
Radiogenomic Analysis of Oncological Data: A Technical Survey

 ,
,  , and
, and
Abstract
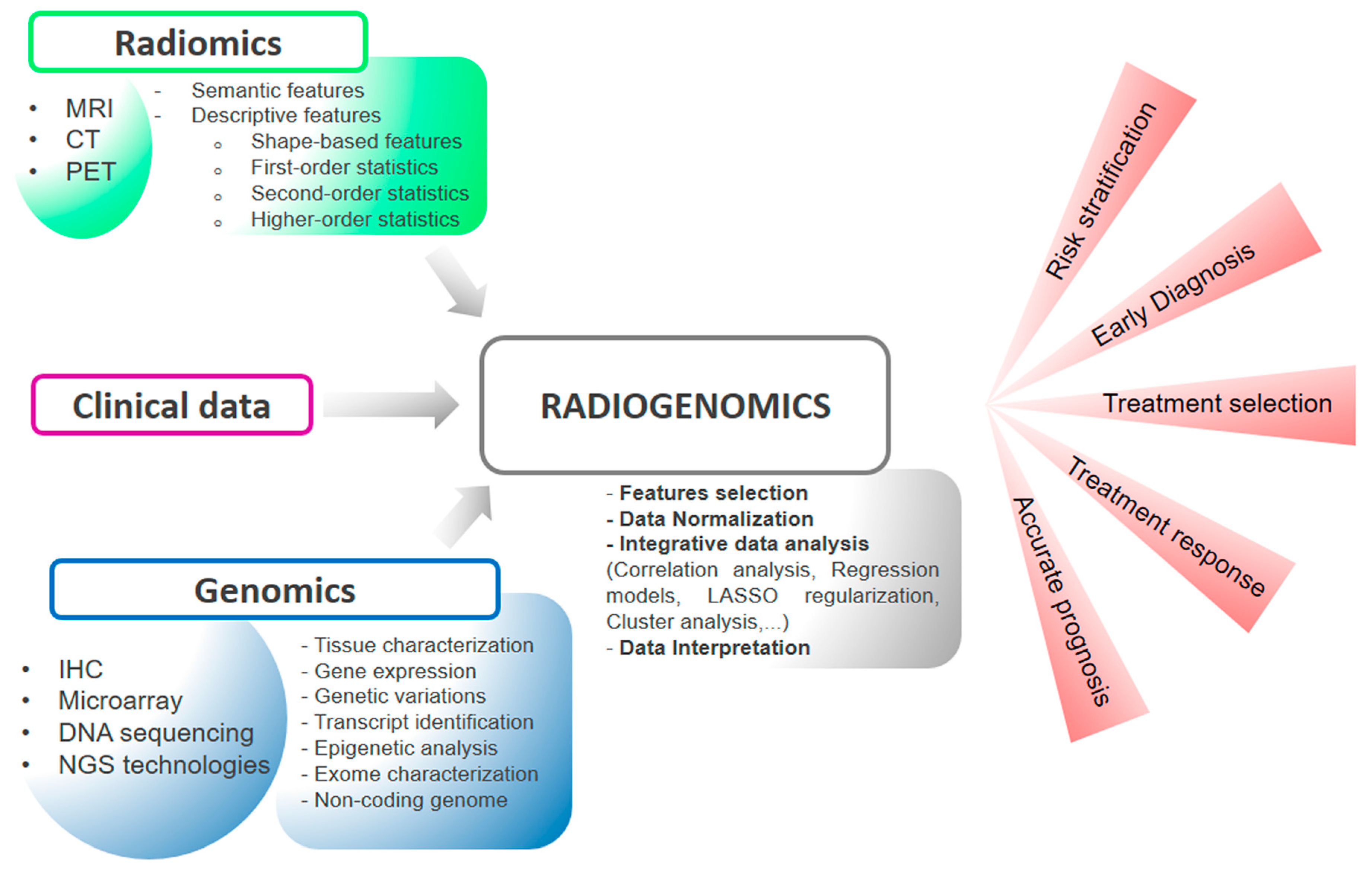
:1. Introduction
2. Methodologies
2.1. Radiomics
- Acquiring the images
- Segmenting the regions of interest (ROIs)
- Estimating descriptive features.
2.1.1. Image Acquisition
Computed Tomography (CT)
Positron Emission Tomography (PET)
Magnetic Resonance (MR)
2.1.2. Region of Interest Segmentation
2.1.3. Descriptive Features
Shape-Based Features
- Volume:where N is the number of voxels within a segmented volume of interest, and vs is the voxel size of the acquisition.V = N∙vs
- Surface area:NT is the number of triangles obtained from the triangulation of a tumour surface; , and are the vertices of the i-th triangle. The surface area, together with the volume, and eventually, the maximum diameter, provide information on the size of a lesion.
- Compactness:This factor measures how much a lesion is different from a sphere, indicating, consequently, its irregularity.
First-Order Statistics
- Mean: shows the average intensity value and is given by:X(i) is the gray value of the i-th voxels within a region of interest. Other estimates of the central tendency, used in descriptive statistics, can be computed, such as the mode and median.
- Standard deviation: indicates how widely intensity values vary, and is computed as:Other measures of histogram dispersions are the variance and the mean absolute deviation. The variability within a volume can also be indicated by common statistics, such as minimum, maximum and range values.
- Entropy: a statistical measure of randomness within a data sample, given by:where P is the first-order histogram of the volume of interest, computed on Nlbins. Additionally, uniformity and energy can be used to measure the randomness of a volume histogram.
- Skewness: a parameter that describes the asymmetry of a histogram around the mean, calculated as:
- Kurtosis: a parameter that depicts the degree of peakedness (broad or narrow) of a histogram and is given by:
Second-Order Statistics
- Gray Level Co-occurrence Matrix: These matrices determine how often a pixel of intensity i finds itself within a certain relationship to another pixel of intensity j. A GLCM is a joint probability function, defined as P (i,j;d,a), where the elements (i,j) represent the number of times that intensity levels i and j occur in two voxels separated by distance d in the direction a. The matrix size depends on the intensity levels within a segmented lesion and the number of matrices on the chosen d and a. For each matrix, several features can be extracted, and the final value for each d considered is obtained as the mean of the feature over the directions. Examples of characteristics that are mineable from each matrix are: Mean, standard deviation, and entropy for the joint and marginal probabilities, autocorrelation, cluster prominence, cluster shade and tendency, contrast, correlation, difference entropy, dissimilarity, energy, homogeneity, etc. [36].
- Gray Level Run-Length Matrix: A gray level run is the number of consecutive pixels having the same grey levels. In a GLRLM, defined as p (i,j;a), the row indices represent the discretized gray values and the column indices are the number of consecutive occurrences of the i-th gray value in direction a. The matrix size, consequently, depends on the number of gray values in a lesion (number of rows) and the maximum run length (number of columns). A GLRLM can be obtained for each a, and the textural features can be obtained as the mean over the directions of the values extracted from each matrix. Examples of mineable features are: Short and long run emphasis, gray level non-uniformity, run-length non-uniformity, run percentage, low and high gray level run emphasis, etc. [30].
- Gray Tone Difference Matrix: A column matrix, in which elements s (i) are the sum over the set of pixels having gray tone i, of the difference between the voxels of the set and the mean value, computed over the corresponding neighborhood. Consequently, the matrix depends on the size of the neighborhood. From GTDM, several features can be computed: Coarseness, contrast, busyness, complexity, and strength.
Higher-Order Statistics
- Laplacian of Gaussian filter [32]: This allows the highlighting of structures at a particular scale, corresponding to the width of a filter. Consequently, increasingly coarse texture patterns can be extracted from an image and analyzed using second-order statistics.
- Gabor filters: These allow for edge detection in different directions and widths [37]. For each filtered image, the Gabor energy feature can be extracted as a sum of the square intensity over all tumour pixels.
- Wavelet transform: This decouples textural information by decomposing the input image into low- and high-frequency coefficients without losing spatial localization. In particular, high-frequency coefficients also contain information on texture directionality. If an undecimated scheme is chosen, lesion segmentation, identified in the original image, can be used for computation of the first-order statistics and textural features from the wavelet coefficients.
- Fractal dimensions: These are estimates of object complexity. Fractal dimensions describe the relationship between the change in a measuring scale and the measurement at that scale [31], and can be calculated using a 3D box-counting algorithm [15]. Successively, values, such as mean and standard deviation, can be extracted.
2.2. Genomics
2.2.1. Microarray
2.2.2. Next-Generation Sequencing
RNA-Sequencing
2.2.3. Immunohistochemistry
2.3. Radiogenomic Data Analysis
3. Discussion
3.1. Radiogenomics in Breast Cancer
3.2. Radiogenomics in Glioblastoma Multiforme
3.3. Radiogenomics in Lung Cancer
3.4. Radiogenomics in Kidney Cancer
3.5. Radiogenomics in Prostate Cancer
3.6. Radiogenomics in Liver Cancer
3.7. General Considerations
4. Conclusions
Author Contributions
Conflicts of Interest
References
- Elsheikh, T.M.; Asa, S.L.; Chan, J.K.C.; DeLellis, R.A.; Heffess, C.S.; LiVolsi, V.A.; Wenig, B.M. Interobserver and intraobserver variation among experts in the diagnosis of thyroid follicular lesions with borderline nuclear features of papillary carcinoma. Am. J. Clin. Pathol. 2008, 130, 736–744. [Google Scholar] [CrossRef] [PubMed]
- Hariri, A.R.; Weinberger, D.R. Imaging genomics. Br. Med. Bull. 2003, 65, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Bai, H.X.; Lee, A.M.; Yang, L.; Zhang, P.; Davatzikos, C.; Maris, J.M.; Diskin, S.J. Imaging genomics in cancer research: Limitations and promises. Br. J. Radiol. 2016, 89. [Google Scholar] [CrossRef] [PubMed]
- Obermeyer, Z.; Emanuel, E.J. Predicting the future—Big data, machine learning, and clinical medicine. N. Engl. J. Med. 2016, 375, 1216–1219. [Google Scholar] [CrossRef] [PubMed]
- Gillies, R.J.; Kinahan, P.E.; Hricak, H. Radiomics: Images are more than pictures, they are data. Radiology 2016, 278, 563–577. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Kim, S.; Risacher, S.L.; Nho, K.; Swaminathan, S.; West, J.D.; Foroud, T.; Pankratz, N.; Moore, J.H.; Sloan, C.D.; et al. Whole genome association study of brain-wide imaging phenotypes for identifying quantitative trait loci in MCI and AD: A study of the adni cohort. Neuroimage 2010, 53, 1051–1063. [Google Scholar] [CrossRef] [PubMed]
- Aiello, M.; Cavaliere, C.; Salvatore, M. Hybrid PET/MR imaging and brain connectivity. Front. Neurosci. 2016, 10. [Google Scholar] [CrossRef] [PubMed]
- Mazurowski, M.A. Radiogenomics: What it is and why it is important. J. Am. Coll. Radiol. 2015, 12, 862–866. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Bai, H.X.; Lee, A.M. Leveraging imperfect data sets to draw new conclusions: Radiogenomics’ true value? J. Am. Coll. Radiol. 2016, 13, 120–121. [Google Scholar] [CrossRef] [PubMed]
- Micheel, C.M.; Nass, S.J.; Omenn, G.S. Evolution of Translational Omics: Lessons Learned and the Path Forward; National Academies Press: Washington, DC, USA, 2012. [Google Scholar]
- Lambin, P.; Rios-Velazquez, E.; Leijenaar, R.; Carvalho, S.; van Stiphout, R.; Granton, P.; Zegers, C.M.L.; Gillies, R.; Boellard, R.; Dekker, A.; et al. Radiomics: Extracting more information from medical images using advanced feature analysis. Eur. J. Cancer 2012, 48, 441–446. [Google Scholar] [CrossRef] [PubMed]
- Xydeas, C.S.; Petrovic, V. Objective image fusion performance measure. Electron. Lett. 2000, 36, 308–309. [Google Scholar] [CrossRef]
- Kumar, V.; Gu, Y.H.; Basu, S.; Berglund, A.; Eschrich, S.A.; Schabath, M.B.; Forster, K.; Aerts, H.; Dekker, A.; Fenstermacher, D.; et al. Radiomics: The process and the challenges. Magn. Reson. Imaging 2012, 30, 1234–1248. [Google Scholar] [CrossRef] [PubMed]
- Anselmo, A.C.; Zhang, M.; Kumar, S.; Vogus, D.R.; Menegatti, S.; Helgeson, M.E.; Mitragotri, S. Elasticity of nanopartides influences their blood circulation, phagocytosis, endocytosis, and targeting. ACS Nano 2015, 9, 3169–3177. [Google Scholar] [CrossRef] [PubMed]
- Galavis, P.E.; Hollensen, C.; Jallow, N.; Paliwal, B.; Jeraj, R. Variability of textural features in FDG PET images due to different acquisition modes and reconstruction parameters. Acta Oncol. 2010, 49, 1012–1016. [Google Scholar] [CrossRef] [PubMed]
- Forgacs, A.; Jonsson, H.P.; Dahlbom, M.; Daver, F.; di Franco, M.D.; Opposits, G.; Krizsan, A.K.; Garai, I.; Czernin, J.; Varga, J.; et al. A study on the basic criteria for selecting heterogeneity parameters of F18-FDG PET images. PLoS ONE 2016, 11, e0164113. [Google Scholar] [CrossRef] [PubMed]
- Carlier, T.; Bailly, C. State-of-the-art and recent advances in quantification for therapeutic follow-up in oncology using PET. Front. Med. 2015, 2, 18. [Google Scholar] [CrossRef] [PubMed]
- Leijenaar, R.T.H.; Carvalho, S.; Velazquez, E.R.; Van Elmpt, W.J.C.; Parmar, C.; Hoekstra, O.S.; Hoekstra, C.J.; Boellaard, R.; Dekker, A.; Gillies, R.J.; et al. Stability of FDG-PET radiomics features: An integrated analysis of test-retest and inter-observer variability. Acta Oncol. 2013, 52, 1391–1397. [Google Scholar] [CrossRef] [PubMed]
- Gatenby, R.A.; Grove, O.; Gillies, R.J. Quantitative imaging in cancer evolution and ecology. Radiology 2013, 269, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Hall, L.; Goldgof, D.; Russo, R.; Balagurunathan, Y.; Gillies, R.; Gatenby, R. Radiologically defined ecological dynamics and clinical outcomes in glioblastoma multiforme: Preliminary results. Transl. Oncol. 2014, 7, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Velazquez, E.R.; Aerts, H.; Gu, Y.H.; Goldgof, D.B.; De Ruysscher, D.; Dekker, A.; Korn, R.; Gillies, R.J.; Lambin, P. A semiautomatic CT-based ensemble segmentation of lung tumors: Comparison with oncologists’ delineations and with the surgical specimen. Radiother. Oncol. 2012, 105, 167–173. [Google Scholar] [CrossRef] [PubMed]
- van Dam, I.E.; de Koste, J.R.V.; Hanna, G.G.; Muirhead, R.; Slotman, B.J.; Senan, S. Improving target delineation on 4-dimensional CT scans in stage i nsclc using a deformable registration tool. Radiother. Oncol. 2010, 96, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Aerts, H. The potential of radiomic-based phenotyping in precisionmedicine a review. JAMA Oncol. 2016, 2, 1636–1642. [Google Scholar] [CrossRef] [PubMed]
- Nishino, M.; Dahlberg, S.E.; Fulton, L.E.; Digumarthy, S.R.; Hatabu, H.; Johnson, B.E.; Sequist, L.V. Volumetric tumor response and progression in EGFR-mutant nsclc patients treated with erlotinib or gefitinib. Acad. Radiol. 2016, 23, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Nishino, M.; Guo, M.Y.; Jackman, D.M.; DiPiro, P.J.; Yap, J.T.; Ho, T.K.; Hatabu, H.; Janne, P.A.; Van den Abbeele, A.D.; Johnson, B.E. CT tumor volume measurement in advanced non-small-cell lung cancer: Performance characteristics of an emerging clinical tool. Acad. Radiol. 2011, 18, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Parmar, C.; Velazquez, E.R.; Leijenaar, R.; Jermoumi, M.; Carvalho, S.; Mak, R.H.; Mitra, S.; Shankar, B.U.; Kikinis, R.; Haibe-Kains, B.; et al. Robust radiomics feature quantification using semiautomatic volumetric segmentation. PLoS ONE 2014, 9, e102107. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, J.P.B.; Rose, C.J.; Waterton, J.C.; Carano, R.A.D.; Parker, G.J.M.; Jackson, A. Imaging intratumor heterogeneity: Role in therapy response, resistance, and clinical outcome. Clin. Cancer Res. 2015, 21, 249–257. [Google Scholar] [CrossRef] [PubMed]
- American College of Radiology. Breast Imaging Reporting and Data System Atlas (BI-Rads Atlas); American College of Radiology: Reston, VA, USA, 2003; Volume 98. [Google Scholar]
- Weinreb, J.C.; Barentsz, J.O.; Choyke, P.L.; Cornud, F.; Haider, M.A.; Macura, K.J.; Margolis, D.; Schnall, M.D.; Shtern, F.; Tempany, C.M.; et al. PI-RADS prostate imaging—Reporting and data system: 2015, version 2. Eur. Urol. 2016, 69, 16–40. [Google Scholar] [CrossRef] [PubMed]
- Mazurowski, M.A.; Desjardins, A.; Malof, J.M. Imaging descriptors improve the predictive power of survival models for glioblastoma patients. Neuro. Oncol. 2013, 15, 1389–1394. [Google Scholar] [CrossRef] [PubMed]
- Aerts, H.; Grossmann, P.; Tan, Y.Q.; Oxnard, G.G.; Rizvi, N.; Schwartz, L.H.; Zhao, B.S. Defining a radiomic response phenotype: A pilot study using targeted therapy in NSCLC. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Davnall, F.; Yip, C.S.P.; Ljungqvist, G.; Selmi, M.; Ng, F.; Sanghera, B.; Ganeshan, B.; Miles, K.A.; Cook, G.J.; Goh, V. Assessment of tumor heterogeneity: An emerging imaging tool for clinical practice? Insights Imaging 2012, 3, 573–589. [Google Scholar] [CrossRef] [PubMed]
- Haralick, R.M.; Shanmugam, K. Textural features for image classification. IEEE Trans. Syst. Man Cybern. 1973, 3, 610–621. [Google Scholar] [CrossRef]
- Galloway, M.M. Texture analysis using gray level run lengths. Comput. Graph. Image Process. 1975, 4, 172–179. [Google Scholar] [CrossRef]
- Amadasun, M.; King, R. Textural features corresponding to textural properties. IEEE Trans. Syst. Man Cybern. 1989, 19, 1264–1274. [Google Scholar] [CrossRef]
- Aerts, H.; Velazquez, E.R.; Leijenaar, R.T.H.; Parmar, C.; Grossmann, P.; Cavalho, S.; Bussink, J.; Monshouwer, R.; Haibe-Kains, B.; Rietveld, D.; et al. Decoding tumour phenotype by noninvasive imaging using a quantitative radiomics approach. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef]
- Fogel, I.; Sagi, D. Gabor filters as texture discriminator. Biol. Cybern. 1989, 61, 103–113. [Google Scholar] [CrossRef]
- Poustka, A.; Pohl, T.; Barlow, D.; Zehetner, G.; Craig, A.; Michiels, F.; Ehrich, E.; Frischauf, A.-M.; Lehrach, H. Molecular approaches to mammalian genetics. Cold Spring Harb. Symp. Quant. Biol. 1986, 51, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Cantor, C.R.; Mirzabekov, A.; Southern, E. Report on the sequencing by hybridization workshop. Genomics 1992, 13, 1378–1383. [Google Scholar] [CrossRef]
- Marzancola, M.G.; Sedighi, A.; Li, P.C.H. DNA microarray-based diagnostics. Methods Mol. Biol. 2016, 1368, 161–178. [Google Scholar] [PubMed]
- Jaksik, R.; Iwanaszko, M.; Rzeszowska-Wolny, J.; Kimmel, M. Microarray experiments and factors which affect their reliability. Biol. Direct 2015, 10. [Google Scholar] [CrossRef] [PubMed]
- Schena, M.; Shalon, D.; Davis, R.W.; Brown, P.O. Quantitative monitoring of gene expression patterns with a complementary dna microarray. Science 1995, 270, 467–470. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A.; Baylin, S.B. The epigenomics of cancer. Cell 2007, 128, 683–692. [Google Scholar] [CrossRef] [PubMed]
- Zeller, C.; Masrour, N.; Patel, N.; Dai, W.; Wilhelm-Benartzi, C.; Brown, R. DNA methylation profiling using infinium methylation assay. Bio Protocol. 2013, 3. [Google Scholar] [CrossRef]
- Meldrum, C.; Doyle, M.A.; Tothill, R.W. Next-generation sequencing for cancer diagnostics: A practical perspective. Clin. Biochem. Rev. 2011, 32, 177–195. [Google Scholar] [PubMed]
- Bahassi, E.; Stambrook, P.J. Next-generation sequencing technologies: Breaking the sound barrier of human genetics. Mutagenesis 2014, 29, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Metzker, M.L. Applications of next-generation sequencing sequencing technologies—The next generation. Nat. Rev. Genet. 2010, 11, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Voelkerding, K.V.; Dames, S.A.; Durtschi, J.D. Next-generation sequencing: From basic research to diagnostics. Clin. Chem. 2009, 55, 641–658. [Google Scholar] [CrossRef] [PubMed]
- Wade, J.T. Mapping transcription regulatory networks with ChIP-Seq and RNA-Seq. Adv. Exp. Med. Biol. 2015, 883, 119–134. [Google Scholar] [PubMed]
- Sun, Z.F.; Cunningham, J.; Slager, S.; Kocher, J.P. Base resolution methylome profiling: Considerations in platform selection, data preprocessing and analysis. Epigenomics 2015, 7, 813–828. [Google Scholar] [CrossRef] [PubMed]
- Costa, V.; Angelini, C.; De Feis, I.; Ciccodicola, A. Uncovering the complexity of transcriptomes with RNA-Seq. J. Biomed. Biotechnol. 2010, 2010. [Google Scholar] [CrossRef] [PubMed]
- Sager, M.; Yeat, N.C.; Pajaro-Van der Stadt, S.; Lin, C.; Ren, Q.Y.; Lin, J. Transcriptomics in cancer diagnostics: Developments in technology, clinical research and commercialization. Expert Rev. Mol. Diagn. 2015, 15, 1589–1603. [Google Scholar] [CrossRef] [PubMed]
- Pan, Q.; Shai, O.; Lee, L.J.; Frey, J.; Blencowe, B.J. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008, 40, 1413–1415. [Google Scholar] [CrossRef] [PubMed]
- Guttman, M.; Garber, M.; Levin, J.Z.; Donaghey, J.; Robinson, J.; Adiconis, X.; Fan, L.; Koziol, M.J.; Gnirke, A.; Nusbaum, C.; et al. Ab initio reconstruction of cell type-specific transcriptomes in mouse reveals the conserved multi-exonic structure of lincRNAs. Nat. Biotechnol. 2010, 28, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Rienzo, M.; Costa, V.; Scarpato, M.; Schiano, C.; Casamassimi, A.; Grimaldi, V.; Ciccodicola, A.; Napoli, C. RNA-Seq for the identification of novel mediator transcripts in endothelial progenitor cells. Gene 2014, 547, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Degner, J.F.; Marioni, J.C.; Pai, A.A.; Pickrell, J.K.; Nkadori, E.; Gilad, Y.; Pritchard, J.K. Effect of read-mapping biases on detecting allele-specific expression from RNA-sequencing data. Bioinformatics 2009, 25, 3207–3212. [Google Scholar] [CrossRef] [PubMed]
- Edgren, H.; Murumagi, A.; Kangaspeska, S.; Nicorici, D.; Hongisto, V.; Kleivi, K.; Rye, I.H.; Nyberg, S.; Wolf, M.; Borresen-Dale, A.L.; et al. Identification of fusion genes in breast cancer by paired-end RNA-sequencing. Genome Biol. 2011, 12. [Google Scholar] [CrossRef] [PubMed]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.Y.; Cheng, Y.B.; Tan, B.C.M.; Kang, L.; Tian, Z.J.; Zhu, Y.K.; Zhang, W.W.; Liang, Y.; Hu, X.D.; Tan, X.M.; et al. Comprehensive analysis of RNA-seq data reveals extensive RNA editing in a human transcriptome. Nat. Biotechnol. 2012, 30, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Cloonan, N.; Forrest, A.R.R.; Kolle, G.; Gardiner, B.B.A.; Faulkner, G.J.; Brown, M.K.; Taylor, D.F.; Steptoe, A.L.; Wani, S.; Bethel, G.; et al. Stem cell transcriptome profiling via massive-scale mRNA sequencing. Nat. Methods 2008, 5, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Marioni, J.C.; Mason, C.E.; Mane, S.M.; Stephens, M.; Gilad, Y. RNA-seq: An assessment of technical reproducibility and comparison with gene expression arrays. Genome Res. 2008, 18, 1509–1517. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.G.; Wang, S.Q.; Li, W. Rseqc: Quality control of RNA-seq experiments. Bioinformatics 2012, 28, 2184–2185. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Alcalde, F.; Okonechnikov, K.; Carbonell, J.; Cruz, L.M.; Gotz, S.; Tarazona, S.; Dopazo, J.; Meyer, T.F.; Conesa, A. Qualimap: Evaluating next-generation sequencing alignment data. Bioinformatics 2012, 28, 2678–2679. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.L.; Wu, G.X.; Tang, J.B.; Luo, R.B.; Patterson, J.; Liu, S.L.; Huang, W.H.; He, G.Z.; Gu, S.C.; Li, S.K.; et al. Soapdenovo-trans: De novo transcriptome assembly with short RNA-Seq reads. Bioinformatics 2014, 30, 1660–1666. [Google Scholar] [CrossRef] [PubMed]
- Schulz, M.H.; Zerbino, D.R.; Vingron, M.; Birney, E. Oases: Robust de novo RNA-Seq assembly across the dynamic range of expression levels. Bioinformatics 2012, 28, 1086–1092. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.D.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-Seq using the trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef] [PubMed]
- Steijger, T.; Abril, J.F.; Engstrom, P.G.; Kokocinski, F.; Hubbard, T.J.; Guigo, R.; Harrow, J.; Bertone, P.; Consortium, R. Assessment of transcript reconstruction methods for RNA-Seq. Nat. Methods 2013, 10, 1177–1184. [Google Scholar] [CrossRef] [PubMed]
- Boley, N.; Stoiber, M.H.; Booth, B.W.; Wan, K.H.; Hoskins, R.A.; Bickel, P.J.; Celniker, S.E.; Brown, J.B. Genome-guided transcript assembly by integrative analysis of RNA sequence data. Nat. Biotechnol. 2014, 32, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.; Pimentel, H.; Trapnell, C.; Pachter, L. Identification of novel transcripts in annotated genomes using RNA-Seq. Bioinformatics 2011, 27, 2325–2329. [Google Scholar] [CrossRef] [PubMed]
- Mezlini, A.M.; Smith, E.J.M.; Fiume, M.; Buske, O.; Savich, G.L.; Shah, S.; Aparicio, S.; Chiang, D.Y.; Goldenberg, A.; Brudno, M. Ireckon: Simultaneous isoform discovery and abundance estimation from RNA-Seq data. Genome Res. 2013, 23, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. Stringtie enables improved reconstruction of a transcriptome from RNA-Seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Engstrom, P.G.; Steijger, T.; Sipos, B.; Grant, G.R.; Kahles, A.; Ratsch, G.; Goldman, N.; Hubbard, T.J.; Harrow, J.; Guigo, R.; et al. Systematic evaluation of spliced alignment programs for RNA-Seq data. Nat. Methods 2013, 10, 1185–1191. [Google Scholar] [CrossRef] [PubMed]
- Stanke, M.; Keller, O.; Gunduz, I.; Hayes, A.; Waack, S.; Morgenstern, B. Augustus: Ab initio prediction of alternative transcripts. Nucleic Acids Res. 2006, 34, 435–439. [Google Scholar] [CrossRef] [PubMed]
- Hiller, D.; Wong, W.H. Simultaneous isoform discovery and quantification from RNA-Seq. Stat. Biosci. 2013, 5, 100–118. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq-a Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. Featurecounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. Star: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.R.; Neff, N.F.; Kalisky, T.; Dalerba, P.; Treutlein, B.; Rothenberg, M.E.; Mburu, F.M.; Mantalas, G.L.; Sim, S.; Clarke, M.F.; et al. Quantitative assessment of single-cell RNA-sequencing methods. Nat. Methods 2014, 11, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Madrigal, P.; Tarazona, S.; Gomez-Cabrero, D.; Cervera, A.; McPherson, A.; Szczesniak, M.W.; Gaffney, D.J.; Elo, L.L.; Zhang, X.G.; et al. A survey of best practices for RNA-Seq data analysis. Genome Biol. 2016, 17. [Google Scholar] [CrossRef] [PubMed]
- Wilkerson, M.D.; Cabanski, C.R.; Sun, W.; Hoadley, K.A.; Walter, V.; Mose, L.E.; Troester, M.A.; Hammerman, P.S.; Parker, J.S.; Perou, C.M.; et al. Integrated RNA and DNA sequencing improves mutation detection in low purity tumors. Nucleic Acids Res. 2014, 42. [Google Scholar] [CrossRef] [PubMed]
- Ke, R.Q.; Mignardi, M.; Pacureanu, A.; Svedlund, J.; Botling, J.; Wahlby, C.; Nilsson, M. In situ sequencing for rna analysis in preserved tissue and cells. Nat. Methods 2013, 10, 857–860. [Google Scholar] [CrossRef] [PubMed]
- Hrdlickova, R.; Toloue, M.; Tian, B. RNA-Seq methods for transcriptome analysis. Wiley Interdiscip. Rev. RNA 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Vara, J.A. Principles and methods of immunohistochemistry. Drug Saf. Eval. Methods Protoc. 2011, 691, 83–96. [Google Scholar]
- Kuo, M.D.; Jamshidi, N. Behind the numbers: Decoding molecular phenotypes with radiogenomics-guiding principles and technical considerations. Radiology 2014, 270, 320–325. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Chang, K.; Ramkissoon, S.; Tanguturi, S.; Bi, W.L.; Reardon, D.A.; Ligon, K.L.; Alexander, B.M.; Wen, P.Y.; Huang, R.Y. Multimodal MRI features predict isocitrate dehydrogenase genotype in high-grade gliomas. Neuro. Oncol. 2017, 19, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.T.; Li, H.; Guo, W.T.; Drukker, K.; Lan, L.; Giger, M.L.; Ji, Y. Deciphering genomic underpinnings of quantitative MRI-based radiomic phenotypes of invasive breast carcinoma. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.H. On bias, variance, 0/1—Loss, and the curse-of-dimensionality. Data Min. Know. Discov. 1997, 1, 55–77. [Google Scholar] [CrossRef]
- Balagurunathan, Y.; Kumar, V.; Gu, Y.H.; Kim, J.; Wang, H.; Liu, Y.; Goldgof, D.B.; Hall, L.O.; Korn, R.; Zhao, B.S.; et al. Test-retest reproducibility analysis of lung CT image features. J. Digit. Imaging 2014, 27, 805–823. [Google Scholar] [CrossRef] [PubMed]
- Kuo, M.D.; Gollub, J.; Sirlin, C.B.; Ooi, C.; Chen, X. Radiogenomic analysis to identify imaging phenotypes associated with drug response gene expression programs in hepatocellular carcinoma. J. Vasc. Interv. Radiol. 2007, 18, 821–831. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.M.; Parmar, C.; Grossmann, P.; Quackenbush, J.; Lambin, P.; Bussink, J.; Mak, R.; Aerts, H. Exploratory study to identify radiomics classifiers for lung cancer histology. Front. Oncol. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Parmar, C.; Grossmann, P.; Bussink, J.; Lambin, P.; Aerts, H. Machine learning methods for quantitative radiomic biomarkers. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.T.; Li, H.; Zhu, Y.T.; Lan, L.; Yang, S.J.; Drukker, K.; Morris, E.; Burnside, E.; Whitman, G.; Giger, M.L.; et al. Prediction of clinical phenotypes in invasive breast carcinomas from the integration of radiomics and genomics data. J. Med. Imaging 2015, 2. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.T.; Chen, Y.F.; Hastie, T.; Sobel, E.; Lange, K. Genome-wide association analysis by lasso penalized logistic regression. Bioinformatics 2009, 25, 714–721. [Google Scholar] [CrossRef] [PubMed]
- Jamshidi, N.; Jonasch, E.; Zapala, M.; Korn, R.L.; Aganovic, L.; Zhao, H.J.; Sitaram, R.T.; Tibshirani, R.J.; Banerjee, S.; Brooks, J.D.; et al. The radiogenomic risk score: Construction of a prognostic quantitative, noninvasive image-based molecular assay for renal cell carcinoma. Radiology 2015, 277, 114–123. [Google Scholar] [CrossRef] [PubMed]
- Leung, M.K.K.; Delong, A.; Alipanahi, B.; Frey, B.J. Machine learning in genomic medicine: A review of computational problems and data sets. Proc. IEEE 2016, 104, 176–197. [Google Scholar] [CrossRef]
- Deng, L.; Yu, D. Deep learning: Methods and applications. Found. Trends Signal Proc. 2014, 7, 197–387. [Google Scholar] [CrossRef]
- Panth, K.M.; Leijenaar, R.T.H.; Carvalho, S.; Lieuwes, N.G.; Yaromina, A.; Dubois, L.; Lambin, P. Is there a causal relationship between genetic changes and radiomics-based image features? An in vivo preclinical experiment with doxycycline inducible GADD34 tumor cells. Radiother. Oncol. 2015, 116, 462–466. [Google Scholar] [CrossRef] [PubMed]
- Grimm, L.J.; Zhang, J.; Mazurowski, M.A. Computational approach to radiogenomics of breast cancer: Luminal A and luminal B molecular subtypes are associated with imaging features on routine breast mri extracted using computer vision algorithms. J. Magn. Reson. Imaging 2015, 42, 902–907. [Google Scholar] [CrossRef] [PubMed]
- Mazurowski, M.A.; Zhang, J.; Grimm, L.J.; Yoon, S.C.; Silber, J.I. Radiogenomic analysis of breast cancer: Luminal b molecular subtype is associated with enhancement dynamics at MR imaging. Radiology 2014, 273, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Sung, J.S.; Jochelson, M.S.; Brennan, S.; Joo, S.; Wen, Y.H.; Moskowitz, C.; Zheng, J.T.; Dershaw, D.D.; Morris, E.A. MR imaging features of triple-negative breast cancers. Breast J. 2013, 19, 643–649. [Google Scholar] [CrossRef] [PubMed]
- Sutton, E.J.; Oh, J.H.; Dashevsky, B.Z.; Veeraraghavan, H.; Apte, A.P.; Thakur, S.B.; Deasy, J.O.; Morris, E.A. Breast cancer subtype intertumor heterogeneity: MRI-based features predict results of a genomic assay. J. Magn. Reson. Imaging 2015, 42, 1398–1406. [Google Scholar] [CrossRef] [PubMed]
- Sutton, E.J.; Dashevsky, B.Z.; Oh, J.H.; Veeraraghavan, H.; Apte, A.P.; Thakur, S.B.; Morris, E.A.; Deasy, J.O. Breast cancer molecular subtype classifier that incorporates MRI features. J. Magn. Reson. Imaging 2016, 44, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Uematsu, T.; Kasami, M.; Yuen, S. Triple-negative breast cancer: Correlation between MR imaging and pathologic findings. Radiology 2009, 250, 638–647. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.; Maki, D.D.; Korn, R.L.; Kuo, M.D. Radiogenomic analysis of breast cancer using MRI: A preliminary study to define the landscape. Am. J. Roentgenol. 2012, 199, 654–663. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.; Han, W.; Kim, Y.; Du, L.T.; Jamshidi, N.; Huang, D.S.; Kim, J.H.; Kuo, M.D. Breast cancer: Radiogenomic biomarker reveals associations among dynamic contrast-enhanced MR imaging, long noncoding rna, and metastasis. Radiology 2015, 275, 384–392. [Google Scholar] [CrossRef] [PubMed]
- Cipolla, V.; Santucci, D.; Guerrieri, D.; Drudi, F.M.; Meggiorini, M.L.; de Felice, C. Correlation between 3T apparent diffusion coefficient values and grading of invasive breast carcinoma. Eur. J. Radiol. 2014, 83, 2144–2150. [Google Scholar] [CrossRef] [PubMed]
- Molinari, C.; Clauser, P.; Girometti, R.; Linda, A.; Cimino, E.; Puglisi, F.; Zuiani, C.; Bazzocchi, M. MR mammography using diffusion-weighted imaging in evaluating breast cancer: A correlation with proliferation index. Radiol. Med. 2015, 120, 911–918. [Google Scholar] [CrossRef] [PubMed]
- An, Y.Y.; Kim, S.H.; Kang, B.J.; Park, C.S.; Jung, N.Y.; Kim, J.Y. Breast cancer in very young women (<30 years): Correlation of imaging features with clinicopathological features and immunohistochemical subtypes. Eur. J. Radiol. 2015, 84, 1894–1902. [Google Scholar] [CrossRef] [PubMed]
- Bae, M.S.; Seo, M.; Kim, K.G.; Park, I.A.; Moon, W.K. Quantitative mri morphology of invasive breast cancer: Correlation with immunohistochemical biomarkers and subtypes. Acta Radiol. 2015, 56, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.J.; Park, J.Y.; Shin, K.C.; Kim, H.H.; Cha, J.H.; Chae, E.Y.; Choi, W.J. Characterization of tumor and adjacent peritumoral stroma in patients with breast cancer using high-resolution diffusion-weighted imaging: Correlation with pathologic biomarkers. Eur. J. Radiol. 2016, 85, 1004–1011. [Google Scholar] [CrossRef] [PubMed]
- Kitajim, K.; Yamano, T.; Fukushima, K.; Miyoshi, Y.; Hirota, S.; Kawanaka, Y.; Miya, M.; Doi, H.; Yamakado, K. Correlation of the SUVmax of FDG-PET and ADC values of diffusion-weighted mr imaging with pathologic prognostic factors in breast carcinoma. Eur. J. Radiol. 2016, 85, 943–949. [Google Scholar] [CrossRef] [PubMed]
- Koo, H.R.; Park, J.S.; Kang, K.W.; Han, W.; Park, I.A.; Moon, W.K. Correlation between F-18-FDG uptake on PET/CT and prognostic factors in triple-negative breast cancer. Eur. Radiol. 2015, 25, 3314–3321. [Google Scholar] [CrossRef] [PubMed]
- Koo, H.R.; Park, J.S.; Kang, K.W.; Cho, N.; Chang, J.M.; Bae, M.S.; Kim, W.H.; Lee, S.H.; Kim, M.Y.; Kim, J.Y.; et al. F-18-FDG uptake in breast cancer correlates with immunohistochemically defined subtypes. Eur. Radiol. 2014, 24, 610–618. [Google Scholar] [CrossRef] [PubMed]
- Tchou, J.; Sonnad, S.S.; Bergey, M.R.; Basu, S.; Tomaszewski, J.; Alavi, A.; Schnall, M. Degree of tumor FDG uptake correlates with proliferation index in triple negative breast cancer. Mol. Imaging Biol. 2010, 12, 657–662. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, K.; Abe, H.; Newstead, G.M.; Egashira, R.; Nakazono, T.; Imaizumi, T.; Irie, H. Intratumoral heterogeneity of the distribution of kinetic parameters in breast cancer: Comparison based on the molecular subtypes of invasive breast cancer. Breast Cancer 2015, 22, 496–502. [Google Scholar] [CrossRef] [PubMed]
- Mehta, S.; Hughes, N.P.; Li, S.; Jubb, A.; Adams, R.; Lord, S.; Koumakis, L.; van Stiphout, R.; Padhani, A.; Makris, A.; et al. Radiogenomics monitoring in breast cancer identifies metabolism and immune checkpoints as early actionable mechanisms of resistance to anti-angiogenic treatment. Ebiomedicine 2016, 10, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Osborne, J.R.; Port, E.; Gonen, M.; Doane, A.S.; Yeung, H.; Gerald, W.; Cook, J.B.; Larson, S. F-18-FDG PET of locally invasive breast cancer and association of estrogen receptor status with standardized uptake value: Microarray and immunohistochemical analysis. J. Nucl. Med. 2010, 51, 543–550. [Google Scholar] [CrossRef] [PubMed]
- Palaskas, N.; Larson, S.M.; Schultz, N.; Komisopoulou, E.; Wong, J.; Rohle, D.; Campos, C.; Yannuzzi, N.; Osborne, J.R.; Linkov, I.; et al. F-18-fluorodeoxy-glucose positron emission tomography marks MYC-overexpressing human basal-like breast cancers. Cancer Res. 2011, 71, 5164–5174. [Google Scholar] [CrossRef] [PubMed]
- Diehn, M.; Nardini, C.; Wang, D.S.; McGovern, S.; Jayaraman, M.; Liang, Y.; Alclape, K.; Cha, S.; Kuo, M.D. Identification of noninvasive imaging surrogates for brain tumor gene-expression modules. Proc. Natl. Acad. Sci. USA 2008, 105, 5213–5218. [Google Scholar] [CrossRef] [PubMed]
- Zinn, P.O.; Majadan, B.; Sathyan, P.; Singh, S.K.; Majumder, S.; Jolesz, F.A.; Colen, R.R. Radiogenomic mapping of edema/cellular invasion MRI-phenotypes in glioblastoma multiforme. PLoS ONE 2011, 6, e25451. [Google Scholar] [CrossRef] [PubMed]
- Jamshidi, N.; Diehn, M.; Bredel, M.; Kuo, M.D. Illuminating radiogenomic characteristics of glioblastoma multiforme through integration of MR imaging, messenger RNA expression, and DNA copy number variation. Radiology 2014, 270, 212–222. [Google Scholar] [CrossRef] [PubMed]
- Gutman, D.A.; Dunn, W.D.; Grossmann, P.; Cooper, L.A.D.; Holder, C.A.; Ligon, K.L.; Alexander, B.M.; Aerts, H. Somatic mutations associated with MRI-derived volumetric features in glioblastoma. Neuroradiology 2015, 57, 1227–1237. [Google Scholar] [CrossRef] [PubMed]
- Rao, A.; Rao, G.; Gutman, D.A.; Flanders, A.E.; Hwang, S.N.; Rubin, D.L.; Colen, R.R.; Zinn, P.O.; Jain, R.; Wintermark, M.; et al. A combinatorial radiographic phenotype may stratify patient survival and be associated with invasion and proliferation characteristics in glioblastoma. J. Neurosurg. 2016, 124, 1008–1017. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.S.; Ning, S.; Eschbacher, J.M.; Baxter, L.C.; Gaw, N.; Ranjbar, S.; Plasencia, J.; Dueck, A.C.; Peng, S.; Smith, K.A. Radiogenomics to characterize regional genetic heterogeneity in glioblastoma. Neuro. Oncol. 2017, 19, 128–137. [Google Scholar] [CrossRef] [PubMed]
- Gevaert, O.; Mitchell, L.A.; Achrol, A.S.; Xu, J.J.; Echegaray, S.; Steinberg, G.K.; Cheshier, S.H.; Napel, S.; Zaharchuk, G.; Plevritis, S.K. Glioblastoma multiforme: Exploratory radiogenomic analysis by using quantitative image features. Radiology 2014, 273, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Heiland, D.H.; Demerath, T.; Kellner, E.; Kiselev, V.G.; Pfeifer, D.; Schnell, O.; Staszewski, O.; Urbach, H.; Weyerbrock, A.; Mader, I. Molecular differences between cerebral blood volume and vessel size in glioblastoma multiforme. Oncotarget 2016. [Google Scholar] [CrossRef] [PubMed]
- Kickingereder, P.; Bonekamp, D.; Nowosielski, M.; Kratz, A.; Sill, M.; Burth, S.; Wick, A.; Eidel, O.; Schlemmer, H.P.; Radbruch, A.; et al. Radiogenomics of glioblastoma: Machine learning-based classification of molecular characteristics by using multiparametric and multiregional MR imaging features. Radiology 2016, 281, 907–918. [Google Scholar] [CrossRef] [PubMed]
- Barajas, R.F.; Hodgson, J.G.; Chang, J.S.; Vandenberg, S.R.; Yeh, R.F.; Parsa, A.T.; McDermott, M.W.; Berger, M.S.; Dillon, W.P.; Cha, S. Glioblastoma multiforme regional genetic and cellular expression patterns: Influence on anatomic and physiologic MR imaging. Radiology 2010, 254, 564–576. [Google Scholar] [CrossRef] [PubMed]
- Pope, W.B.; Chen, J.H.; Dong, J.; Carlson, M.R.J.; Perlina, A.; Cloughesy, T.F.; Liau, L.M.; Mischel, P.S.; Nghiemphu, P.; Lai, A.; et al. Relationship between gene expression and enhancement in glioblastoma multiforme: Exploratory DNA microarray analysis. Radiology 2008, 249, 268–277. [Google Scholar] [CrossRef] [PubMed]
- Zinn, P.O.; Sathyan, P.; Mahajan, B.; Bruyere, J.; Hegi, M.; Majumder, S.; Colen, R.R. A novel volume-age-KPS (VAK) glioblastoma classification identifies a prognostic cognate microrna-gene signature. PLoS ONE 2012, 7, e41522. [Google Scholar] [CrossRef] [PubMed]
- Carlson, M.R.J.; Pope, W.B.; Horvath, S.; Braunstein, J.G.; Nghiemphu, P.; Tso, C.L.; Mellinghoff, I.; Lai, A.; Liau, L.M.; Mischel, P.S.; et al. Relationship between survival and edema in malignant gliomas: Role of vascular endothelial growth factor and neuronal pentraxin 2. Clin. Cancer Res. 2007, 13, 2592–2598. [Google Scholar] [CrossRef] [PubMed]
- Colen, R.R.; Vangel, M.; Wang, J.X.; Gutman, D.A.; Hwang, S.N.; Wintermark, M.; Jain, R.; Jilwan-Nicolas, M.; Chen, J.Y.; Raghavan, P.; et al. Imaging genomic mapping of an invasive mri phenotype predicts patient outcome and metabolic dysfunction: A TCGA glioma phenotype research group project. BMC Med. Genom. 2014, 7. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.; Poisson, L.; Narang, J.; Scarpace, L.; Rosenblum, M.L.; Rempel, S.; Mikkelsen, T. Correlation of perfusion parameters with genes related to angiogenesis regulation in glioblastoma: A feasibility study. Am. J. Neuroradiol. 2012, 33, 1343–1348. [Google Scholar] [CrossRef] [PubMed]
- Naeini, K.M.; Pope, W.B.; Cloughesy, T.F.; Harris, R.J.; Lai, A.; Eskin, A.; Chowdhury, R.; Phillips, H.S.; Nghiemphu, P.L.; Behbahanian, Y.; et al. Identifying the mesenchymal molecular subtype of glioblastoma using quantitative volumetric analysis of anatomic magnetic resonance images. Neuro Oncol. 2013, 15, 626–634. [Google Scholar] [CrossRef] [PubMed]
- Nicolasjilwan, M.; Hu, Y.; Yan, C.H.; Meerzaman, D.; Holder, C.A.; Gutman, D.; Jain, R.; Colen, R.; Rubin, D.L.; Zinn, P.O.; et al. Addition of MR imaging features and genetic biomarkers strengthens glioblastoma survival prediction in TCGA patients. J. Neuroradiol. 2015, 42, 212–221. [Google Scholar] [CrossRef] [PubMed]
- Pope, W.B.; Prins, R.M.; Thomas, M.A.; Nagarajan, R.; Yen, K.E.; Bittinger, M.A.; Salamon, N.; Chou, A.P.; Yong, W.H.; Soto, H.; et al. Non-invasive detection of 2-hydroxyglutarate and other metabolites in IDH1 mutant glioma patients using magnetic resonance spectroscopy. J. Neuro Oncol. 2012, 107, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Demerath, T.; Simon-Gabriel, C.P.; Kellner, E.; Schwarzwald, R.; Lange, T.; Heiland, D.H.; Reinacher, P.; Staszewski, O.; Mast, H.; Kiselev, V.G.; et al. Mesoscopic imaging of glioblastomas: Are diffusion, perfusion and spectroscopic measures influenced by the radiogenetic phenotype? Neuroradiol. J. 2017, 30, 36–47. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Ren, S.; Tha, K.K.; Wu, J.; Shirato, H.; Li, R. Volume of high-risk intratumoral subregions at multi-parametric MR imaging predicts overall survival and complements molecular analysis of glioblastoma. Eur. Radiol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Gevaert, O.; Echegaray, S.; Khuong, A.; Hoang, C.D.; Shrager, J.B.; Jensen, K.C.; Berry, G.J.; Guo, H.H.; Lau, C.; Plevritis, S.K.; et al. Predictive radiogenomics modeling of EGFR mutation status in lung cancer. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Gevaert, O.; Xu, J.J.; Hoang, C.D.; Leung, A.N.; Xu, Y.; Quon, A.; Rubin, D.L.; Napel, S.; Plevritis, S.K. Non-small cell lung cancer: Identifying prognostic imaging biomarkers by leveraging public gene expression microarray data-methods and preliminary results. Radiology 2012, 264, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Nair, V.S.; Gevaert, O.; Davidzon, G.; Plevritis, S.K.; West, R. NF-κB protein expression associates with F-18-FDG PET tumor uptake in non-small cell lung cancer: A radiogenomics validation study to understand tumor metabolism. Lung Cancer 2014, 83, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Karlo, C.A.; Di Paolo, P.L.; Chaim, J.; Hakimi, A.A.; Ostrovnaya, I.; Russo, P.; Hricak, H.; Motzer, R.; Hsieh, J.J.; Akin, O. Radiogenomics of clear cell renal cell carcinoma: Associations between CT imaging features and mutations. Radiology 2014, 270, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Shinagare, A.B.; Vikram, R.; Jaffe, C.; Akin, O.; Kirby, J.; Huang, E.; Freymann, J.; Sainani, N.I.; Sadow, C.A.; Bathala, T.K.; et al. Radiogenomics of clear cell renal cell carcinoma: Preliminary findings of the cancer genome Atlas-Renal Cell Carcinoma (TCGA-RCC) imaging research group. Abdom. Imaging 2015, 40, 1684–1692. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhou, Z.; Thomas, K.; Wang, J. Mo-de-207b-05: Predicting gene mutations in renal cell carcinoma based on CT imaging features: Validation using TCGA-TCIA datasets. Med. Phys. 2016, 43, 3705. [Google Scholar] [CrossRef]
- Segal, E.; Sirlin, C.B.; Ooi, C.; Adler, A.S.; Gollub, J.; Chen, X.; Chan, B.K.; Matcuk, G.R.; Barry, C.T.; Chang, H.Y.; et al. Decoding global gene expression programs in liver cancer by noninvasive imaging. Nat. Biotechnol. 2007, 25, 675–680. [Google Scholar] [CrossRef] [PubMed]
- Miura, T.; Ban, D.; Tanaka, S.; Mogushi, K.; Kudo, A.; Matsumura, S.; Mitsunori, Y.; Ochiai, T.; Tanaka, H.; Tanabe, M. Distinct clinicopathological phenotype of hepatocellular carcinoma with ethoxybenzyl-magnetic resonance imaging hyperintensity: Association with gene expression signature. Am. J. Surg. 2015, 210, 561–569. [Google Scholar] [CrossRef] [PubMed]
- McCann, S.M.; Jiang, Y.L.; Fan, X.B.; Wang, J.N.; Antic, T.; Prior, F.; VanderWeele, D.; Oto, A. Quantitative multiparametric MRI features and pten expression of peripheral zone prostate cancer: A pilot study. Am. J. Roentgenol. 2016, 206, 559–565. [Google Scholar] [CrossRef] [PubMed]
- Stoyanova, R.; Pollack, A.; Takhar, M.; Lynne, C.; Parra, N.; Lam, L.L.C.; Alshalalfa, M.; Buerki, C.; Castillo, R.; Jorda, M.; et al. Association of multiparametric MRI quantitative imaging features with prostate cancer gene expression in MRI-targeted prostate biopsies. Oncotarget 2016, 7, 53362–53376. [Google Scholar] [CrossRef] [PubMed]
- Untch, M.; Gerber, B.; Harbeck, N.; Jackisch, C.; Marschner, N.; Mobus, V.; von Minckwitz, G.; Loibl, S.; Beckmann, M.W.; Blohmer, J.U.; et al. 13th St. Gallen international breast cancer conference 2013: Primary therapy of early breast cancer evidence, controversies, consensus—Opinion of a german team of experts (Zurich 2013). Breast Care 2013, 8, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Mirabelli, P.; Incoronato, M.; Coppola, L.; Infante, T.; Parente, C.A.; Nicolai, E.; Soricelli, A.; Salvatore, M. SDN biobank: Bioresource of human samples associated with functional and/or morphological bioimaging results for the study of oncological, cardiological, neurological, and metabolic diseases. Open J. Bioresour. 2017, 4. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Tumour type | Rationale of the Study | Number of sample | Imaging data | Imaging Features | Segmentation | Genomic Features | Statistical Analysis | Ref. |
|---|---|---|---|---|---|---|---|---|
| BREAST CANCER (BC) | Correlation | 275 | MRI (T1WI, T2WI, DCE) | Shape-based features, second- and higher-order statistics, kinetic parameters | Semi-automated | IHC | Binary multivariate logistic regression model and univariate models | [103] |
| Prediction | 91 | MRI (DCE) | Shape-based features, second-order statistics, kinetic parameters | Semi-automated | RNA Seq Microarray (TCGA) | Logistic regression with LASSO regularization and ROC analysis | [97] | |
| Correlation | 48 | MRI (T1WI, T2WI, DCE) | Shape-based features, second-order statistics, kinetic parameters | Semi-automated | IHC | Multivariate logistic regression models | [104] | |
| Correlation | 221 | MRI (T1WI, T2WI, DCE) | Semantic features | Manual | IHC | Wilcoxon test and Fisher’s tests | [105] | |
| Correlation Prediction | 95 | MRI (T1WI, T2WI, DCE) | Shape-based features, first- and second-order statistics | Manual | IHC Microarray | Multiple linear regression analysis and Spearman’s rank correlation | [106] | |
| Correlation | 178 | MRI (T1WI, T2WI, DCE) | Shape-based features, first- and second-order statistics | Manual | IHC | Multiclass support vector machines with the a leave-one-out cross-validation approach | [107] | |
| Correlation | 176 | MRI (T2WI DCE) | Semantic features | Manual | IHC | Chi-squared and Fisher’s tests | [108] | |
| Correlation | 353 | MRI (T1WI, DCE) | Semantic features | Manual | Microarray | Spearman rank-correlation | [109] | |
| Correlation | 109 | MRI (T1WI, DCE) | Shape-based features, first- and second-order statistics, kinetic parameters | Automated | RNA Seq | Cox regression analysis | [110] | |
| Correlation | 92 | MRI (T2WI, DWI, DCE) | Semantic features, ADC | Manual | IHC | Mann–Whitney U and Kruskal–Wallis H tests | [111] | |
| Correlation | 115 | MRI (T1WI, T2WI, DWI, DCE) | ADC | Manual | IHC | Mann–Whitney U and Kruskal–Wallis H tests | [112] | |
| Prediction | 50 | MRI (T1WI, T2WI, DCE) | Semantic features | Manual | IHC | Student’s unpaired t-test, one-way ANOVA, Chi-squared and Fisher’s test | [113] | |
| Correlation | 282 | MRI (T1WI, T2WI, DCE) | Shape-based features | Manual | IHC | Multiple linear regression analysis | [114] | |
| Prediction | 96 | MRI (T1WI, T2WI, DWI, DCE) | Shape-based features, ADC | Manual | IHC | Multivariate logistic regression analysis | [115] | |
| Prediction | 214 | MRI (T1WI, T2WI, DWI, DCE) PET/CT | ADC, SUV | Manual | IHC | Mann–Whitney U and Kruskal–Wallis H tests | [116] | |
| Correlation | 103 | PET/CT | SUV | Manual | IHC | Chi-squared test, Fisher’s and Wilcoxon tests | [117] | |
| Correlation | 552 | PET/CT | SUV | Manual | IHC | Univariate and multiple linear regression analysis | [118] | |
| Correlation | 82 | PET/CT | SUV | Manual | IHC | Chi-squared test, Fisher’s and Mann Whitney tests | [119] | |
| Correlation | 91 | MRI (DCE) | Shape-based features, second-order statistics, kinetic parameters | Semi-automated | Microarray (TGCA) | Regression and clustering analysis | [91] | |
| Correlation | 228 | MRI (T2WI, DCE) | Kinetic parameters | Semi-automated | IHC | Kruskal–Wallis H test | [120] | |
| Prediction | 36 | MRI (DCE) | Kinetic parameters | Manual | IHC Microarray | Wilcoxon test, Spearman’s rank correlation, and Kruskal–Wallis H test | [121] | |
| Correlation | 36 | PET | SUV | Manual | IHC Microarray | Two-way unsupervised hierarchic clustering and Spearman’s rank correlation | [122] | |
| Correlation | 18 | PET | SUV | Manual | Microarray | Rank-rank hypergeometric overlap | [123] | |
| GLIOBLASTOMA (GBM) | Correlation Prediction | 25 | MRI | Semantic features | Manual | Microarray | Correlation analysis | [124] |
| Correlation | 78 | MRI-FLAIR, T1-c | Size, volume | Automated | TCGA | Pathways genomic analysis | [125] | |
| Correlation | 23 | MRI (T1-c, DSC) | Semantic features | Manual | Microarray (GSEA) | Correlation analysis | [126] | |
| Correlation Prediction | 76 | MRI (TCIA) MRI (T1-c, FLAIR) | Semantic features | Semi-automated | Microarray (TCGA) | Student’s t-test ,ROC AUC analysis | [127] | |
| Correlation Prediction | 92 | MRI (TCIA) | Semantic features | Manual | Microarray (TCGA) | Hierarchical clustering and survival analysis | [128] | |
| Correlation | 48 | MRI anatomical | Second-order statistics | NA | CGH array exome sequencing | Multivariate predictive decision-tree models | [129] | |
| Correlation Prediction | 55 | MRI (TCIA) | Semantic features | Manual | Microarray (TCGA) | Cox proportional hazards modeling and correlation analysis | [130] | |
| Correlation | 21 | MRI (DSC) | Mean values | Manual | Microarray | Cox regression analysis | [131] | |
| Correlation | 152 | MRI (DWI,DSC, SWI,T1WI,T2W2) | First-order statistics, Semantic features | Manual | Microarray | Hierarchical clustering | [132] | |
| Correlation | 13 | MRI (DWI, DSC) | Mean values | Manual | Microarray | Correlation analysis | [133] | |
| Correlation Prediction | 52 | MRI (T1-c,DSC) | Clinical scores | NA | Microarray | Univariate Cox proportional hazard models | [134] | |
| Prediction | 78 | MRI (T1-c, DSC) | Semantic features | Manual | Microarray (TGCA) | Proportional Hazards Model | [135] | |
| Prediction | 71 | MRI | Clinical scores | NA | Microarray | Multivariate Cox proportional hazard models | [136] | |
| Prediction | 104 | MRI (T1, T2 CE) | Semantic features | Manual | Microarray (TGCA) | Univariate proportional hazards regression | [137] | |
| Correlation | 18 | perfusion CT | Perfusion parameters | Manual | Microarray (TGCA) | Correlation analysis | [138] | |
| Correlation | 46 | MRI (DCE, FLAIR) | Semantic features | Manual | Microarray | Kruskal – Wallis H test | [139] | |
| Prediction | 68 | MRI (DCE, DWI, anatomy ) | Semantic features | Manual | Microarray (TGCA) | Univariate Cox Regression models | [140] | |
| Correlation | 27 | MRS | Metabolite concentration | Manual | IHC, PCR | Correlation analysis | [141] | |
| Correlation | 26 | MRI (DCE, DWI, DSC, MRS) | Semantic features | Manual | IHC | Correlation analysis | [142] | |
| Prediction | 108 | MRI (DCE, DWI) | Semantic features | Manual | Microarray (TGCA) | NA | [143] | |
| LUNG | Prediction | 186 | CT | Semantic features | Manual | PCR | Univariate analysis and multivariate decision tree models | [144] |
| Prediction | 138 | PET/CT | Shape-based feature, second-order statistics, semantic features | Manual | Microarray | Generalized linear regression with LASSO regularization | [145] | |
| Correlation Prediction | 355 | PET/CT | SUV | Manual | Microarray | Student’s t-test, Wilcoxon test, Chi-squared and Fisher’s test | [146] | |
| Prediction | 422 | CT | Shape-based features, first-, second- and higher-order statistics | Manual | Microarray | Intraclass correlation coefficient, Friedman test | [36] | |
| KIDNEY | Prediction | 70 | CT | First-order statistics, semantic features | Manual | Microarray | Multivariate linear regression | [99] |
| Correlation | 233 | CT | Shape-based features, first-order statistics, Semantic features | Manual | DNA-Seq (TCGA) | Fisher’s tests | [147] | |
| Correlation | 103 | CT and MRI | Shape-based features, first-order statistics, Semantic features | Manual | Microarray (TCGA) | Pearson’s test and Mann–Whitney U test | [148] | |
| Prediction | 58 | CT (TCIA) | Shape-based features, first- and second-order statistics | Manual | Microarray (TCGA) | Support vector machine classifier | [149] | |
| LIVER (HCC) | Correlation | 30 | DCE-CT | Semantic features | NA | Microarray | Correlation analysis | [94] |
| Correlation Prediction | 47 | three-phase contrast enhanced CT | Semantic features | NA | Microarray | Bayesian models | [150] | |
| Correlation | 77 | Liver-specific contrast enhanced-MRI | Clinical scores | NA | IHC Microarray | Student’s t-test | [151] | |
| PROSTATE | Correlation | 45 | MRI (T1WI, T2WI, DWI, DCE) | First-order statistics, kinetic parameters, ADC | Manual | IHC | Spearman’s rank correlation coefficient | [152] |
| Prediction | 17 | MRI (T2WI, DWI, DCE) | First-order statistics, kinetic parameters, ADC | Semi-automated | Microarray | Pearson’s correlation, two-way hierarchical clustering | [153] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Incoronato, M.; Aiello, M.; Infante, T.; Cavaliere, C.; Grimaldi, A.M.; Mirabelli, P.; Monti, S.; Salvatore, M. Radiogenomic Analysis of Oncological Data: A Technical Survey. Int. J. Mol. Sci. 2017, 18, 805. https://doi.org/10.3390/ijms18040805
Incoronato M, Aiello M, Infante T, Cavaliere C, Grimaldi AM, Mirabelli P, Monti S, Salvatore M. Radiogenomic Analysis of Oncological Data: A Technical Survey. International Journal of Molecular Sciences. 2017; 18(4):805. https://doi.org/10.3390/ijms18040805
Chicago/Turabian StyleIncoronato, Mariarosaria, Marco Aiello, Teresa Infante, Carlo Cavaliere, Anna Maria Grimaldi, Peppino Mirabelli, Serena Monti, and Marco Salvatore. 2017. "Radiogenomic Analysis of Oncological Data: A Technical Survey" International Journal of Molecular Sciences 18, no. 4: 805. https://doi.org/10.3390/ijms18040805
APA StyleIncoronato, M., Aiello, M., Infante, T., Cavaliere, C., Grimaldi, A. M., Mirabelli, P., Monti, S., & Salvatore, M. (2017). Radiogenomic Analysis of Oncological Data: A Technical Survey. International Journal of Molecular Sciences, 18(4), 805. https://doi.org/10.3390/ijms18040805