2013. [Google Scholar] [CrossRef] [PubMed]
- Cominacini, L.; Pasini, A.F.; Garbin, U.; Pastorino, A.; Rigoni, A.; Nava, C.; Davoli, A.; Cascio, V.L.; Sawamura, T. The Platelet-Endothelium Interaction Mediated by Lectin-Like Oxidized Low-Density Lipoprotein Receptor-1 Reduces the Intracellular Concentration of Nitric Oxide in Endothelial Cells. J. Am. Coll. Cardiol. 2003, 41, 499–507. [Google Scholar] [CrossRef]
- Kakutani, M.; Masaki, T.; Sawamura, T. A Platelet–Endothelium Interaction Mediated by Lectin-Like Oxidized Low-Density Lipoprotein Receptor-1. Proc. Natl. Acad. Sci. USA 2000, 97, 360–364. [Google Scholar] [CrossRef] [PubMed]
- Li, L.X.; Chen, J.X.; Liao, D.F.; Yu, L. Probucol Inhibits Oxidized-Low Density Lipoprotein-Induced Adhesion of Monocytes to Endothelial Cells by Reducing P-Selectin Synthesis in Vitro. Endothelium 1998, 6, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Thorin, E.; Hamilton, C.A.; Dominiczak, M.H.; Reid, J.L. Chronic Exposure of Cultured Bovine Endothelial Cells to Oxidized LDL Abolishes Prostacyclin Release. Arterioscler. Thromb. 1994, 14, 453–459. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Tsai, S.F.; Kuo, Y.M. The Therapeutic Potential of Anti-Inflammatory Exerkines in the Treatment of Atherosclerosis. Int. J. Mol. Sci. 2017, 18, 1260. [Google Scholar] [CrossRef] [PubMed]
- Uribarri, J.; Woodruff, S.; Goodman, S.; Cai, W.; Chen, X.; Pyzik, R.; Yong, A.; Striker, G.E.; Vlassara, H. Advanced Glycation End Products in Foods and a Practical Guide to Their Reduction in the Diet. J. Am. Diet. Assoc. 2010, 110, 911–916. [Google Scholar] [CrossRef] [PubMed]
- Orekhov, A.N.; Bobryshev, Y.V.; Sobenin, I.A.; Melnichenko, A.A.; Chistiakov, D.A. Modified Low Density Lipoprotein and Lipoprotein-Containing Circulating Immune Complexes as Diagnostic and Prognostic Biomarkers of Atherosclerosis and Type 1 Diabetes Macrovascular Disease. Int. J. Mol. Sci. 2014, 15, 12807. [Google Scholar] [CrossRef] [PubMed]
- Steyers, C.; Miller, F. Endothelial Dysfunction in Chronic Inflammatory Diseases. Int. J. Mol. Sci. 2014, 15, 11324. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Wang, Z.; Zhang, L.; Wang, Y. Roles of Cells from the Arterial Vessel Wall in Atherosclerosis. Mediat. Inflamm. 2017. [Google Scholar] [CrossRef] [PubMed]
- Napoli, C.; de Nigris, F.; Williams-Ignarro, S.; Pignalosa, O.; Sica, V.; Ignarro, L.J. Nitric Oxide and Atherosclerosis: An Update. Nitric Oxide 2006, 15, 265–279. [Google Scholar] [CrossRef] [PubMed]
- Ignarro, L.J.; Napoli, C. Novel Features of Nitric Oxide, Endothelial Nitric Oxide Synthase, and Atherosclerosis. Curr. Atheroscler. Rep. 2005, 5, 17–23. [Google Scholar] [CrossRef]
- Moncada, S.; Higgs, A. The L-Arginine-Nitric Oxide Pathway. N. Engl. J. Med. 1993, 329, 2002–2012. [Google Scholar] [PubMed]
- Stuehr, D.J. Mammalian Nitric Oxide Synthases. Biochim. Biophys. Acta 1999, 1411, 217–230. [Google Scholar] [CrossRef]
- Moncada, S.; Palmer, R.M.; Higgs, E.A. Nitric Oxide: Physiology, Pathophysiology, and Pharmacology. Pharmacol. Rev. 1991, 43, 109–142. [Google Scholar] [PubMed]
- Nathan, C.; Xie, Q.W. Regulation of Biosynthesis of Nitric Oxide. J. Biol. Chem. 1994, 269, 13725–13728. [Google Scholar] [PubMed]
- Kawashima, S.; Mitsuhiro, Y. Dysfunction of Endothelial Nitric Oxide Synthase and Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 998–1005. [Google Scholar] [CrossRef] [PubMed]
- Laursen, J.B.; Somers, M.; Kurz, S.; McCann, L.; Warnholtz, A.; Freeman, B.A.; Tarpey, M.; Fukai, T.; Harrison, D.G. Endothelial Regulation of Vasomotion in ApoE-Deficient Mice: Implications for Interactions between Peroxynitrite and Tetrahydrobiopterin. Circulation 2001, 103, 1282–1288. [Google Scholar] [CrossRef] [PubMed]
- Lunte, C.E.; Kissinger, P.T. Determination of Quinonoid Dihydrobiopterin by Liquid Chromatography and Electrochemical Detection. J. Chromatogr. 1984, 317, 407–412. [Google Scholar] [CrossRef]
- Vann, L.R.; Payne, S.G.; Edsall, L.C.; Twitty, S.; Spiegel, S.; Milstien, S. Involvement of Sphingosine Kinase in TNF-α-Stimulated Tetrahydrobiopterin Biosynthesis in C6 Glioma Cells. J. Biol. Chem. 2002, 277, 12649–12656. [Google Scholar] [CrossRef] [PubMed]
- SenBanerjee, S.; Lin, Z.; Atkins, G.B.; Greif, D.M.; Rao, R.M.; Kumar, A.; Feinberg, M.W.; Chen, Z.; Simon, D.I.; Luscinskas, F.W.; et al. KLF2 Is a Novel Transcriptional Regulator of Endothelial Proinflammatory Activation. J. Exp. Med. 2004, 199, 1305–1315. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Unoki, H.; Iwasa, S.; Watanabe, T. Role of Endothelin-1 in Atherosclerosis. Ann. N. Y. Acad. Sci. 2000, 902, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Pernow, J.; Shemyakin, A.; Bohm, F. New Perspectives on Endothelin-1 in Atherosclerosis and Diabetes Mellitus. Life Sci. 2012, 91, 507–516. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, T.; Yanagisawa, M.; Masaki, T. Molecular Characterization of Endothelin Receptors. Trends Pharmacol. Sci. 1992, 13, 103–108. [Google Scholar] [PubMed]
- Schiffrin, E.L.; Touyz, R.M. Vascular Biology of Endothelin. J. Cardiovasc. Pharmacol. 1997, 32, S2–S13. [Google Scholar]
- Yanagisawa, M.; Masaki, T. Molecular Biology and Biochemistry of the Endothelins. Trends Pharmacol. Sci. 1989, 10, 374–378. [Google Scholar] [PubMed]
- Böhm, F.; Pernow, J. The Importance of Endothelin-1 for Vascular Dysfunction in Cardiovascular Disease. Cardiovasc. Res. 2007, 76, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Ito, H.; Hirata, Y.; Adachi, S.; Tanaka, M.; Tsujino, M.; Koike, A.; Nogami, A.; Murumo, F.; Hiroe, M. Endothelin-1 Is an Autocrine/Paracrine Factor in the Mechanism of Angiotensin II-Induced Hypertrophy in Cultured Rat Cardiomyocytes. J. Clin. Investig. 1993, 92, 398–403. [Google Scholar] [CrossRef] [PubMed]
- Iwasa, S.; Fan, J.; Shimokama, T.; Nagata, M.; Watanabe, T. Increased Immunoreactivity of Endothelin-1 and Endothelin B Receptor in Human Atherosclerotic Lesions. A Possible Role in Atherogenesis. Atherosclerosis 1999, 146, 93–100. [Google Scholar] [CrossRef]
- Ivanova, E.A.; Orekhov, A.N. The Role of Endoplasmic Reticulum Stress and Unfolded Protein Response in Atherosclerosis. Int. J. Mol. Sci. 2016, 17, 193. [Google Scholar] [CrossRef] [PubMed]
- Filep, J.G.; Sirois, M.G.; Foldes-Filep, E.; Rousseau, A.; Plante, G.E.; Fournier, A.; Yano, M.; Sirois, P. Enhancement by Endothelin-1 of Microvascular Permeability Via the Activation of ETA Receptors. Br. J. Pharmacol. 1993, 109, 880–886. [Google Scholar] [CrossRef] [PubMed]
- Swerlick, A.R.; Lawley, T.J. Role of Microvascular Endothelial Cells in Inflammation. J. Investig. Dermatol. 1993, 100, S111–S115. [Google Scholar] [CrossRef]
- Hansson, G.K. Inflammation, Atherosclerosis, and Coronary Artery Disease. N. Engl. J. Med. 2005, 352, 1685–1695. [Google Scholar] [CrossRef] [PubMed]
- Kume, N.; Cybulsky, M.I.; Gimbrone, M.A., Jr. Lysophosphatidylcholine, a Component of Atherogenic Lipoproteins, Induces Mononuclear Leukocyte Adhesion Molecules in Cultured Human and Rabbit Arterial Endothelial Cells. J. Clin. Investig. 1992, 90, 1138–1144. [Google Scholar] [CrossRef] [PubMed]
- Dai, G.; Kaazempur-Mofrad, M.R.; Natarajan, S.; Zhang, Y.; Vaughn, S.; Blackman, B.R.; Kamm, R.D.; Garcia-Cardena, G.; Gimbrone, M.A., Jr. Distinct Endothelial Phenotypes Evoked by Arterial Waveforms Derived from Atherosclerosis-Susceptible and -Resistant Regions of Human Vasculature. Proc. Natl. Acad. Sci. USA 2004, 101, 14871–14876. [Google Scholar] [CrossRef] [PubMed]
- Boring, L.; Gosling, J.; Cleary, M.; Charo, I.F. Decreased Lesion Formation in CCR2-/- Mice Reveals a Role for Chemokines in the Initiation of Atherosclerosis. Nature 1998, 394, 894–897. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Okada, Y.; Clinton, S.K.; Gerard, C.; Sukhova, G.K.; Libby, P.; Rollins, B.J. Absence of Monocyte Chemoattractant Protein-1 Reduces Atherosclerosis in Low Density Lipoprotein Receptor-Deficient Mice. Mol. Cell 1998, 2, 275–281. [Google Scholar] [CrossRef]
- Haley, K.J.; Lilly, C.M.; Yang, J.H.; Feng, Y.; Kennedy, S.P.; Turi, T.G.; Thompson, J.F.; Sukhova, G.H.; Libby, P.; Lee, R.T. Overexpression of Eotaxin and the CCR3 Receptor in Human Atherosclerosis: Using Genomic Technology to Identify a Potential Novel Pathway of Vascular Inflammation. Circulation 2000, 102, 2185–2189. [Google Scholar] [CrossRef] [PubMed]
- Mach, F.; Sauty, A.; Iarossi, A.S.; Sukhova, G.K.; Neote, K.; Libby, P.; Luster, A.D. Differential Expression of Three T Lymphocyte-Activating CXC Chemokines by Human Atheroma-Associated Cells. J. Clin. Investig. 1999, 104, 1041–1050. [Google Scholar] [CrossRef] [PubMed]
- Lesnik, P.; Haskell, C.A.; Charo, I.F. Decreased Atherosclerosis in CX3CR1-/- Mice Reveals a Role for Fractalkine in Atherogenesis. J. Clin. Investig. 2003, 111, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Hansson, G.K. Immune Mechanisms in Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 1876–1890. [Google Scholar] [CrossRef] [PubMed]
- Mestas, J.; Ley, K. Monocyte-Endothelial Cell Interactions in the Development of Atherosclerosis. Trends Cardiovasc. Med. 2008, 18, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.J.; Tabas, I. Macrophages in the Pathogenesis of Atherosclerosis. Cell 2011, 145, 341–355. [Google Scholar] [CrossRef] [PubMed]
- Rizzacasa, B.; Morini, E.; Pucci, S.; Murdocca, M.; Novelli, G.; Amati, F. LOX-1 and Its Splice Variants: A New Challenge for Atherosclerosis and Cancer-Targeted Therapies. Int. J. Mol. Sci. 2017, 18, 290. [Google Scholar] [CrossRef] [PubMed]
- Kruth, H.S. The Fate of Lipoprotein Cholesterol Entering the Arterial Wall. Curr. Opin. Lipidol. 1997, 8, 246–252. [Google Scholar] [CrossRef] [PubMed]
- Alberts-Grill, N.; Timothy, L.D.; Rezvan, A.; Jo, H. The Role of the Vascular Dendritic Cell Network in Atherosclerosis. Am. J. Phys. 2013, 305, C1–C21. [Google Scholar] [CrossRef] [PubMed]
- Hansson, G.K.; Robertson, A.K. TGF-β in Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2004, 24, e137–e138. [Google Scholar] [CrossRef] [PubMed]
- Grainger, D.J. Transforming Growth Factor β and Atherosclerosis: So Far, So Good for the Protective Cytokine Hypothesis. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 399–404. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Liu, A.C.; Gotlieb, A.I. Common Pathogenic Features of Atherosclerosis and Calcific Aortic Stenosis: Role of Transforming Growth Factor-β. Cardiovasc. Pathol. 2010, 19, 236–247. [Google Scholar] [CrossRef] [PubMed]
- Grainger, D.J. TGF-β and Atherosclerosis in Man. Cardiovasc. Res. 2007, 74, 213–222. [Google Scholar] [CrossRef] [PubMed]
- McCaffrey, T.A. TGF-βs and TGF-β Receptors in Atherosclerosis. Cytokine. Growth. Factor. Rev. 2000, 11, 103–114. [Google Scholar] [CrossRef]
- Falck-Hansen, M.; Christina, K.; Claudia, M. Toll-Like Receptors in Atherosclerosis. Int. J. Mol. Sci. 2013, 14, 14008–14023. [Google Scholar] [CrossRef] [PubMed]
- Galkina, E.; Ley, K. Immune and Inflammatory Mechanisms of Atherosclerosis. Annu. Rev. Immunol. 2009, 27, 165–197. [Google Scholar] [CrossRef] [PubMed]
- Van Vré, E.A.; Van Brussel, I.; Bosmans, J.M.; Vrints, C.J.; Bult, H. Dendritic Cells in Human Atherosclerosis: From Circulation to Atherosclerotic Plaques. Mediat. Inflamm. 2011. [Google Scholar] [CrossRef] [PubMed]
- Aliberti, J.; Schulz, O.; Pennington, D.J.; Tsujimura, H.; e Sousa, C.R.; Ozato, K.; Sher, A. Essential Role for ICSBP in the in vivo Development of Murine CD8α+ Dendritic Cells. Blood 2003, 101, 305–310. [Google Scholar] [CrossRef] [PubMed]
- Hacker, C.; Kirsch, R.D.; Ju, X.S.; Hieronymus, T.; Gust, T.C.; Kuhl, C.; Jorgas, T.; Kurz, S.M.; Rose-John, S.; Yokota, Y.; et al. Transcriptional Profiling Identifies ID2 Function in Dendritic Cell Development. Nat. Immunol. 2003, 4, 380–386. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, D.; Miller, J.; Merad, M. Dendritic Cell and Macrophage Heterogeneity in vivo. Immunity 2011, 35, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Hildner, K.; Edelson, B.T.; Purtha, W.E.; Diamond, M.; Matsushita, H.; Kohyama, M.; Calderon, B.; Schraml, B.U.; Unanue, E.R.; Diamond, M.S.; et al. BATF3 Deficiency Reveals a Critical Role for CD8α+ Dendritic Cells in Cytotoxic T Cell Immunity. Science 2008, 322, 1097–1100. [Google Scholar] [CrossRef] [PubMed]
- Schiavoni, G.; Mattei, F.; Sestili, P.; Borghi, P.; Venditti, M.; Morse, H.C.; Belardelli, F.; Gabriele, L. ICSBP Is Essential for the Development of Mouse Type I Interferon-Producing Cells and for the Generation and Activation of CD8α+ Dendritic Cells. J. Exp. Med. 2002, 196, 1415–1425. [Google Scholar] [CrossRef] [PubMed]
- Tailor, P.; Tamura, T.; Morse, H.C.; Ozato, K. The BXH2 Mutation in IRF8 Differentially Impairs Dendritic Cell Subset Development in the Mouse. Blood 2008, 111, 1942–1945. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, M.; Tabas, I. Dendritic Cells in Atherosclerosis. In Semin Immunopathol; Springer: Berlin/Heidelberg, Germany, 2014; Volume 36, pp. 93–102. [Google Scholar]
- Niessner, A.; Weyand, C.M. Dendritic Cells in Atherosclerotic Disease. Clin. Immunol. 2010, 134, 25. [Google Scholar] [CrossRef] [PubMed]
- Koltsova, E.K.; Ley, K. How Dendritic Cells Shape Atherosclerosis. Trends Immunol. 2011, 32, 540–547. [Google Scholar] [CrossRef] [PubMed]
- Zernecke, A. Dendritic Cells in Atherosclerosis: Evidence in Mice and Humans. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 763–770. [Google Scholar] [CrossRef] [PubMed]
- Tabas, I. Macrophage Death and Defective Inflammation Resolution in Atherosclerosis. Nat. Rev. Immunol. 2010, 10, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Maganto-Garcia, E.; Tarrio, M.L.; Grabie, N.; Bu, D.X.; Lichtman, A.H. Dynamic Changes in Regulatory T Cells Are Linked to Levels of Diet-Induced Hypercholesterolemia. Circulation 2011, 124, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Weber, C.; Meiler, S.; Döring, Y.; Koch, M.; Drechsler, M.; Megens, R.T.; Rowinska, Z.; Bidzhekov, K.; Fecher, C.; Ribechini, E.; et al. CCL17-Expressing Dendritic Cells Drive Atherosclerosis by Restraining Regulatory T Cell Homeostasis in Mice. J. Clin. Investig. 2011, 121, 2898–2910. [Google Scholar] [CrossRef] [PubMed]
- Jonasson, L.; Holm, J.; Skalli, O.; Bondjers, G.; Hansson, G.K. Regional Accumulations of T Cells, Macrophages, and Smooth Muscle Cells in the Human Atherosclerotic Plaque. Arteriosclerosis 1986, 6, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Kleindienst, R.; Xu, Q.; Willeit, J.; Waldenberger, F.R.; Weimann, S.; Wick, G. Immunology of Atherosclerosis. Demonstration of Heat Shock Protein 60 Expression and T Lymphocytes Bearing α/β or γ/δ Receptor in Human Atherosclerotic Lesions. Am. J. Pathol. 1993, 142, 1927–1937. [Google Scholar] [PubMed]
- Stemme, S.; Holm, J.; Hansson, G.K. T Lymphocytes in Human Atherosclerotic Plaques Are Memory Cells Expressing CD45RO and the Integrin VLA-1. Arterioscler. Thromb. 1992, 12, 206–211. [Google Scholar] [CrossRef] [PubMed]
- Van der Wal, A.C.; Das, P.K.; van de Berg, D.B.; van der Loos, C.M.; Becker, A.E. Atherosclerotic Lesions in Humans. in situ Immunophenotypic Analysis Suggesting an Immune Mediated Response. Lab. Investig. 1989, 61, 166–170. [Google Scholar] [PubMed]
- Wick, G.; Jakic, B.; Buszko, M.; Wick, M.C.; Grundtman, C. The Role of Heat Shock Proteins in Atherosclerosis. Nat. Rev. Cardiol. 2014, 11, 516–529. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Ridker, P.M.; Maseri, A. Inflammation and Atherosclerosis. Annu. Rev. Pathol. 2002, 1, 297–329. [Google Scholar]
- Ashkar, S.; Weber, G.F.; Panoutsakopoulou, V.; Sanchirico, M.E.; Jansson, M.; Zawaideh, S.; Rittling, S.R.; Denhardt, D.T.; Glimcher, M.J.; Cantor, H. Eta-1 (Osteopontin): An Early Component of Type-1 (Cell-Mediated) Immunity. Science 2000, 287, 860–864. [Google Scholar] [CrossRef] [PubMed]
- Giachelli, C.M.; Bae, N.; Almeida, M.; Denhardt, D.T.; Alpers, C.E.; Schwartz, S.M. Osteopontin Is Elevated During Neointima Formation in Rat Arteries and Is a Novel Component of Human Atherosclerotic Plaques. J. Clin. Investig. 1993, 92, 1686–1696. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, E.R.; Garvin, M.R.; Stewart, D.K.; Hinohara, T.; Simpson, J.B.; Schwartz, S.M.; Giachelli, C.M. Osteopontin Is Synthesized by Macrophage, Smooth Muscle, and Endothelial Cells in Primary and Restenotic Human Coronary Atherosclerotic Plaques. Arterioscler. Thromb. 1994, 14, 1648–1656. [Google Scholar] [CrossRef] [PubMed]
- Uyemura, K.; Demer, L.L.; Castle, S.C.; Jullien, D.; Berliner, J.A.; Gately, M.K.; Warrier, R.R.; Pham, N.; Fogelman, A.M.; Modlin, R.L. Cross-Regulatory Roles of Interleukin (IL)-12 and IL-10 in Atherosclerosis. J. Clin. Investig. 1996, 97, 2130–2138. [Google Scholar] [CrossRef] [PubMed]
- Peilot, H.; Rosengren, B.; Bondjers, G.; Hurt-Camejo, E. Interferon-γ Induces Secretory Group IIA Phospholipase A2 in Human Arterial Smooth Muscle Cells. Involvement of Cell Differentiation, STAT-3 Activation, and Modulation by Other Cytokines. J. Biol. Chem. 2000, 275, 22895–22904. [Google Scholar] [CrossRef] [PubMed]
- Friesel, R.; Komoriya, A.; Maciag, T. Inhibition of Endothelial Cell Proliferation by γ-Interferon. J. Cell Biol. 1987, 104, 689–696. [Google Scholar] [CrossRef] [PubMed]
- Hansson, G.K.; Hellstrand, M.; Rymo, L.; Rubbia, L.; Gabbiani, G. Interferon γ Inhibits both Proliferation and Expression of Differentiation-Specific α-Smooth Muscle Actin in Arterial Smooth Muscle Cells. J. Exp. Med. 1989, 170, 1595–1608. [Google Scholar] [CrossRef] [PubMed]
- Hansson, G.K.; Holm, J. Interferon-Γ Inhibits Arterial Stenosis after Injury. Circulation 1991, 84, 1266–1272. [Google Scholar] [CrossRef] [PubMed]
- Szabo, S.J.; Sullivan, B.M.; Peng, S.L.; Glimcher, L.H. Molecular Mechanisms Regulating Th1 Immune Responses. Annu. Rev. Immunol. 2003, 21, 713–758. [Google Scholar] [CrossRef] [PubMed]
- Gewurz, H.; Zhang, X.H.; Lint, T.F. Structure and Function of the Pentraxins. Curr. Opin. Immunol. 1995, 7, 54–64. [Google Scholar] [CrossRef]
- Ikeda, U.; Ikeda, M.; Seino, Y.; Takahashi, M.; Kano, S.; Shimada, K. Interleukin 6 Gene Transcripts Are Expressed in Atherosclerotic Lesions of Genetically Hyperlipidemic Rabbits. Atherosclerosis 1992, 92, 213–218. [Google Scholar] [CrossRef]
- Loppnow, H.; Libby, P. Proliferating or Interleukin 1-Activated Human Vascular Smooth Muscle Cells Secrete Copious Interleukin 6. J. Clin. Investig. 1990, 85, 731–738. [Google Scholar] [CrossRef] [PubMed]
- Mallat, Z.; Heymes, C.; Ohan, J.; Faggin, E.; Leseche, G.; Tedgui, A. Expression of Interleukin-10 in Advanced Human Atherosclerotic Plaques: Relation to Inducible Nitric Oxide Synthase Expression and Cell Death. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 611–616. [Google Scholar] [CrossRef] [PubMed]
- Binder, C.J.; Hartvigsen, K.; Chang, M.K.; Miller, M.; Broide, D.; Palinski, W.; Curtiss, L.K.; Corr, M.; Witztum, J.L. IL-5 Links Adaptive and Natural Immunity Specific for Epitopes of Oxidized LDL and Protects from Atherosclerosis. J. Clin. Investig. 2004, 114, 427–437. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, K.; Shichiri, M.; Libby, P.; Lee, R.T.; Mitchell, R.N. Th2-Predominant Inflammation and Blockade of IFN-γ Signaling Induce Aneurysms in Allografted Aortas. J. Clin. Investig. 2004, 114, 300–308. [Google Scholar] [CrossRef] [PubMed]
- Geng, Y.J.; Henderson, L.E.; Levesque, E.B.; Muszynski, M.; Libby, P. Fas Is Expressed in Human Atherosclerotic Intima and Promotes Apoptosis of Cytokine-Primed Human Vascular Smooth Muscle Cells. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 2200–2208. [Google Scholar] [CrossRef] [PubMed]
- Major, A.S.; Wilson, M.T.; McCaleb, J.L.; Su, Y.R.; Stanic, A.K.; Joyce, S.; van Kaer, L.; Fazio, S.; Linton, M.F. Quantitative and Qualitative Differences in Proatherogenic NKT Cells in Apolipoprotein E-Deficient Mice. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 2351–2357. [Google Scholar] [CrossRef] [PubMed]
- Melian, A.; Geng, Y.J.; Sukhova, G.K.; Libby, P.; Porcelli, S.A. Cd1 Expression in Human Atherosclerosis. A Potential Mechanism for T Cell Activation by Foam Cells. Am. J. Pathol. 1999, 155, 775–786. [Google Scholar] [PubMed]
- Paulsson, G.; Zhou, X.; Tornquist, E.; Hansson, G.K. Oligoclonal T Cell Expansions in Atherosclerotic Lesions of Apolipoprotein E-Deficient Mice. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Tupin, E.; Nicoletti, A.; Elhage, R.; Rudling, M.; Ljunggren, H.G.; Hansson, G.K.; Berne, G.P. CD1D-Dependent Activation of NKT Cells Aggravates Atherosclerosis. J. Exp. Med. 2004, 199, 417–422. [Google Scholar] [CrossRef] [PubMed]
- Bot, I.; de Jager, S.C.; Zernecke, A.; Lindstedt, K.A.; van Berkel, T.J.; Weber, C.; Biessen, E.A. Perivascular Mast Cells Promote Atherogenesis and Induce Plaque Destabilization in Apolipoprotein E–Deficient Mice. Circulation 2007, 115, 2516–2525. [Google Scholar] [CrossRef] [PubMed]
- Lindstedt, K.A.; Mäyränpää, M.I.; Kovanen, P.T. Mast Cells in Vulnerable Atherosclerotic Plaques—A View to a Kill. J. Cell Mol. Med. 2007, 11, 739–758. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Calabresi, L.; Chiesa, G.; Franceschini, G.; Kovanen, P.T. Mast Cell Chymase Degrades ApoE and ApoA-II in ApoA-I–Knockout Mouse Plasma and Reduces Its Ability to Promote Cellular Cholesterol Efflux. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 1475–1481. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Sukhova, G.K.; Wolters, P.J.; Yang, M.; Kitamoto, S.; Libby, P.; MacFarlane, L.A.; Clair, J.M.; Shi, G.P. Mast Cells Promote Atherosclerosis by Releasing Proinflammatory Cytokines. Nat. Med. 2007, 13, 719–724. [Google Scholar] [CrossRef] [PubMed]
- Binder, C.J.; Shaw, P.X.; Chang, M.K.; Boullier, A.; Hartvigsen, K.; Horkko, S.; Miller, Y.I.; Woelkers, D.A.; Corr, M.; Witztum, J.L. The Role of Natural Antibodies in Atherogenesis. J. Lipid Res. 2005, 46, 1353–1363. [Google Scholar] [CrossRef] [PubMed]
- Caligiuri, G.; Nicoletti, A.; Poirier, B.; Hansson, G.K. Protective Immunity against Atherosclerosis Carried by B Cells of Hypercholesterolemic Mice. J. Clin. Investig. 2002, 109, 745–753. [Google Scholar] [CrossRef] [PubMed]
- Major, A.S.; Fazio, S.; Linton, M.F. B-Lymphocyte Deficiency Increases Atherosclerosis in LDL Receptor-Null Mice. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 1892–1898. [Google Scholar] [CrossRef] [PubMed]
- Shaw, P.X.; Horkko, S.; Chang, M.K.; Curtiss, L.K.; Palinski, W.; Silverman, G.J.; Witztum, J.L. Natural Antibodies with the T15 Idiotype May Act in Atherosclerosis, Apoptotic Clearance, and Protective Immunity. J. Clin. Investig. 2000, 105, 1731–1740. [Google Scholar] [CrossRef] [PubMed]
- Schulz, C.; Massberg, S. Platelets in Atherosclerosis and Thrombosis. Handb. Exp. Pharmacol. 2012, 111–133. [Google Scholar]
- Lievens, D.; von Hundelshausen, P. Platelets in Atherosclerosis. Thromb. Haemost. 2011, 106, 827–838. [Google Scholar] [CrossRef] [PubMed]
- Huo, Y.; Ley, K.F. Role of Platelets in the Development of Atherosclerosis. Trends Cardiovasc. Med. 2004, 14, 18–22. [Google Scholar] [CrossRef] [PubMed]
- Badimon, L.; Padró, T.; Vilahur, G. Atherosclerosis, Platelets and Thrombosis in Acute Ischaemic Heart Disease. Eur. Heart J. Acute Cardiovasc. Care 2012, 1, 60–74. [Google Scholar] [CrossRef] [PubMed]
- Koltai, K.; Kesmarky, G.; Feher, G.; Tibold, A.; Toth, K. Platelet Aggregometry Testing: Molecular Mechanisms, Techniques and Clinical Implications. Int. J. Mol. Sci. 2017, 18, 1803. [Google Scholar] [CrossRef] [PubMed]
- Andrews, R.K.; Berndt, M.C. Platelet Physiology and Thrombosis. Thromb. Res. 2004, 114, 447–453. [Google Scholar] [CrossRef] [PubMed]
- Ruggeri, Z.M.; Bader, R.; de Marco, L. Glanzmann Thrombasthenia: Deficient Binding of Von Willebrand Factor to Thrombin-Stimulated Platelets. Proc. Natl. Acad. Sci. USA 1982, 79, 6038–6041. [Google Scholar] [CrossRef] [PubMed]
- Ni, H.; Denis, C.V.; Subbarao, S.; Degen, J.L.; Sato, T.N.; Hynes, R.O.; Wagner, D.D. Persistence of Platelet Thrombus Formation in Arterioles of Mice Lacking Both Von Willebrand Factor and Fibrinogen. J. Clin. Investig. 2000, 106, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Hantgan, R.R. Fibrin Protofibril and Fibrinogen Binding to ADP-Stimulated Platelets: Evidence for a Common Mechanism. Biochim. Biophys. Acta 1988, 968, 24–35. [Google Scholar] [CrossRef]
- Bennett, J.S.; Vilaire, G. Exposure of Platelet Fibrinogen Receptors by ADP and Epinephrine. J. Clin. Investig. 1979, 64, 1393–1401. [Google Scholar] [CrossRef] [PubMed]
- Das, U.N. Atherosclerosis and Prostaglandins. Int. J. Tissue React. 1982, 4, 127–132. [Google Scholar] [PubMed]
- Martínez-Sánchez, S.M.; Minguela, A.; Prieto-Merino, D.; Zafrilla-Rentero, M.P.; Abellán-Alemán, J.; Montoro-García, S. The Effect of Regular Intake of Dry-Cured Ham Rich in Bioactive Peptides on Inflammation, Platelet and Monocyte Activation Markers in Humans. Nutrients 2017, 9, 321. [Google Scholar] [CrossRef] [PubMed]
- Molica, F.; Stierlin, F.B.; Fontana, P.; Kwak, B.R. Pannexin- and Connexin-Mediated Intercellular Communication in Platelet Function. Int. J. Mol. Sci. 2017, 18, 850. [Google Scholar] [CrossRef] [PubMed]
- Virmani, R.; Burke, A.P.; Farb, A. Plaque Rupture and Plaque Erosion. Thromb. Haemost. 1999, 82, 1–3. [Google Scholar] [PubMed]
- Lafont, A. Basic Aspects of Plaque Vulnerability. Heart 2003, 89, 1262–1267. [Google Scholar] [CrossRef] [PubMed]
- Hansson, G.K.; Libby, P.; Tabas, I. Inflammation and Plaque Vulnerability. J. Int. Med. 2015, 278, 483–493. [Google Scholar] [CrossRef] [PubMed]
- Shah, P.K. Mechanisms of Plaque Vulnerability and Rupture. J. Am. Coll. Cardiol. 2003, 41, S15–S22. [Google Scholar] [CrossRef]



© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, M.-Y.; Li, C.-J.; Hou, M.-F.; Chu, P.-Y. New Insights into the Role of Inflammation in the Pathogenesis of Atherosclerosis. Int. J. Mol. Sci. 2017, 18, 2034. https://doi.org/10.3390/ijms18102034
Wu M-Y, Li C-J, Hou M-F, Chu P-Y. New Insights into the Role of Inflammation in the Pathogenesis of Atherosclerosis. International Journal of Molecular Sciences. 2017; 18(10):2034. https://doi.org/10.3390/ijms18102034
Chicago/Turabian StyleWu, Meng-Yu, Chia-Jung Li, Ming-Feng Hou, and Pei-Yi Chu. 2017. "New Insights into the Role of Inflammation in the Pathogenesis of Atherosclerosis" International Journal of Molecular Sciences 18, no. 10: 2034. https://doi.org/10.3390/ijms18102034
APA StyleWu, M.-Y., Li, C.-J., Hou, M.-F., & Chu, P.-Y. (2017). New Insights into the Role of Inflammation in the Pathogenesis of Atherosclerosis. International Journal of Molecular Sciences, 18(10), 2034. https://doi.org/10.3390/ijms18102034