Regulation of DNA Repair Mechanisms: How the Chromatin Environment Regulates the DNA Damage Response


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Repairing Damaged DNA: NER and DSB Repair
3. Phosphorylation Cascades Regulate the DDR
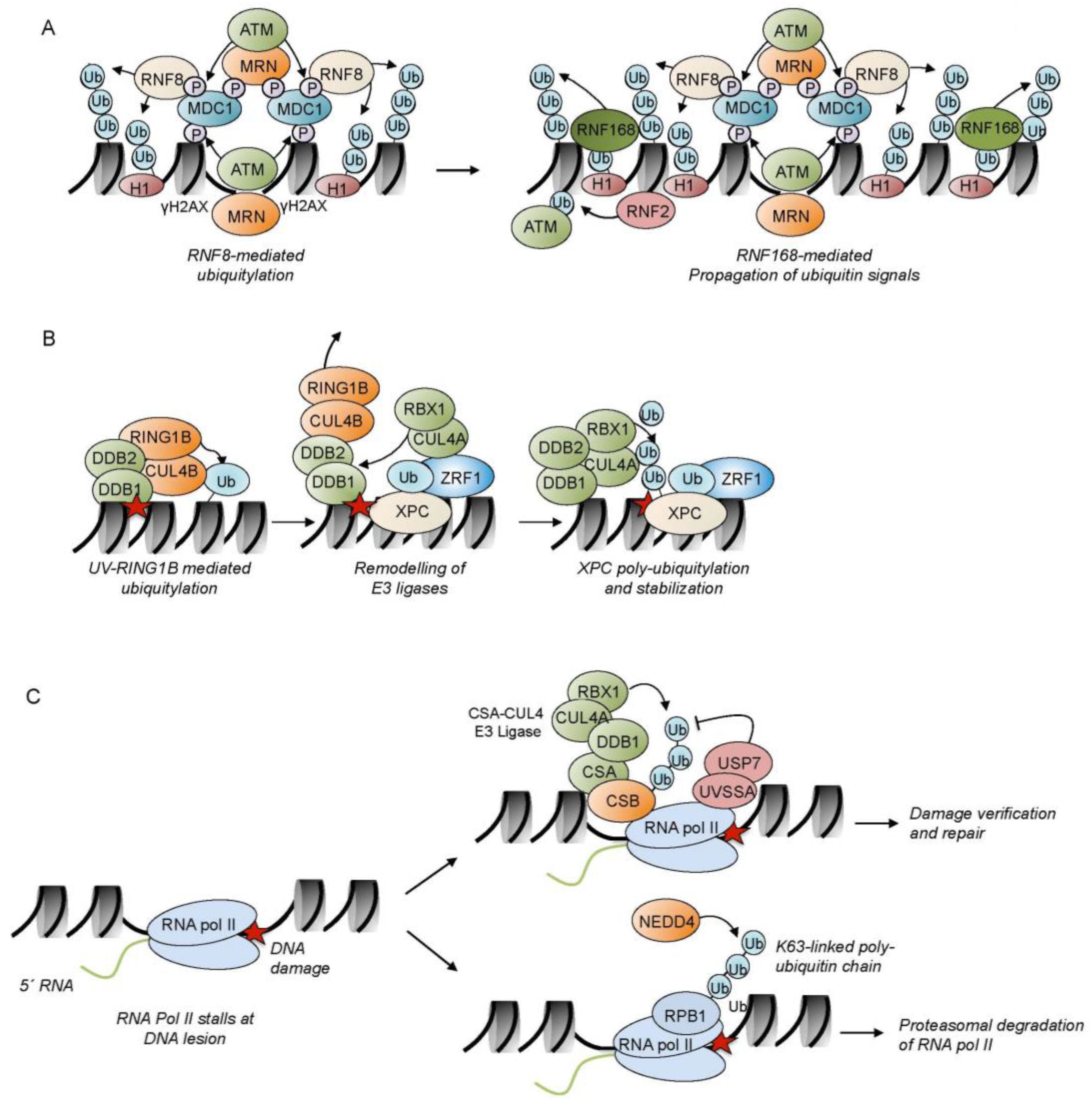
4. Driving the DDR by Ubiquitylation
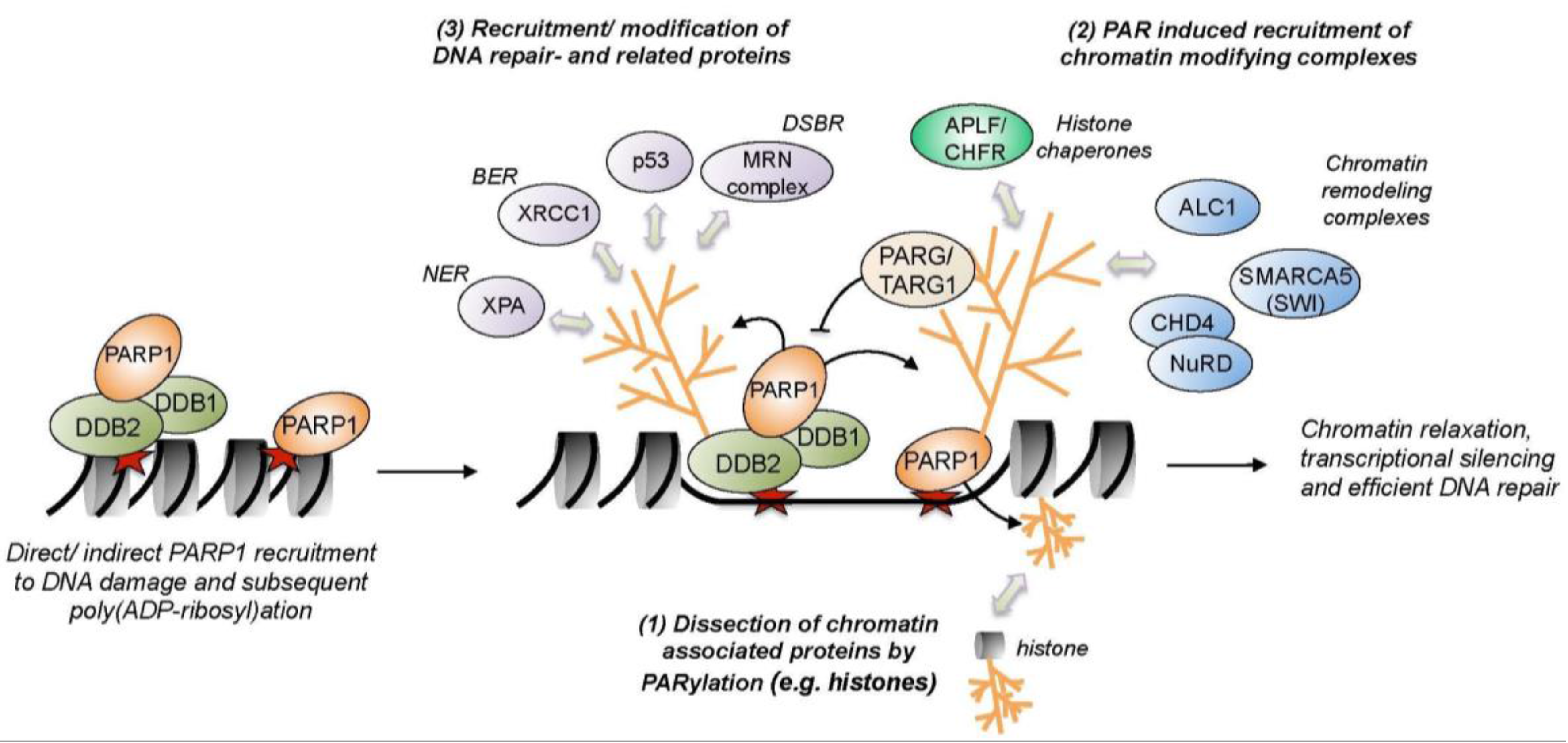
5. Impact of Chromatin Remodeling and PARylation on DNA Repair
6. Role of ncRNAs and DICER
7. Concluding Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ciccia, A.; Elledge, S.J. The DNA Damage Response: Making It Safe to Play with Knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- De Graaf, C.A.; van Steensel, B. Chromatin organization: Form to function. Curr. Opin. Genet. Dev. 2013, 23, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Smerdon, M.J. DNA repair and the role of chromatin structure. Curr. Opin. Cell Biol. 1991, 3, 422–428. [Google Scholar] [CrossRef]
- Soria, G.; Polo, S.E.; Almouzni, G. Prime, repair, restore: The active role of chromatin in the DNA damage response. Mol. Cell 2012, 46, 722–734. [Google Scholar] [CrossRef] [PubMed]
- Cleaver, J.E. Nucleosome structure controls rates of excision repair in DNA of human cells. Nature 1977, 270, 451–453. [Google Scholar] [CrossRef] [PubMed]
- Smerdon, M.J.; Lieberman, M.W. Nucleosome rearrangement in human chromatin during UV-induced DNA-reapir synthesis. Proc. Natl. Acad. Sci. USA 1978, 75, 4238–4241. [Google Scholar] [CrossRef] [PubMed]
- Adam, S.; Dabin, J.; Polo, S.E. Chromatin plasticity in response to DNA damage: The shape of things to come. DNA Repair 2015, 32, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Clapier, C.R.; Iwasa, J.; Cairns, B.R.; Peterson, C.L. Mechanisms of action and regulation of ATP-dependent chromatin-remodelling complexes. Nat. Rev. Mol. Cell Biol. 2017, 18, 407–422. [Google Scholar] [CrossRef] [PubMed]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [PubMed]
- De Laat, W.L.; Jaspers, N.G.; Hoeijmakers, J.H. Molecular mechanism of nucleotide excision repair. Genes Dev. 1999, 13, 768–785. [Google Scholar] [CrossRef] [PubMed]
- Fousteri, M.; Mullenders, L.H. Transcription-coupled nucleotide excision repair in mammalian cells: Molecular mechanisms and biological effects. Cell Res. 2008, 18, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Marteijn, J.A.; Lans, H.; Vermeulen, W.; Hoeijmakers, J.H. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat. Rev. Mol. Cell Biol. 2014, 15, 465–481. [Google Scholar] [CrossRef] [PubMed]
- Marnef, A.; Legube, G. Organizing DNA repair in the nucleus: DSBs hit the road. Curr. Opin. Cell Biol. 2017, 46, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Meek, K.; Dang, V.; Lees-Miller, S.P. DNA-PK: The means to justify the ends? Adv. Immunol. 2008, 99, 33–58. [Google Scholar] [PubMed]
- Mari, P.O.; Florea, B.I.; Persengiev, S.P.; Verkaik, N.S.; Brüggenwirth, H.T.; Modesti, M.; Giglia-Mari, G.; Bezstarosti, K.; Demmers, J.A.; Luider, T.M. Dynamic assembly of end-joining complexes requires interaction between Ku70/80 and XRCC4. Proc. Natl. Acad. Sci. USA 2006, 103, 18597–18602. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.J.; Chen, D.J. DNA double strand break repair via non-homologous end-joining. Transl. Cancer Res. 2013, 2, 130–143. [Google Scholar] [PubMed]
- Lieber, M.R. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu. Rev. Biochem. 2010, 79, 181–211. [Google Scholar] [CrossRef] [PubMed]
- Kakarougkas, A.; Jeggo, P.A. DNA DSB repair pathway choice: An orchestrated handover mechanism. Br. J. Radiol. 2014, 87, 20130685. [Google Scholar] [CrossRef] [PubMed]
- Ceccaldi, R.; Rondinelli, B.; D’Andrea, A.D. Repair Pathway Choices and Consequences at the Double-Strand Break. Trends Cell Biol. 2016, 26, 52–64. [Google Scholar] [CrossRef] [PubMed]
- Bothmer, A.; Robbiani, D.F.; Feldhahn, N.; Gazumyan, A.; Nussenzweig, A.; Nussenzweig, M.C. 53BP1 regulates DNA resection and the choice between classical and alternative end joining during class switch recombination. J. Exp. Med. 2010, 207, 855–865. [Google Scholar] [CrossRef] [PubMed]
- Marechal, A.; Zou, L. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb. Perspect. Biol. 2013, 5, a012716. [Google Scholar] [CrossRef] [PubMed]
- Paull, T.T.; Deshpande, R.A. The Mre11/Rad50/Nbs1 complex: Recent insights into catalytic activities and ATP-driven conformational changes. Exp. Cell Res. 2014, 329, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Haince, J.F.; McDonald, D.; Rodrigue, A.; Déry, U.; Masson, J.Y.; Hendzel, M.J.; Poirier, G.G. PARP1-dependent kinetics of recruitment of MRE11 and NBS1 proteins to multiple DNA damage sites. J. Biol. Chem. 2008, 283, 1197–1208. [Google Scholar] [CrossRef] [PubMed]
- Stucki, M.; Clapperton, J.A.; Mohammad, D.; Yaffe, M.B.; Smerdon, S.J.; Jackson, S.P. MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell 2005, 123, 1213–1226. [Google Scholar] [CrossRef] [PubMed]
- Melander, F.; Bekker-Jensen, S.; Falck, J.; Bartek, J.; Mailand, N.; Lukas, J. Phosphorylation of SDT repeats in the MDC1 N terminus triggers retention of NBS1 at the DNA damage-modified chromatin. J. Cell Biol. 2008, 181, 213–226. [Google Scholar] [CrossRef] [PubMed]
- Bonner, W.M.; Redon, C.E.; Dickey, J.S.; Nakamura, A.J.; Sedelnikova, O.A.; Solier, S.; Pommier, Y. GammaH2AX and cancer. Nat. Rev. Cancer 2008, 8, 957–967. [Google Scholar] [CrossRef] [PubMed]
- Doil, C.; Mailand, N.; Bekker-Jensen, S.; Menard, P.; Larsen, D.H.; Pepperkok, R.; Ellenberg, J.; Panier, S.; Durocher, D.; Bartek, J. RNF168 binds and amplifies ubiquitin conjugates on damaged chromosomes to allow accumulation of repair proteins. Cell 2009, 136, 435–446. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Durocher, D. Regulation of DNA Damage Responses by Ubiquitin and SUMO. Mol. Cell 2013, 49, 795–807. [Google Scholar] [CrossRef] [PubMed]
- Yazinski, S.A.; Zou, L. Functions, Regulation, and Therapeutic Implications of the ATR Checkpoint Pathway. Annu. Rev. Genet. 2016, 50, 155–173. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Lisby, M.; Symington, L.S. RPA coordinates DNA end resection and prevents formation of DNA hairpins. Mol. Cell 2013, 50, 589–600. [Google Scholar] [CrossRef] [PubMed]
- Zou, L.; Elledge, S.J. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 2003, 300, 1542–1548. [Google Scholar] [CrossRef] [PubMed]
- Branzei, D.; Foiani, M. Regulation of DNA repair throughout the cell cycle. Nat. Rev. Mol. Cell Biol. 2008, 9, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Auclair, Y.; Rouget, R.; Affar el, B.; Drobetsky, E.A. ATR kinase is required for global genomic nucleotide excision repair exclusively during S phase in human cells. Proc. Natl. Acad. Sci. USA 2008, 105, 17896–17901. [Google Scholar] [CrossRef] [PubMed]
- Heffernan, T.P.; Simpson, D.A.; Frank, A.R.; Heinloth, A.N.; Paules, R.S.; Cordeiro-Stone, M.; Kaufmann, W.K. An ATR- and Chk1-dependent S checkpoint inhibits replicon initiation following UVC-induced DNA damage. Mol. Cell. Biol. 2002, 22, 8552–8561. [Google Scholar] [CrossRef] [PubMed]
- Yajima, H.; Lee, K.J.; Zhang, S.; Kobayashi, J.; Chen, B.P. DNA double-strand break formation upon UV-induced replication stress activates ATM and DNA-PKcs kinases. J. Mol. Biol. 2009, 385, 800–810. [Google Scholar] [CrossRef] [PubMed]
- Ray, A.; Milum, K.; Battu, A.; Wani, G.; Wani, A.A. NER initiation factors, DDB2 and XPC, regulate UV radiation response by recruiting ATR and ATM kinases to DNA damage sites. DNA Repair 2013, 12, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Wakasugi, M.; Sasaki, T.; Matsumoto, M.; Nagaoka, M.; Inoue, K.; Inobe, M.; Horibata, K.; Tanaka, K.; Matsunaga, T. Nucleotide excision repair-dependent DNA double-strand break formation and ATM signaling activation in mammalian quiescent cells. J. Biol. Chem. 2014, 289, 28730–28737. [Google Scholar] [CrossRef] [PubMed]
- Glickman, M.H.; Ciechanover, A. The ubiquitin-proteasome proteolytic pathway: Destruction for the sake of construction. Physiol. Rev. 2002, 82, 373–428. [Google Scholar] [CrossRef] [PubMed]
- Schnell, J.D.; Hicke, L. Non-traditional functions of ubiquitin and ubiquitin-binding proteins. J. Biol. Chem. 2003, 278, 35857–35860. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, D.; Riezman, H. Proteasome-independent functions of ubiquitin in endocytosis and signaling. Science 2007, 315, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Mailand, N.; Bekker-Jensen, S.; Faustrup, H.; Melander, F.; Bartek, J.; Lukas, C.; Lukas, J. RNF8 ubiquitylates histones at DNA double-strand breaks and promotes assembly of repair proteins. Cell 2007, 131, 887–900. [Google Scholar] [CrossRef] [PubMed]
- Pan, M.R.; Peng, G.; Hung, W.C.; Lin, S.Y. Monoubiquitination of H2AX protein regulates DNA damage response signaling. J. Biol. Chem. 2011, 286, 28599–28607. [Google Scholar] [CrossRef] [PubMed]
- Thorslund, T.; Ripplinger, A.; Hoffmann, S.; Wild, T.; Uckelmann, M.; Villumsen, B.; Narita, T.; Sixma, T.K.; Choudhary, C.; Bekker-Jensen, S. Histone H1 couples initiation and amplification of ubiquitin signalling after DNA damage. Nature 2015, 527, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Mattiroli, F.; Vissers, J.H.; van Dijk, W.J.; Ikpa, P.; Citterio, E.; Vermeulen, W.; Marteijn, J.A.; Sixma, T.K. RNF168 ubiquitinates K13–15 on H2A/H2AX to drive DNA damage signaling. Cell 2012, 150, 1182–1195. [Google Scholar] [CrossRef] [PubMed]
- Sobhian, B.; Shao, G.; Lilli, D.R.; Culhane, A.C.; Moreau, L.A.; Xia, B.; Livingston, D.M.; Greenberg, R.A. RAP80 targets BRCA1 to specific ubiquitin structures at DNA damage sites. Science 2007, 316, 1198–1202. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Chen, J.; Yu, X. Ubiquitin-binding protein RAP80 mediates BRCA1-dependent DNA damage response. Science 2007, 316, 1202–1205. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Matsuoka, S.; Ballif, B.A.; Zhang, D.; Smogorzewska, A.; Gygi, S.P.; Elledge, S.J. Abraxas and RAP80 form a BRCA1 protein complex required for the DNA damage response. Science 2007, 316, 1194–1198. [Google Scholar] [CrossRef] [PubMed]
- Panier, S.; Boulton, S.J. Double-strand break repair: 53BP1 comes into focus. Nat. Rev. Mol. Cell Biol. 2014, 15, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Fradet-Turcotte, A.; Canny, M.D.; Escribano-Díaz, C.; Orthwein, A.; Leung, C.C.; Huang, H.; Landry, M.C.; Kitevski-LeBlanc, J.; Noordermeer, S.M.; Sicheri, F. 53BP1 is a reader of the DNA-damage-induced H2A Lys 15 ubiquitin mark. Nature 2013, 499, 50–54. [Google Scholar] [CrossRef] [PubMed]
- Pesavento, J.J.; Yang, H.; Kelleher, N.L.; Mizzen, C.A. Certain and progressive methylation of histone H4 at lysine 20 during the cell cycle. Mol. Cell. Biol. 2008, 28, 468–486. [Google Scholar] [CrossRef] [PubMed]
- Pei, H.; Zhang, L.; Luo, K.; Qin, Y.; Chesi, M.; Fei, F.; Bergsagel, P.L.; Wang, L.; You, Z.; Lou, Z. MMSET regulates histone H4K20 methylation and 53BP1 accumulation at DNA damage sites. Nature 2011, 470, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Ismail, I.H.; Andrin, C.; McDonald, D.; Hendzel, M.J. BMI1-mediated histone ubiquitylation promotes DNA double-strand break repair. J. Cell Biol. 2010, 191, 45–60. [Google Scholar] [CrossRef] [PubMed]
- Ginjala, V.; Nacerddine, K.; Kulkarni, A.; Oza, J.; Hill, S.J.; Yao, M.; Citterio, E.; van Lohuizen, M.; Ganesan, S. BMI1 is recruited to DNA breaks and contributes to DNA damage-induced H2A ubiquitination and repair. Mol. Cell. Biol. 2011, 31, 1972–1982. [Google Scholar] [CrossRef] [PubMed]
- Chagraoui, J.; Hebert, J.; Girard, S.; Sauvageau, G. An anticlastogenic function for the Polycomb Group gene Bmi1. Proc. Natl. Acad. Sci. USA 2011, 108, 5284–5289. [Google Scholar] [CrossRef] [PubMed]
- Ui, A.; Nagaura, Y.; Yasui, A. Transcriptional elongation factor ENL phosphorylated by ATM recruits polycomb and switches off transcription for DSB repair. Mol. Cell 2015, 58, 468–482. [Google Scholar] [CrossRef] [PubMed]
- Bergink, S.; Salomons, F.A.; Hoogstraten, D.; Groothuis, T.A.; de Waard, H.; Wu, J.; Yuan, L.; Citterio, E.; Houtsmuller, A.B.; Neefjes, J. DNA damage triggers nucleotide excision repair-dependent monoubiquitylation of histone H2A. Genes Dev. 2006, 20, 1343–1352. [Google Scholar] [CrossRef] [PubMed]
- Kapetanaki, M.G.; Guerrero-Santoro, J.; Bisi, D.C.; Hsieh, C.L.; Rapić-Otrin, V.; Levine, A.S. The DDB1-CUL4ADDB2 ubiquitin ligase is deficient in xeroderma pigmentosum group E and targets histone H2A at UV-damaged DNA sites. Proc. Natl. Acad. Sci. USA 2006, 103, 2588–2593. [Google Scholar] [CrossRef] [PubMed]
- Marteijn, J.A.; Bekker-Jensen, S.; Mailand, N.; Lans, H.; Schwertman, P.; Gourdin, A.M.; Dantuma, N.P.; Lukas, J.; Vermeulen, W. Nucleotide excision repair-induced H2A ubiquitination is dependent on MDC1 and RNF8 and reveals a universal DNA damage response. J. Cell Biol. 2009, 186, 835–847. [Google Scholar] [CrossRef] [PubMed]
- Guerrero-Santoro, J.; Kapetanaki, M.G.; Hsieh, C.L.; Gorbachinsky, I.; Levine, A.S.; Rapić-Otrin, V. The cullin 4B-based UV-damaged DNA-binding protein ligase binds to UV-damaged chromatin and ubiquitinates histone H2A. Cancer Res. 2008, 68, 5014–5022. [Google Scholar] [CrossRef] [PubMed]
- Gracheva, E.; Chitale, S.; Wilhelm, T.; Rapp, A.; Byrne, J.; Stadler, J.; Medina, R.; Cardoso, M.C.; Richly, H. ZRF1 mediates remodeling of E3 ligases at DNA lesion sites during nucleotide excision repair. J. Cell Biol. 2016, 213, 185–200. [Google Scholar] [CrossRef] [PubMed]
- Sugasawa, K.; Okuda, Y.; Saijo, M.; Nishi, R.; Matsuda, N.; Chu, G.; Mori, T.; Iwai, S.; Tanaka, K.; Tanaka, K. UV-induced ubiquitylation of XPC protein mediated by UV-DDB-ubiquitin ligase complex. Cell 2005, 121, 387–400. [Google Scholar] [CrossRef] [PubMed]
- Chitale, S.; Richly, H. Timing of DNA lesion recognition: Ubiquitin signaling in the NER pathway. Cell cycle 2017, 16, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Marnef, A.; Cohen, S.; Legube, G. Transcription-Coupled DNA Double-Strand Break Repair: Active Genes Need Special Care. J. Mol. Biol. 2017, 429, 1277–1288. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Horibata, K.; Saijo, M.; Ishigami, C.; Ukai, A.; Kanno, S.I.; Tahara, H.; Neilan, E.G.; Honma, M.; Nohmi, T. Mutations in UVSSA cause UV-sensitive syndrome and destabilize ERCC6 in transcription-coupled DNA repair. Nat. Genet. 2012, 44, 593–597. [Google Scholar] [CrossRef] [PubMed]
- Schwertman, P.; Lagarou, A.; Dekkers, D.H.; Raams, A.; van der Hoek, A.C.; Laffeber, C.; Hoeijmakers, J.H.; Demmers, J.A.; Fousteri, M.; Vermeulen, W. UV-sensitive syndrome protein UVSSA recruits USP7 to regulate transcription-coupled repair. Nat. Genet. 2012, 44, 598–602. [Google Scholar] [CrossRef] [PubMed]
- Sarasin, A. UVSSA and USP7: New players regulating transcription-coupled nucleotide excision repair in human cells. Genome Med. 2012, 4, 44. [Google Scholar] [CrossRef] [PubMed]
- Yasukawa, T.; Kamura, T.; Kitajima, S.; Conaway, R.C.; Conaway, J.W.; Aso, T. Mammalian Elongin A complex mediates DNA-damage-induced ubiquitylation and degradation of Rpb1. EMBO J. 2008, 27, 3256–3266. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.D.; Harreman, M.; Svejstrup, J.Q. Ubiquitylation and degradation of elongating RNA polymerase II: The last resort. Biochim. Biophys. Acta Gene Regul. Mech. 2013, 1829, 151–157. [Google Scholar] [CrossRef] [PubMed]
- De Vos, M.; Schreiber, V.; Dantzer, F. The diverse roles and clinical relevance of PARPs in DNA damage repair: Current state of the art. Biochem. Pharmacol. 2012, 84, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Robu, M.; Shah, R.G.; Petitclerc, N.; Brind’Amour, J.; Kandan-Kulangara, F.; Shah, G.M. Role of poly(ADP-ribose) polymerase-1 in the removal of UV-induced DNA lesions by nucleotide excision repair. Proc. Natl. Acad. Sci. USA 2013, 110, 1658–1663. [Google Scholar] [CrossRef] [PubMed]
- Chou, D.M.; Adamson, B.; Dephoure, N.E.; Tan, X.; Nottke, A.C.; Hurov, K.E.; Gygi, S.P.; Colaiácovo, M.P.; Elledge, S.J. A chromatin localization screen reveals poly (ADP ribose)-regulated recruitment of the repressive polycomb and NuRD complexes to sites of DNA damage. Proc. Natl. Acad. Sci. USA 2010, 107, 18475–18480. [Google Scholar] [CrossRef] [PubMed]
- Polo, S.E.; Kaidi, A.; Baskcomb, L.; Galanty, Y.; Jackson, S.P. Regulation of DNA-damage responses and cell-cycle progression by the chromatin remodelling factor CHD4. EMBO J. 2010, 29, 3130–3139. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.P.; Ryan, D.P.; Galanty, Y.; Low, J.K.; Vandevenne, M.; Jackson, S.P.; Mackay, J.P. The N-terminal Region of Chromodomain Helicase DNA-binding Protein 4 (CHD4) Is Essential for Activity and Contains a High Mobility Group (HMG) Box-like-domain That Can Bind Poly(ADP-ribose). J. Biol. Chem. 2016, 291, 924–938. [Google Scholar] [CrossRef] [PubMed]
- Larsen, D.H.; Poinsignon, C.; Gudjonsson, T.; Dinant, C.; Payne, M.R.; Hari, F.J.; Danielsen, J.M.R.; Menard, P.; Sand, J.C.; Stucki, M. The chromatin-remodeling factor CHD4 coordinates signaling and repair after DNA damage. J. Cell Biol. 2010, 190, 731–740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smeenk, G.; Wiegant, W.W.; Vrolijk, H.; Solari, A.P.; Pastink, A.; van Attikum, H. The NuRD chromatin-remodeling complex regulates signaling and repair of DNA damage. J. Cell Biol. 2010, 190, 741–749. [Google Scholar] [CrossRef] [PubMed]
- Ahel, D.; Hořejší, Z.; Wiechens, N.; Polo, S.E.; Garcia-Wilson, E.; Ahel, I.; Flynn, H.; Skehel, M.; West, S.C.; Jackson, S.P. Poly(ADP-ribose)-dependent regulation of DNA repair by the chromatin remodeling enzyme ALC1. Science 2009, 325, 1240–1243. [Google Scholar] [CrossRef] [PubMed]
- Gottschalk, A.J.; Timinszky, G.; Kong, S.E.; Jin, J.; Cai, Y.; Swanson, S.K.; Washburn, M.P.; Florens, L.; Ladurner, A.G.; Conaway, J.W. Poly(ADP-ribosyl)ation directs recruitment and activation of an ATP-dependent chromatin remodeler. Proc. Natl. Acad. Sci. USA 2009, 106, 13770–13774. [Google Scholar] [CrossRef] [PubMed]
- Downs, J.A.; Allard, S.; Jobin-Robitaille, O.; Javaheri, A.; Auger, A.; Bouchard, N.; Kron, S.J.; Jackson, S.P.; Côté, J. Binding of chromatin-modifying activities to phosphorylated histone H2A at DNA damage sites. Mol. Cell 2004, 16, 979–990. [Google Scholar] [CrossRef] [PubMed]
- Price, B.D.; D’Andrea, A.D. Chromatin remodeling at DNA double-strand breaks. Cell 2013, 152, 1344–1354. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Sun, Y.; Jiang, X.; Ayrapetov, M.K.; Moskwa, P.; Yang, S.; Weinstock, D.M.; Price, B.D. The p400 ATPase regulates nucleosome stability and chromatin ubiquitination during DNA repair. J. Cell Biol. 2010, 191, 31–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bird, A.W.; Yu, D.Y.; Pray-Grant, M.G.; Qiu, Q. Acetylation of histone H4 by Esa1 is required for DNA double-strand break repair. Nature 2002, 419, 411–415. [Google Scholar] [CrossRef] [PubMed]
- Ikura, T.; Tashiro, S.; Kakino, A.; Shima, H.; Jacob, N.; Amunugama, R.; Yoder, K.; Izumi, S.; Kuraoka, I.; Tanaka, K. DNA damage-dependent acetylation and ubiquitination of H2AX enhances chromatin dynamics. Mol. Cell. Biol. 2007, 27, 7028–7040. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Jiang, X.; Chen, S.; Fernandes, N.; Price, B.D. A role for the Tip60 histone acetyltransferase in the acetylation and activation of ATM. Proc. Natl. Acad. Sci. USA 2005, 102, 13182–13187. [Google Scholar] [CrossRef] [PubMed]
- Pines, A.; Vrouwe, M.G.; Marteijn, J.A.; Typas, D.; Luijsterburg, M.S.; Cansoy, M.; Hensbergen, P.; Deelder, A.; de Groot, A.; Matsumoto, S. PARP1 promotes nucleotide excision repair through DDB2 stabilization and recruitment of ALC1. J. Cell Biol. 2012, 199, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Maltseva, E.A.; Rechkunova, N.I.; Sukhanova, M.V.; Lavrik, O.I. Poly(ADP-ribose) Polymerase 1 Modulates Interaction of the Nucleotide Excision Repair Factor XPC-RAD23B with DNA via Poly(ADP-ribosyl)ation. J. Biol. Chem. 2015, 290, 21811–21820. [Google Scholar] [CrossRef] [PubMed]
- Fischer, J.M.; Popp, O.; Gebhard, D.; Veith, S.; Fischbach, A.; Beneke, S.; Leitenstorfer, A.; Bergemann, J.; Scheffner, M.; Ferrando-May, E. Poly(ADP-ribose)-mediated interplay of XPA and PARP1 leads to reciprocal regulation of protein function. FEBS J. 2014, 281, 3625–3641. [Google Scholar] [CrossRef] [PubMed]
- Thorslund, T.; von Kobbe, C.; Harrigan, J.A.; Indig, F.E.; Christiansen, M.; Stevnsner, T.; Bohr, V.A. Cooperation of the Cockayne syndrome group B protein and poly(ADP-ribose) polymerase 1 in the response to oxidative stress. Mol. Cell. Biol. 2005, 25, 7625–7636. [Google Scholar] [CrossRef] [PubMed]
- Fousteri, M.; Vermeulen, W.; van Zeeland, A.A.; Mullenders, L.H. Cockayne syndrome A and B proteins differentially regulate recruitment of chromatin remodeling and repair factors to stalled RNA polymerase II in vivo. Mol. Cell 2006, 23, 471–482. [Google Scholar] [CrossRef] [PubMed]
- Dinant, C.; Ampatziadis-Michailidis, G.; Lans, H.; Tresini, M.; Lagarou, A.; Grosbart, M.; Theil, A.F.; van Cappellen, W.A.; Kimura, H.; Bartek, J. Enhanced chromatin dynamics by FACT promotes transcriptional restart after UV-induced DNA damage. Mol. Cell 2013, 51, 469–479. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, K.L.; Smith, J.J.; Dasgupta, A.; Maqani, N.; Grant, P.; Auble, D.T. The NEF4 complex regulates Rad4 levels and utilizes Snf2/Swi2-related ATPase activity for nucleotide excision repair. Mol. Cell. Biol. 2004, 24, 6362–6378. [Google Scholar] [CrossRef] [PubMed]
- Jin, Q.; Yu, L.R.; Wang, L.; Zhang, Z.; Kasper, L.H.; Lee, J.E.; Wang, C.; Brindle, P.K.; Dent, S.Y.; Ge, K. Distinct roles of GCN5/PCAF-mediated H3K9ac and CBP/p300-mediated H3K18/27ac in nuclear receptor transactivation. EMBO J. 2011, 30, 249–262. [Google Scholar] [CrossRef] [PubMed]
- Gong, F.; Fahy, D.; Smerdon, M.J. Rad4-Rad23 interaction with SWI/SNF links ATP-dependent chromatin remodeling with nucleotide excision repair. Nat. Struct. Mol. Biol. 2006, 13, 902–907. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Wang, X.; Bao, S.; Guo, R.; Johnson, D.G.; Shen, X.; Li, L. INO80 chromatin remodeling complex promotes the removal of UV lesions by the nucleotide excision repair pathway. Proc. Natl. Acad. Sci. USA 2010, 107, 17274–17279. [Google Scholar] [CrossRef] [PubMed]
- Holoch, D.; Moazed, D. RNA-mediated epigenetic regulation of gene expression. Nat. Rev. Genet. 2015, 16, 71–84. [Google Scholar] [CrossRef] [PubMed]
- Francia, S.; Michelini, F.; Saxena, A.; Tang, D.; de Hoon, M.; Anelli, V.; Mione, M.; Carninci, P.; di Fagagna, F.D.A. Site-specific DICER and DROSHA RNA products control the DNA-damage response. Nature 2012, 488, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Ba, Z.; Gao, M.; Wu, Y.; Ma, Y.; Amiard, S.; White, C.I.; Danielsen, J.M.R.; Yang, Y.G.; Qi, Y. A role for small RNAs in DNA double-strand break repair. Cell 2012, 149, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Michalik, K.M.; Bottcher, R.; Forstemann, K. A small RNA response at DNA ends in Drosophila. Nucleic Acids Res. 2012, 40, 9596–9603. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Wei, W.; Li, M.M.; Wu, Y.S.; Ba, Z.; Jin, K.X.; Li, M.M.; Liao, Y.Q.; Adhikari, S.; Chong, Z. Ago2 facilitates Rad51 recruitment and DNA double-strand break repair by homologous recombination. Cell Res. 2014, 24, 532–541. [Google Scholar] [CrossRef] [PubMed]
- Francia, S.; Cabrini, M.; Matti, V.; Oldani, A.; di Fagagna, F.D.A. DICER, DROSHA and DNA damage response RNAs are necessary for the secondary recruitment of DNA damage response factors. J. Cell Sci. 2016, 129, 1468–1476. [Google Scholar] [CrossRef] [PubMed]
- Chitale, S.; Richly, H. DICER and ZRF1 contribute to chromatin decondensation during nucleotide excision repair. Nucleic Acids Res. 2017, 45, 5901–5912. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulou, T.; Kaymak, A.; Sayols, S.; Richly, H. Dual role of Med12 in PRC1-dependent gene repression and ncRNA-mediated transcriptional activation. Cell Cycle 2016, 15, 1479–1493. [Google Scholar] [CrossRef] [PubMed]



© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stadler, J.; Richly, H. Regulation of DNA Repair Mechanisms: How the Chromatin Environment Regulates the DNA Damage Response. Int. J. Mol. Sci. 2017, 18, 1715. https://doi.org/10.3390/ijms18081715
Stadler J, Richly H. Regulation of DNA Repair Mechanisms: How the Chromatin Environment Regulates the DNA Damage Response. International Journal of Molecular Sciences. 2017; 18(8):1715. https://doi.org/10.3390/ijms18081715
Chicago/Turabian StyleStadler, Jens, and Holger Richly. 2017. "Regulation of DNA Repair Mechanisms: How the Chromatin Environment Regulates the DNA Damage Response" International Journal of Molecular Sciences 18, no. 8: 1715. https://doi.org/10.3390/ijms18081715
APA StyleStadler, J., & Richly, H. (2017). Regulation of DNA Repair Mechanisms: How the Chromatin Environment Regulates the DNA Damage Response. International Journal of Molecular Sciences, 18(8), 1715. https://doi.org/10.3390/ijms18081715